

Abstract
LPS challenge causes potent activation of innate immunity. Because LPS is ubiquitously present in ambient air, repeated inhalation may lead to activation of the pulmonary immune response. If this activation is unregulated, chronic LPS inhalation would lead to persistent inflammation and organ damage. We hypothesized that the lung uses the mechanism of LPS tolerance to maintain the balance between hypoinflammatory and hyperinflammatory states. We developed a model of chronic pulmonary LPS tolerance induced by pulmonary exposure to 1 μg LPS for 4 consecutive days. Mice were challenged with 10 μg of LPS 24 h later. TNF-α protein was significantly decreased in the bronchoalveolar lavage fluid of tolerant versus nontolerant mice, whereas IL-6 levels were significantly increased in the tolerant group. Tolerant mice were also protected from airway hyperresponsiveness. M2 and M3 muscarinic receptor mRNA was significantly decreased in the lungs of tolerant mice, suggesting a mechanism for the decreased airway hyperresponsiveness. CXCL2 was significantly reduced in tolerant mice, but CXCL1 was equivalent between groups. No difference was seen in neutrophil recruitment to the alveolar space. Interestingly, LPS tolerance does not confer cross-tolerance to the Toll-like receptor (TLR) 2 stimulus Pam3Cys. TNF-α and IL-6 concentrations were significantly increased in LPS-tolerant mice challenged with Pam3Cys; however, chemokine concentrations were unaffected. Our data show that repeated LPS inhalation results in differential regulation of cytokines but does not inhibit neutrophil recruitment. This unrestricted neutrophil recruitment may represent a mechanism by which individuals may be protected from pulmonary bacterial infection and pneumonia.
Keywords: LPS, tolerance, lung, pneumonia, cytokines, airway hyperresponsiveness
INTRODUCTION
LPS, a cell wall component of Gram-negative bacteria, is a potent inducer of the innate inflammatory response. Recognition of LPS by Toll-like receptor 4 (TLR-4) results in the production of proinflammatory cytokines such as TNF-α and IL-6, as well as the neutrophil chemotactic cytokines KC (CXCL1) and macrophage inflammatory protein (MIP) 2 (CXCL2) (1). Pneumonia caused by both Gram-positive and Gram-negative bacteria is a significant cause of hospitalizations, multiorgan dysfunction, and sepsis (2). Systemic infection results in approximately 50% of those hospitalized after infection, making the underlying mechanism of susceptibility an important area of study (3). LPS is ubiquitously present in the environment and in ambient air; however, little is known regarding the effect of repeated LPS inhalation in the absence of underlying disease (4). We hypothesized that the lung must use compensatory mechanisms to avoid excessive inflammation and potential organ damage while retaining the ability to fight infection. LPS tolerance may represent one such adaptive mechanism (5).
LPS tolerance is described as refractoriness to further LPS stimulation that results from an initial LPS exposure (6). This phenomenon has been largely studied in ex vivo macrophage cultures that show decreased production of TNF-α mRNA and protein in response to repeated LPS stimulation (7). Tolerance to LPS can also confer a survival advantage in animal models of septic shock (8). Furthermore, LPS-stimulated whole blood of septic patients produce significantly less TNF-α than healthy controls (9). Despite these significant findings, little is known regarding the effect of repeated LPS exposure in the lung microenvironment.
Here, we present a clinically relevant model of chronic LPS exposure that more accurately recapitulates repeated inhalation in the environmental setting. In this model, we observed differential regulation of acute phase cytokines and CXC chemokines. Importantly, we demonstrate that neutrophil recruitment to the lung is not impaired in mice rendered tolerant to LPS, and that this tolerance does not confer cross-tolerance to the TLR-2 stimulus Pam3Cys (PAM). Therefore, LPS tolerance may serve to attenuate pneumonia severity, thereby protecting against systemic bacterial infection and sepsis.
MATERIALS AND METHODS
Animals
Female BALB/c mice 9 to 12 weeks old were purchased from Jackson Laboratories (Bar Harbor, Maine) and maintained under standard laboratory conditions. The mice were housed in a temperature- and humidity-controlled room with 12-h light/dark cycles and allowed food and water ad libitum. All experiments were performed according to the National Institutes of Health guidelines and were approved by the Boston University Institutional Animal Care and Use Committee or the University of Michigan Committee on Use and Care of Animals.
Induction of chronic pulmonary LPS tolerance
Mice were exposed to either phosphate-buffered saline (PBS) or 1 μg Escherichia coli LPS O111:B5 (Sigma-Aldrich, cat. no. 62325) in a total volume of 50 μL PBS for 4 consecutive days by direct pulmonary installation while under isofluorane anesthesia (10). This strain of LPS has been used extensively in the investigation of the inflammatory response to LPS and has been shown to be an effective immune activator in vivo (11). Briefly, mice were lightly anesthetized and suspended on a vertical board by their front incisors. The tails were taped to the board to support the body weight. The tongue was gently extended and the liquid placed at the base of the oropharynx so that it was inhaled. Tolerant mice were challenged with LPS for 4 consecutive days, whereas nontolerant mice were challenged with PBS. Both groups were challenged with 10 μg LPS 24 h later. Animals were killed and samples collected at various time points indicated in the figure legends.
Measurement of physiological parameters
For determination of total peripheral white blood cell, lymphocyte numbers, and hemoglobin concentrations, 20 μL of blood was collected from the tail vein, and an automated differential was performed using a Hemavet Mascot (Drew Scientific, Ramsey, Minn). Body weight was determined daily.
Bronchoalveolar lavage
Mice were euthanized by cervical dislocation. For bronchoalveolar lavage (BAL), the trachea was cannulated, and the lung was lavaged with two 1-mL aliquots of warm Hank’s balanced salt solution (Gibco, Grand Island, NY). Both aliquots were centrifuged. The supernatant of the first wash was removed and frozen at −20°C for later cytokine analysis. The supernatant from the second wash was discarded, and the cell pellets from both aliquots were resuspended and combined. Total cell counts were obtained using a Beckman-Coulter particle counter model ZF (Coulter Electronics Inc., Hialeah, Fla). Cytospin preparations were stained with Diff-Quick, and 300 cell differential counts were performed to determine the absolute numbers of neutrophils. The right lung was removed, placed in ice-cold protease inhibitor cocktail (Roche, Indianapolis, Ind) containing Triton X in PBS and homogenized with three 10-s passes in a Brinkmann Polytron PT3000 homogenizer. The mixture was then sonicated in hexadecyltrimethylammonium bromide buffer and centrifuged at 15,000 × g for 15 min. The supernatant was used immediately for myeloperoxidase (MPO) assay.
Enzyme-linked immunosorbent assay
Cytokines, chemokines, and TNF-soluble receptors were measured by sandwich enzyme-linked immunosorbent assay (ELISA) as previously described (12). All matched antibody pairs were purchased from R&D Systems (Minneapolis, Minn). Lung homogenate samples were assayed with the addition of 20% normal lung homogenate to the standards to adjust for the increased background caused by nonspecific matrix effects. Mouse tryptase was measured by direct ELISA. Briefly, plates were coated with serially diluted recombinant mouse tryptase-β1 and samples and incubated overnight at 4°C. Plates were washed using the same buffer as the sandwich ELISA. Nonspecific binding sites were blocked with Blocker Casein in PBS (Pierce, Rockford, Ill) for 1 h. Plates were washed and incubated with anti-tryptase-β1capture antibody for 2 h, washed again and incubated with horseradish peroxidase–conjugated mouse anti-rat immunoglobulin G for 30 min. Plates were developed with 3,3′,5,5′ tetramethylbenzidine as described in the sandwich ELISA protocol.
MPO assay
The MPO assay was performed as previously described (13). Briefly, 20 μL of the sonicated mixture was pipetted in duplicate into wells of a 96-well plate. Assay buffer (200 μL) containing o-dianisidine HCl (Sigma D-3252), 87.8 mM monobasic potassium phosphate, 12.3 mM dibasic potassium phosphate, and 0.005% hydrogen peroxide was added to each well, and kinetic measurements were taken at 465 nm every 10 s. Six total readings were taken, and MPO activity is expressed as the slope of the linear plot of these data.
mRNA isolation and reverse-transcriptase–polymerase chain reaction
mRNA was extracted using Trizol reagent (Invitrogen, Grand Island, NY). Polymerase chain reaction (PCR) reactions were performed using an iScript OneStep PCR kit with Sybr Green (BioRad, Hercules, Calif) using the following primer pairs: TNF-α: sense, 5′-ACTCCCAGACCCGGTATCTT-3′; antisense, 5′-CTGCAATTGACCGTCTTCT-3′; IL-6: sense, 5′-ACGGCCTTCCCTACTTCACA-3′; antisense, 5′-TCCAGAAGACCAGAGGAAATTTT-3′; KC: sense, 5′-TCAAGAACATCCAGAGCTTGAAG-3′; antisense, 5′-GGACACCTTTTAGCATCTTTTGG-3′; MIP-2: sense, 5′-CACCAACCACCAGGCTACAG-3′; antisense, 5′-CAGTTAGCCTTGCCTTTGTTCA-3′; M2 receptor: sense, 5′-TAAAGTCAACCGCCACCTTC-3′; antisense, 5′-ATAACGGAGGCATTGCTGAC-3′; M3 receptor: sense, 5′-GCTCAGAGACCAGAGCCTATC-3′; antisense, 5′-ACAGTTGTCACGGTCATCCA-3′; glyceraldehyde 3-phosphate dehydrogenase: sense, 5′-AACGACCCCTTCATTGAC-3′; anti-sense, 5′-TCCACGACATACTCAGCAC-3′. Real-time PCR reactions were performed on a BioRad iQ5 light cycler. Data were normalized to housekeeping gene and expressed as fold increase above the 0-hour nontolerant group using the 2−(ΔΔCt) method (14).
Airway hyperresponsiveness
Airway hyperresponsiveness was measured using unrestrained whole body plethysmography (Buxco Systems, Troy, NY). Mice were placed in the instrument chamber and allowed to acclimate for 5 min. Baseline measurements were recorded for 5 min. Mice were challenged for 2 min with aerosolized PBS and increasing doses of methacholine (Sigma, St. Louis, Mo). Each aerosol was followed by 5 min of monitoring and data collection. The partial pressure difference between the experimental and reference chambers represented the PenH parameter, which corresponds closely with invasive measurements (15, 16). The data are represented as the percent increase in PenH above PBS aerosol.
Cysteinyl-leukotriene immunoassay
Cysteinyl leukotrienes levels in the BAL fluid were measured by an ELISA kit (Cayman Chemicals, Ann Arbor, Mich) according to the manufacturer instructions. All samples were run at two dilutions. Only %B/B0 values in the linear range of the standard curve were accepted. Sample values falling out of this range were further diluted and rerun.
Determination of cross-tolerance to Pam3CSK4
A dose-response study was performed to verify that PAM (Alexis Biochemicals, cat. no. ALX-165-066-M002) induced a robust inflammatory response in the lung and to determine an appropriate dose for tolerance experiments. Differing doses of PAM (2.5, 25, or 250 μg) were delivered via direct pulmonary installation, and BAL was performed 2 h later. For tolerance experiments, mice were challenged with 1 μg LPS for 4 consecutive days and received 25 μg PAM 24 h later for the final challenge. Control groups received PBS for the first four challenges and 25 μg PAM 24 h later. Mice were euthanized, and BAL was performed at 2 h post–final challenge.
Statistical analysis
All data are represented as mean ± SEM. Statistical significance was determined by Student t test using GraphPad Prism version 4.0.3. (GraphPad Software, San Diego, Calif). Statistical significance was achieved when P ≤ 0.05 at the 95% confidence interval.
RESULTS
Body weight and hematology after induction of chronic LPS tolerance
To induce chronic LPS tolerance, mice received direct pulmonary installation of 1 μg LPS for 4 consecutive days and then received 10 μg LPS 24 h later. Nontolerant mice received PBS for the first four challenges and 10 μg LPS 24 h later. This model uses relatively high doses of LPS for the induction of tolerance. As such, we determined whether repeated inhalation of high-dose LPS causes systemic physiological changes. Although the body weight did decrease slightly over time, there were no differences between nontolerant and tolerant animals (Fig. 1A). Day 0 represents the day before the first pulmonary challenge. Because either stress or LPS may induce lymphocyte apoptosis, we measured total circulating white blood cells and lymphocyte numbers and found no significant alteration in these parameters as a result of repeated LPS inhalation (Fig. 1, B and C). Finally, hemoglobin concentrations were also found to be unchanged after induction of LPS tolerance (Fig. 1D). These data indicate that the model used in these studies does not cause systemic illness, and that the subsequent phenotypes examined are a result of local pulmonary immune modulation.
Fig. 1. Physiologic parameters in response to LPS tolerance.
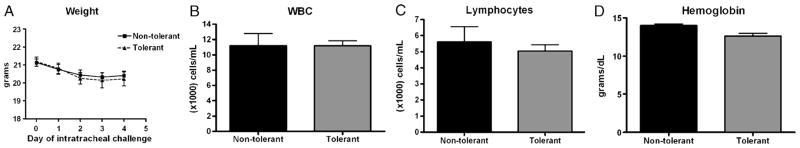
A, Mouse weights were measured the day before, and each day, mice received intratracheal challenges. Complete differential was performed on blood collected from the tail vein and total white blood cell counts (B), lymphocyte counts (C), and hemoglobin concentrations (D) are represented. Data are expressed as mean ± SEM (n = 6–12 mice per group). There were no differences between the tolerant and nontolerant animals in any of the measured parameters.
Acute-phase cytokine expression after induction of chronic LPS tolerance
We have shown that induction of acute pulmonary LPS tolerance results in significant reductions in both TNF-α protein and IL-6 (17). Induction of chronic LPS tolerance also resulted in significant reduction in TNF-α protein (~56%) at both 2 and 24 h post–final challenge along with a significant reduction in TNF-α mRNA expression in the lung tissue 1 h post–final challenge (Fig. 2, A and B). We have previously validated that the ELISA used to measure TNF-α detects this cytokine both free and in complex with both soluble receptors, verifying these results are physiologically relevant (17). In contrast to TNF, chronic LPS tolerance did not result in reduced IL-6 expression. This is in marked contrast to our previous observation in the acute LPS tolerance model showing that IL-6 was significantly reduced in the BAL fluid (17). In the chronic tolerance model, we saw that BAL fluid IL-6 protein at 2 h post–final challenge was significantly increased (~53%) in LPS-tolerant mice compared with the nontolerant group (Fig. 2). This was accompanied by a statistically insignificant increase in IL-6 mRNA expression in lung tissue. Interestingly, pulmonary IL-6 production in response to LPS has been shown to regulate LPB binding protein production in the liver and may regulate pneumonia mortality in patients with preexisting liver injury (18).
Fig. 2. Acute-phase cytokine expression after induction LPS tolerance.
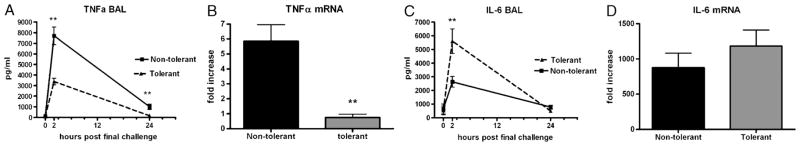
A, Bronchoalveolar lavage TNF-α concentrations at the indicated time points post–final challenge and lung tissue TNF-α mRNA expression 1 h post–final challenge (B). C, Bronchoalveolar lavage IL-6 concentrations at the indicated time points post–final challenge and lung tissue IL-6 mRNA expression 1 h post–final challenge (D). Zero-hour samples were harvested immediately before the final LPS challenge. mRNA data were calculated as fold increase over 0-h nontolerant group and are expressed as mean ± SEM (n = 6–10 mice per group). **P < 0.01 compared with nontolerant group.
Production of TNF-soluble receptors after induction of chronic LPS tolerance
We assessed whether chronic LPS inhalation can up-regulate compensatory anti-inflammatory proteins such as TNF-soluble receptors. As seen in Figure 3, repeated LPS inhalation induces significant expression of both TNF-SRI and TNF-SRII. Although no TNF-α is present at time 0 (Fig. 2A), both TNF-soluble receptors are substantially up-regulated. We suspect that TNF-SRII plays a more potent physiological role in LPS tolerance given that it is produced at roughly 20 times higher concentrations. The therapeutic TNF-α inhibitor Etancercept, a TNF-SRII-immunoglobulin G fusion protein, is widely used to treat excessive inflammation in several chronic inflammatory conditions (19). Our data demonstrate that inducing inflammation causes the physiological up-regulation of a compound proven effective for treatment of chronic inflammation.
Fig. 3. TNF-soluble receptor expression in response to induction of LPS tolerance.

TNF-SRI (A) and TNF-SRII (B) concentrations in BAL fluid at the indicated time points post–final challenge. Zero-hour samples were harvested immediately before the final LPS challenge. Data are expressed as mean ± SEM (n = 6–10 mice per group). **P < 0.01 and ***P < 0.0001 compared with the nontolerant group.
AHR and mediators of AHR
Pulmonary bacterial infections have been shown to increase airway resistance and decrease lung function in various animal models (20). We measured AHR in response to increasing doses of aerosolized methacholine at 4 h post–final challenge in both nontolerant and tolerant mice and found that induction of LPS tolerance is protective against AHR severity (Fig. 4A). We measured no differences in baseline PenH values between tolerant and nontolerant mice (data not shown). We then assayed for the mediators of AHR, cysteinyl leukotrienes, and tryptase as possible mechanisms governing the decrease in AHR seen in LPS-tolerant mice. Surprisingly, both cysteinyl leukotrienes and tryptase measured in the BAL fluid 2 h post–final challenge were significantly elevated in LPS-tolerant mice (Fig. 4, B and C). This indicates that, although LPS exposure in the LPS-tolerant group may induce production of the mediators of AHR, a divergent mechanism must be present that governs the methacholine-induced AHR assayed in this experiment.
Fig. 4. Airway hyperresponsiveness and mediators of AHR in response to LPS tolerance.

A, Airway hyperresponsiveness in response to increasing doses of aerosolized methacholine at 4 h post–final challenge. B, Bronchoalveolar lavage cysteinyl leukotriene concentrations at 2 h post–final challenge. C, Bronchoalveolar lavage tryptase concentrations at 2 h post–final challenge. Data are expressed as mean ± SEM (n = 6–10 mice per group). *P < 0.05 and **P < 0.01 compared with the nontolerant group.
Muscarinic receptor expression in response to chronic LPS tolerance
The finding that methacholine-induced AHR was attenuated in LPS-tolerant mice, even in the presence of significant concentrations of potent mediators of AHR, led us to investigate the expression of pulmonary methacholine receptors as a possible mechanism underlying the protection from AHR. Methacholine is a nonspecific muscarinic receptor agonist that causes bronchial smooth muscle contraction (21). We measured mRNA levels of the M2 and M3 muscarinic receptors. Our data show decreased expression of both M2 and M3 receptors in the lung tissue at 1 h post–final challenge in LPS-tolerant mice compared with nontolerant mice (Fig. 5). Furthermore, these data are expressed as fold change relative to the 0-h nontolerant group. These mice have been exposed only to the mechanical stress of aspirating 50 μL of liquid. A fold increase of 1.0 would represent basal expression equivalent to that observed after this mechanical stress (22). However, in the tolerant group, both M2 and M3 receptors were decreased below basal expression levels, suggesting that down-regulation of these receptors occurs upon induction of LPS tolerance.
Fig. 5. mRNA expression of M2 and M3 muscarinic acetylcholine receptors in nontolerant and tolerant mice at 1 h post–final challenge.

Data are calculated as fold increase over 0-h nontolerant group and are expressed as mean ± SEM (n = 6 mice per group). *P < 0.05 and **P < 0.01 compared with the nontolerant group.
Although LPS has been shown to potentiate smooth muscle responses to cholinergic stimuli, we provide the first evidence that LPS challenge can affect the level of expression of either of these muscarinic cholinergic receptors (23). Although we have shown mRNA down-regulation, but not receptor expression, our data demonstrate a clear dissociation between the mediators causing AHR (increased) and AHR itself (decreased). The likely bridge between the methacholine challenge and the induction of AHR lies in the ability of methacholine to activate the appropriate receptors.
Neutrophil recruitment and expression of CXC chemokines in response to LPS tolerance
Cytospin preparations and 300 cell differential counts were made from cells recovered in the BAL fluid at 24 h post–final challenge. In contrast to the predominance of macrophages present in naive, unchallenged mice, the cytospin preparations demonstrate comparable numbers of neutrophils recruited into the alveolar space in nontolerant and tolerant mice (Fig. 6).
Fig. 6. Cytospin preparations of cells recovered from BAL fluid of naïve (unchallenged) (A), nontolerant (B), and LPS-tolerant (C) mice at 24 h post–final LPS challenge.

Each is represented at 1,000× magnification. The cells from naïve mice are virtually all macrophages, whereas numerous neutrophils are observed in the other groups.
We found that the number of neutrophils recruited to the alveolar space was not affected by repeated LPS inhalation. At early time points (0 and 2 h) LPS-tolerant mice have significantly more neutrophils in the BAL fluid, which were recruited in response to the first four LPS exposures. By 24 h, however, both groups recruited comparable numbers of neutrophils into the BAL fluid (Fig. 7A). Linear regression analysis of both groups between 2 and 24 h shows no significant difference between the slopes of the regression lines, indicating that the capacity of LPS-tolerant animals to respond to a high-dose LPS challenge was not compromised. Myeloperoxidase activity was measured in the lung tissue of mice previously subjected to BAL. At 24 h post–final challenge, no significant difference in MPO activity was measured between groups, suggesting that comparable numbers of neutrophils are sequestered in the lung tissue of nontolerant and tolerant mice (Fig. 7B).
Fig. 7. Neutrophil influx and MPO activity in response to LPS tolerance.

A, Bronchoalveolar lavage neutrophil numbers at various time points post–final challenge. Cytospins were prepared and quantified for collected cells, and data are represented as absolute cell number per mouse. B, After BAL, the right lung was homogenized and sonicated for MPO assay. Data are expressed as mean ± SEM (n = 6–10 mice per group). **P < 0.01 compared with the nontolerant group.
We further assayed CXC chemokine production as a potential mechanism responsible for recruitment of neutrophils. No biologically significant difference in KC (CXCL1) concentrations were measured in the BAL fluid between nontolerant and tolerant mice (Fig. 8A). However, a statistically significant reduction in CXCL1 mRNA in the tolerant mice was measured in the lung tissue at 1 h post–final challenge (Fig. 8B). Interestingly, a specific reduction in BAL MIP-2 (CXCL2) expression was measured at 2 h post–final challenge in the LPS-tolerant mice. This was accompanied by a significant reduction on CXCL2 mRNA in the lung tissue (Fig. 8, C and D). Although there was a reduction in these CXC chemokines, based on the data in Figure 7, the levels must have been sufficient to still recruit neutrophils to the lung.
Fig. 8. Neutrophil chemotactic protein expression after induction LPS tolerance.
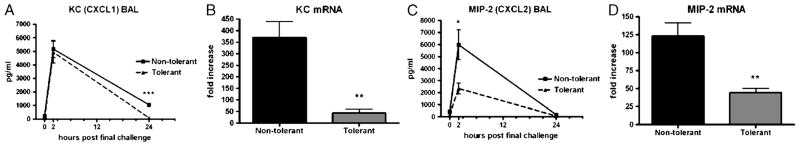
A, Bronchoalveolar lavage KC (CXCL1) concentrations at the indicated time points post–final challenge and (B) lung tissue KC (CXCL1) mRNA expression 1 h post–final challenge. C, Bronchoalveolar lavage MIP-2 (CXCL2) concentrations at the indicated time points post–final challenge and lung tissue MIP-2 (CXCL2) mRNA expression (D) 1 h post–final challenge. Zero-hour samples were harvested immediately before the final LPS challenge. mRNA data are expressed as fold increase above naïve mice and are represented as mean ± SEM (n = 6–10 mice per group). *P < 0.05 and **P < 0.01 compared with the nontolerant group.
Determination of pulmonary cross-tolerance to TLR-2 stimulus
To determine whether repeated inhalation of LPS can lead to cross-tolerance to other TLR stimuli, we used the synthetic lipopeptide TLR-2 agonist PAM. Initial dose-response experiments were performed using a single pulmonary installation of 2.5, 25, or 250 μg PAM, followed by BAL at 2 h. Measurement of TNF-α, IL-6, CXCL1, and CXCL2 all showed clear dose-dependent increases in response to the varying doses of PAM (data not shown). To test the induction of cross-tolerance, LPS tolerance was induced as described in the “Materials and methods” section. For the final challenge, mice received 25 μg PAM, and BAL was performed 2 h later. Interestingly, LPS-tolerant mice produced a more robust cytokine response to the final PAM challenge compared with non–LPS-tolerant mice (Fig. 9, A and B). However, there was no significant difference in CXC chemokine production between tolerant and nontolerant mice receiving a PAM challenge (Fig. 9, C and D). We also observed PAM to be a significantly weaker inducer of inflammation as seen in the significantly lower concentrations of both cytokines and chemokines measured in the cross-tolerance studies.
Fig. 9. Cross-tolerance to TLR-2 ligand PAM.
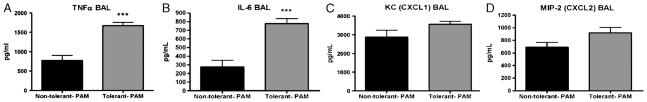
LPS tolerance was induced by direct pulmonary exposure to LPS for 4 consecutive days (tolerant PAM). Mice were challenged with 25 μg PAM 24 h later. Control mice received PBS for 4 consecutive days and 25 μg PAM 24 h later (nontolerant PAM). TNF (A), IL-6 (B), KC (CXCL1) (C), MIP-2 (CXCL2) (D) expression in BAL fluid 2 h post-PAM challenge. Data are expressed as mean ± SEM (n = 6 mice per group). ***P < 0.0001 compared with the nontolerant group.
DISCUSSION
Severe bacterial pneumonia can lead to multiple organ dysfunction and sepsis in roughly half of those infected. It is conceivable that the local lung immune environment at the time of bacterial encounter plays a role in bacterial clearance and can influence the severity of infection and the onset of sepsis. LPS is ubiquitously present in the environment; however, the effect of repeated LPS exposure in the lung microenvironment has been a largely neglected field of study. Because the lung is constantly exposed to environmental pathogens, the local immune environment requires tight regulation, and the mechanism governing this regulation may affect the lung’s ability to fight infection while avoiding excessive inflammation (24).
Our findings demonstrate that after chronic exposure to high levels of LPS, down-regulation of both TNF-α protein and mRNA still occurs. This, coupled with the finding that IL-6 levels are increased in the BAL fluid of LPS-tolerant mice, indicates that pulmonary tolerance is not the result of global immunosuppression (Fig. 2). Several studies have implicated the IL-1 receptor–associated kinase (IRAK) family in the regulation of LPS tolerance. Models of endotoxemia have demonstrated up-regulation of IRAK-M (produced by monocytes) and down-regulation of IRAK-1 in LPS preconditioned peripheral blood mononuclear cells restimulated with LPS (25). It is possible that IRAK-M down-regulation plays a role in suppression of macrophage TNF-α in this model. Furthermore, other cell types such as epithelial cells and fibroblasts may have decreased susceptibility to become tolerized, accounting for the significant production of IL-6 and CXCL1.
Adenosine receptors have been shown to play a role in the differential regulation of TNF-α and IL-6 in rat cardiomyocytes (26). Activation of the A2a receptor can inhibit TNF-α production by macrophages and inhibit tissue damage in models of acute lung injury in response to LPS and models of lung I/R injury (27, 28). A2a receptor knockout mice are also more susceptible to adult respiratory distress syndrome–induced lung injury (29). As such, adenosine receptors may play an important role in the differential expression of TNF-α and IL-6 observed in this model and may also modulate lung injury resulting from repeated LPS inhalation.
These studies show that LPS tolerance protects against methacholine-induced AHR even in the presence of significantly higher concentrations of both cysteinyl leukotrienes and tryptase. Because both of these molecules were produced in response to LPS challenge and are primary mediators of AHR, we sought to determine if the mechanism responsible for the protection from AHR we observed was due to an alteration in the response to methacholine itself (30). Methacholine, a synthetic choline ester, is a nonspecific agonist for muscarinic cholinergic receptors (31). Both M2 and M3 muscarinic receptors are expressed in the lung and bronchi (22). Upon methacholine challenge, M3 muscarinic receptors cause smooth muscle contraction, whereas M2 receptors inhibit smooth muscle relaxation (32). Decreased expression of both receptors would manifest as an attenuated airway response to inhaled methacholine. Our data demonstrated the first evidence that LPS challenge can alter muscarinic receptor expression and suggest that the mechanism of reduced AHR seen in LPS tolerant animals may be a reduction in expression of both M2 and M3 muscarinic receptors.
Community-acquired pneumonia (CAP) results in hospitalization of roughly 500,000 people in North America annually and has a mortality rate of up to 50% in individuals hospitalized with severe CAP (33). Furthermore, multiorgan failure and sepsis occurs in approximately 50% of hospitalized CAP patients, making this condition a significant health care concern due to its high morbidity and mortality rates (2). Neutrophil recruitment into the lung is a key event required for bacterial clearance (34). In this study, we show that LPS-tolerant mice recruit equivalent numbers of neutrophils into the alveolar space in response to the final LPS challenge. More importantly, linear regression analysis of the time frame between 2 and 24 h post–final challenge showed no significant difference between groups in the slope of the regression lines. This indicates that, although tolerant animals have a significant number of neutrophils already present in the BAL fluid at 0 h, their capacity to recruit neutrophils in response to the final LPS challenge was not impaired. Although we have used Gram-negative LPS in these studies, we recognize that bacterial pneumonia generally arises from Gram-positive infections (2). Nonetheless, the ubiquitous presence of Gram-negative LPS in the environment makes it an important factor to study when considering the lung immune environment at the time of infection. Clearly, further studies must be performed to determine whether Gram-positive stimuli can induce pulmonary tolerance.
We further sought to elucidate the mechanism responsible for the recruitment of neutrophils. We found no statistical difference in KC (CXCL1) production between nontolerant and tolerant mice, whereas a significant decrease in mRNA was measured at 1 h post–final challenge. We hypothesize that induction of LPS tolerance may affect mRNA stability, possibly by increased deadenylation of the mRNA body, promoting subsequent degradation (35). Additionally, we have previously shown that CXCL1 mRNA production peaks at 2 h post–final challenge in the lung tissue. LPS tolerance may also affect the rate at which CXCL1 mRNA is transcribed (17). We also measured a significant reduction in MIP-2 (CXCL2) expression in the BAL fluid, which was accompanied by a significant decrease in mRNA. To our knowledge, differential regulation of CXCL1 and CXCL2 in the lung in response to LPS challenge has not previously been reported.
Exposure to LPS has been shown to induce cross-tolerance to TLR-2 ligands such as LTA (36). We used the TLR-2 agonist PAM to study cross-tolerance in the lung. Our studies showed that cross-tolerance to PAM is not induced in the lung. Chronic LPS inhalation potentiated the cytokine response, resulting in increased TNF-α and IL-6 production in response to PAM challenge. However, the expression of both CXCL1 and CXCL2 was equivalent in LPS-tolerant and nontolerant mice. LPS challenge has been shown to increase the expression of TLR-2 on lung endothelial cells in a TLR-4/MyD88-dependent manner. Furthermore, nicotinamide adenine dinucleotide phosphate (reduced form) oxidase produced by neutrophils can regulate TLR-2 expression (37, 38). Therefore, the increased cytokine production may be the result of higher TLR-2 expression induced by LPS and the persistent presence of neutrophils in LPS-tolerant mice.
Epidemiologic data have demonstrated both positive and negative correlations between LPS inhalation and disease. High levels of environmental LPS such as those present on farms and in agricultural and textile mills correlate with reduced incidence of atopy and asthma (39). Furthermore, individuals residing or working in these environments can have reduced incidences of certain lung cancers (40). Conversely, LPS inhalation has been correlated with increased incidence and severity of asthma, as well as with causing persistent inflammation and decreased lung function (41). These data may also suggest the mechanism underlying the decreased incidence of asthma and atopy seen in farm communities and in the developing world; however, further studies are required to determine whether individuals exposed to high levels of inhalational LPS are protected from atopy and asthma.
This study demonstrates a new model of chronic pulmonary LPS tolerance and demonstrates a novel mechanism of immune regulation in the lung. LPS tolerance does not impair the ability to mount a neutrophilic response to exogenous pathogens. However, one cannot rule out the possibility that the persistence of neutrophils in the lung can lead to organ injury and long-term loss of function. More studies must be performed to determine the role of chronic LPS tolerance on lung injury and also to determine whether differential immune regulation occurs in individuals who continually inhale high levels of LPS.
Acknowledgments
This work was supported by National Institutes of Health (grant no. R01 ES013528).
References
- 1.Miller SI, Ernst RK, Bader MW. LPS, TLR4 and infectious disease diversity. Nat Rev Microbiol. 2005;3(1):36–46. doi: 10.1038/nrmicro1068. [DOI] [PubMed] [Google Scholar]
- 2.Andonegui G, Goring K, Liu D, McCafferty DM, Winston BW. Characterization of S. Pneumoniae pneumonia–induced multiple organ dysfunction syndrome; an experimental mouse model of Gram-positive sepsis. Shock. 2009;31(4):423–428. doi: 10.1097/SHK.0b013e318188c273. [DOI] [PubMed] [Google Scholar]
- 3.Dremsizov T, Clermont G, Kellum JA, Kalassian KG, Fine MJ, Angus DC. Severe sepsis in community-acquired pneumonia: when does it happen, and do systemic inflammatory response syndrome criteria help predict course? Chest. 2006;129(4):968–978. doi: 10.1378/chest.129.4.968. [DOI] [PubMed] [Google Scholar]
- 4.Hackett TL, Holloway R, Holgate ST, Warner JA. Dynamics of pro-inflammatory and anti-inflammatory cytokine release during acute inflammation in chronic obstructive pulmonary disease: an ex vivo study. Respir Res. 2008;9:47. doi: 10.1186/1465-9921-9-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Foster SL, Hargreaves DC, Medzhitov R. Gene-specific control of inflammation by TLR-induced chromatin modifications. Nature. 2007;447(7147):972–978. doi: 10.1038/nature05836. [DOI] [PubMed] [Google Scholar]
- 6.Fraker DL, Stovroff MC, Merino MJ, Norton JA. Tolerance to tumor necrosis factor in rats and the relationship to endotoxin tolerance and toxicity. J Exp Med. 1988;168(1):95–105. doi: 10.1084/jem.168.1.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mathison J, Wolfson E, Steinemann S, Tobias P, Ulevitch R. Lipopolysaccharide (LPS) recognition in macrophages. Participation of LPS-binding protein and CD14 in LPS-induced adaptation in rabbit peritoneal exudate macrophages. J Clin Invest. 1993;92(4):2053–2059. doi: 10.1172/JCI116801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wheeler DS, Lahni PM, Denenberg AG, Poynter SE, Wong HR, Cook JA, Zingarelli B. Induction of endotoxin tolerance enhances bacterial clearance and survival in murine polymicrobial sepsis. Shock. 2008;30(3):267–273. doi: 10.1097/shk.0b013e318162c190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Heagy W, Hansen C, Nieman K, Cohen M, Richardson C, Rodriguez JL, West MA. Impaired ex vivo lipopolysaccharide-stimulated whole blood tumor necrosis factor production may identify “septic” intensive care unit patients. Shock. 2000;14(3):271–276. doi: 10.1097/00024382-200014030-00005. discussion 276–277. [DOI] [PubMed] [Google Scholar]
- 10.Gavett SH, O’Hearn DJ, Li X, Huang SK, Finkelman FD, Wills-Karp M. Interleukin 12 inhibits antigen-induced airway hyperresponsiveness, inflammation, and Th2 cytokine expression in mice. J Exp Med. 1995;182(5):1527–1536. doi: 10.1084/jem.182.5.1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xing L, Remick DG. Mechanisms of dimethyl sulfoxide augmentation of IL-1 beta production. J Immunol. 2005;174(10):6195–6202. doi: 10.4049/jimmunol.174.10.6195. [DOI] [PubMed] [Google Scholar]
- 12.Nemzek JA, Siddiqui J, Remick DG. Development and optimization of cytokine ELISAs using commercial antibody pairs. J Immunol Methods. 2001;255(1–2):149–157. doi: 10.1016/s0022-1759(01)00419-7. [DOI] [PubMed] [Google Scholar]
- 13.Nemzek JA, Ebong SJ, Kim J, Bolgos GL, Remick DG. Keratinocyte growth factor pretreatment is associated with decreased macrophage inflammatory protein–2alpha concentrations and reduced neutrophil recruitment in acid aspiration lung injury. Shock. 2002;18(6):501–506. doi: 10.1097/00024382-200212000-00003. [DOI] [PubMed] [Google Scholar]
- 14.Giulietti A, Overbergh L, Valckx D, Decallonne B, Bouillon R, Mathieu C. An overview of real-time quantitative PCR: applications to quantify cytokine gene expression. Methods. 2001;25(4):386–401. doi: 10.1006/meth.2001.1261. [DOI] [PubMed] [Google Scholar]
- 15.McGraw DW, Almoosa KF, Paul RJ, Kobilka BK, Liggett SB. Antithetic regulation by beta-adrenergic receptors of Gq receptor signaling via phospholipase C underlies the airway beta-agonist paradox. J Clin Invest. 2003;112(4):619–626. doi: 10.1172/JCI18193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hamelmann E, Schwarze J, Takeda K, Oshiba A, Larsen GL, Irvin CG, Gelfand EW. Noninvasive measurement of airway responsiveness in allergic mice using barometric plethysmography. Am J Respir Crit Care Med. 1997;156(3 Pt1):766–775. doi: 10.1164/ajrccm.156.3.9606031. [DOI] [PubMed] [Google Scholar]
- 17.Natarajan S, Kim J, Remick DG. Acute pulmonary lipopolysaccharide tolerance decreases TNF-alpha without reducing neutrophil recruitment. J Immunol. 2008;181(12):8402–8408. doi: 10.4049/jimmunol.181.12.8402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gamble L, Bagby GJ, Quinton LJ, Happel KI, Mizgerd JP, Zhang P, Nelson S. The systemic and pulmonary LPS binding protein response to intratracheal lipopolysaccharide. Shock. 2009;31(2):212–217. doi: 10.1097/SHK.0b013e31817c0d7d. [DOI] [PubMed] [Google Scholar]
- 19.Wong M, Ziring D, Korin Y, Desai S, Kim S, Lin J, Gjertson D, Brawn J, Reed E, Singh RR. TNFalpha blockade in human diseases: mechanisms and future directions. Clin Immunol. 2008;126(2):121–136. doi: 10.1016/j.clim.2007.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kukavica-Ibrulj I, Hamelin ME, Prince GA, Gagnon C, Bergeron Y, Bergeron MG, Boivin G. Infection with human metapneumovirus predisposes mice to severe pneumococcal pneumonia. J Virol. 2009;83(3):1341–1349. doi: 10.1128/JVI.01123-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Birnbaum S, Barreiro TJ. Methacholine challenge testing: identifying its diagnostic role, testing, coding, and reimbursement. Chest. 2007;131(6):1932–1935. doi: 10.1378/chest.06-1385. [DOI] [PubMed] [Google Scholar]
- 22.Emala CW, Aryana A, Levine MA, Yasuda RP, Satkus SA, Wolfe BB, Hirshman CA. Expression of muscarinic receptor subtypes and M2-muscarinic inhibition of adenylyl cyclase in lung. Am J Physiol. 1995;268(1 Pt 1):L101–L107. doi: 10.1152/ajplung.1995.268.1.L101. [DOI] [PubMed] [Google Scholar]
- 23.Yamawaki I, Tamaoki J, Kanemura T, Horii S, Takizawa T. Effects of lipopolysaccharide from Pseudomonas aeruginosa on airway smooth muscle functions in guinea pigs. Respiration. 1990;57(4):268–274. doi: 10.1159/000195854. [DOI] [PubMed] [Google Scholar]
- 24.Thepen T, Hoeben K, Breve J, Kraal G. Alveolar macrophages down-regulate local pulmonary immune responses against intratracheally administered T-cell-dependent, but not T-cell-independent antigens. Immunology. 1992;76(1):60–64. [PMC free article] [PubMed] [Google Scholar]
- 25.Li Y, Howell EA, Lagoo AS, Kuchibhatla M, Pan H, Cohen HJ, Lagoo SA. Differential gene expression of interleukin-1 receptor associated kinase-1 and interleukin-1 receptor associated kinase–m in peripheral blood mononuclear cells of young and aged rats following preconditioning with endotoxin. Shock. 2009;31(1):55–63. doi: 10.1097/SHK.0b013e3181778ab2. [DOI] [PubMed] [Google Scholar]
- 26.Wagner DR, Kubota T, Sanders VJ, McTiernan CF, Feldman AM. Differential regulation of cardiac expression of IL-6 and TNF-alpha by A2- and A3-adenosine receptors. Am J Physiol. 1999;276(6 Pt 2):H2141–H2147. doi: 10.1152/ajpheart.1999.276.6.H2141. [DOI] [PubMed] [Google Scholar]
- 27.Reece TB, Ellman PI, Maxey TS, Crosby IK, Warren PS, Chong TW, Le Gallo RD, Linden J, Kern JA, Tribble CG, et al. Adenosine A2a receptor activation reduces inflammation and preserves pulmonary function in an in vivo model of lung transplantation. J Thorac Cardiovasc Surg. 2005;129(5):1137–1143. doi: 10.1016/j.jtcvs.2004.11.042. [DOI] [PubMed] [Google Scholar]
- 28.Reutershan J, Cagnina RE, Chang D, Linden J, Ley K. Therapeutic anti-inflammatory effects of myeloid cell adenosine receptor A2a stimulation in lipopolysaccharide-induced lung injury. J Immunol. 2007;179(2):1254–1263. doi: 10.4049/jimmunol.179.2.1254. [DOI] [PubMed] [Google Scholar]
- 29.Thiel M, Chouker A, Ohta A, Jackson E, Caldwell C, Smith P, Lukashev D, Bittman I, Sitkovsky MV. Oxygenation inhibits the physiological tissue-protecting mechanism and thereby exacerbates acute inflammatory lung injury. PLoS Biol. 2005;3(6):e174. doi: 10.1371/journal.pbio.0030174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim DC, Hsu FI, Barrett NA, Friend DS, Grenningloh R, Ho IC, Al-Garawi A, Lora JM, Lam BK, Austen KF, et al. Cysteinyl leukotrienes regulate Th2 cell-dependent pulmonary inflammation. J Immunol. 2006;176(7):4440–4448. doi: 10.4049/jimmunol.176.7.4440. [DOI] [PubMed] [Google Scholar]
- 31.Johnson M. Molecular mechanisms of beta(2)-adrenergic receptor function, response, and regulation. J Allergy Clin Immunol. 2006;117(1):18–24. doi: 10.1016/j.jaci.2005.11.012. quiz 25. [DOI] [PubMed] [Google Scholar]
- 32.Carey MA, Card JW, Bradbury JA, Moorman MP, Haykal-Coates N, Gavett SH, Graves JP, Walker VR, Flake GP, Voltz JW, et al. Spontaneous airway hyperresponsiveness in estrogen receptor–alpha–deficient mice. Am J Respir Crit Care Med. 2007;175(2):126–135. doi: 10.1164/rccm.200509-1493OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.File TM., Jr Streptococcus pneumoniae and community-acquired pneumonia: a cause for concern. Am J Med. 2004;117(Suppl 3A):39S–50S. doi: 10.1016/j.amjmed.2004.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wagner JG, Harkema JR, Roth RA. Pulmonary leukostasis and the inhibition of airway neutrophil recruitment are early events in the endotoxemic rat. Shock. 2002;17(2):151–158. doi: 10.1097/00024382-200202000-00012. [DOI] [PubMed] [Google Scholar]
- 35.Datta S, Biswas R, Novotny M, Pavicic PG, Jr, Herjan T, Mandal P, Hamilton TA. Tristetraprolin regulates CXCL1 (KC) mRNA stability. J Immunol. 2008;180(4):2545–2552. doi: 10.4049/jimmunol.180.4.2545. [DOI] [PubMed] [Google Scholar]
- 36.Jacinto R, Hartung T, McCall C, Li L. Lipopolysaccharide- and lipoteichoic acid–induced tolerance and cross-tolerance: distinct alterations in IL-1 receptor–associated kinase. J Immunol. 2002;168(12):6136–6141. doi: 10.4049/jimmunol.168.12.6136. [DOI] [PubMed] [Google Scholar]
- 37.Laroux FS, Romero X, Wetzler L, Engel P, Terhorst C. Cutting edge: Myd88 controls phagocyte NADPH oxidase function and killing of Gram-negative bacteria. J Immunol. 2005;175(9):5596–5600. doi: 10.4049/jimmunol.175.9.5596. [DOI] [PubMed] [Google Scholar]
- 38.Fan J, Frey RS, Malik AB. TLR4 signaling induces TLR2 expression in endothelial cells via neutrophil NADPH oxidase. J Clin Invest. 2003;112(8):1234–1243. doi: 10.1172/JCI18696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Smit LA, Wouters IM, Hobo MM, Eduard W, Doekes G, Heederik D. Agricultural seed dust as a potential cause of organic dust toxic syndrome. Occup Environ Med. 2006;63(1):59–67. doi: 10.1136/oem.2005.021527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lange JH. Reduced cancer rates in agricultural workers: a benefit of environmental and occupational endotoxin exposure. Med Hypotheses. 2000;55(5):383–385. doi: 10.1054/mehy.2000.1072. [DOI] [PubMed] [Google Scholar]
- 41.Liu AH. Endotoxin exposure in allergy and asthma: reconciling a paradox. J Allergy Clin Immunol. 2002;109(3):379–392. doi: 10.1067/mai.2002.122157. [DOI] [PubMed] [Google Scholar]