

Abstract
Somatic mutations in mitochondrial DNA build up in aging tissues and are thought to contribute to physiological aging. Surprisingly, it is not known if these mutations occur early or late in life. A new study looks at mechanisms of accelerated mitochondrial aging in HIV-infected individuals treated with nucleoside analog anti-retroviral drugs and offers support for an early origin of mitochondrial DNA mutations.
Somatic mutations in mitochondrial DNA (mtDNA) build up in somatic tissues with age. However, it is not clear when these mtDNA mutations occur: early or late in life? An early origin of somatic mtDNA mutations implies that exposure to mitochondrial mutagens in early life, in utero or even during oocyte maturation may contribute to the mutational burden in mtDNA at old age. This concept is still not commonly considered: it is usually assumed that most mtDNA mutations are created late in life when mtDNA is more prone to chemical damage. On page XXX of this issue, Patrick Chinnery and colleagues1 investigate accelerated mitochondrial aging in HIV-infected individuals treated with nucleoside analog anti-retroviral drugs (NRTIs), which are powerful inhibitors of mtDNA polymerase. They suggest that the drugs act through multiplication of preexisting mutations rather than by inducing new ones. This result appears to support an early origin for mtDNA mutations.
de novo mutations or expansion of existing ones?
It is commonly thought that mutational load increases when mutations are created de novo, that is, by mutagens or spontaneous error. The mtDNA of a cell is a dynamic population of thousands of individual copies of DNA which are constantly replicated and destroyed or segregated to sister cells at mitosis. Thus, mutant mtDNA can potentially expand at the expense of other mtDNA molecules in a cell and eventually even replace them entirely. Such successes are caused by either a selective advantage conferred by the mutation or by random genetic drift. In conclusion, mtDNA mutations can increase two ways: by de novo mutation or by expansion of preexisting mutations. Which way is mainly responsible for the mutational buildup with age? Previous mathematical simulations2 and analysis of the mutation distribution in tissues3 suggested that, unexpectedly, many mtDNA mutations may originate early in life and may have been primarily created by expanding preexisting mutations, although the relative contributions of de novo and expanded mutations remain uncertain.
Two mechanisms of NRTI toxicity
Patients treated with NRTIs show, among other premature aging symptoms, many COX− muscle fibers (that is, fibers deficient in cytochrome C oxidase (COX), an enzyme partially encoded in mtDNA), which are a hallmark of old age. There were essentially no COX− fibers in the non-treated controls. Chinnery and colleagues1 showed that these mitochondrial defects are caused by pathogenic mtDNA mutations that expanded within these fibers. How do NRTIs cause these mutation expansions? There are two plausible mechanisms. One possibility (Fig. 1a) is that NRTIs are mutagenic and induce seed mutations that initiate expansions that then proceed independently of NRTI exposure. Indeed, as mtDNA polymerase inhibitors, NRTIs could also increase the polymerase’s error rate. The other possible mechanism (Fig. 1b) is that NRTIs greatly accelerate expansion of pre-existing mutations. This scenario is also plausible because inhibition of DNA polymerase by NRTIs results in a decrease of mtDNA copies per cell. Smaller populations should have faster genetic drift and therefore faster expansions of mtDNA mutations. Chinnery and colleagues1 corroborate this expectation using a very elegant mathematical simulation.
Figure 1.
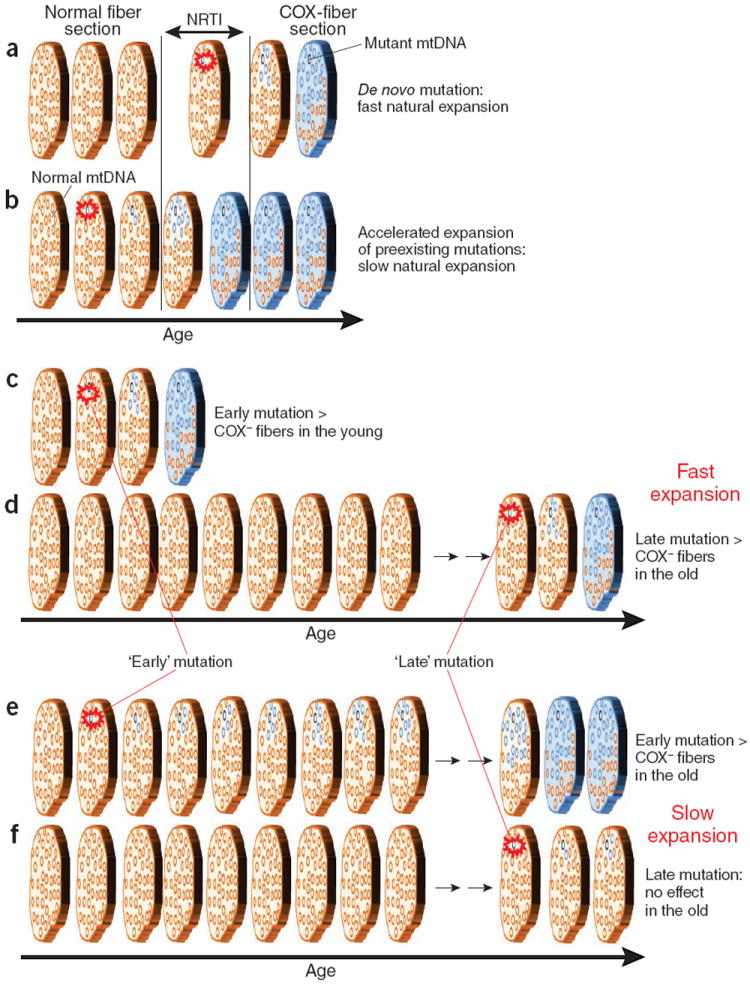
Schematic of plausible mechanisms of mutation expansion and development of mitochondrial deficiency in muscle fibers. Muscle fiber is represented by a short section shown at different time points. Brown represents normal fiber, and blue represents COX− (cytochrome C oxidase-deficient) fiber. mtDNA molecules are represented by the small circles within the fiber section, with brown representing normal fiber and blue representing mutant fiber. The red burst denotes an initial mutational event. NRTIs could induce seed mutations that initiate expansions (a) or accelerate expansion of preexisting mutations (b). If expansion occurs rapidly, early mutations, if they exist, should be observable quickly (c) and COX− fibers in the old originate from recent late mutations (d). If expansion is slow, only early mutations will have an effect on aging (e), not late mutations (f).
Mechanisms of NRTI toxicity are related to expansion rate and the timing of mutations
The importance of determining the mechanism of NRTI toxicity goes beyond potential applications in drug design. It may also help to clarify the mechanism of mitochondrial aging in general. If NRTIs act through inducing new mutations (Fig. 1a), then these new mutations should expand fairly quickly because COX− muscle fibers appear only a few years after NRTI therapy. If expansion occurs on a quick timescale, then any mutations that were acquired early in life should have expanded and be observable soon thereafter as COX− fibers (Fig. 1c). The fact that COX− fibers show up only in very old age implies that these early mutations, if they exist, should be of little physiological importance. Most COX defects in this scenario would be expected to originate in late-age mutations (Fig. 1d). If the second mechanism is correct (NRTIs act by accelerating expansion of preexisting mutations (Fig. 1b)), then the typical expansion rate must be quite slow, otherwise preexisting mutations would have expanded without NRTIs. But if expansion is slow, then only mutations that originate early in life will have time to fully expand, cause COX defects and contribute to physiological aging (Fig. 1e), whereas late mutations will not have enough time to do so (Fig. 1f). In summary, if NRTIs cause COX defects by inducing de novo mutations, then the rate of expansion should be fast, and early life mutations should be of little concern. However, the evidence in Payne et al.1 supports the hypothesis that NRTIs accelerate expansion of preexisting mutations and, thus, implies early life mutations in aging.
Cautions and comparison to other studies
How certain are these conclusions1? They are mainly based on the failure to detect NRTI-induced point mutations. However, a majority of COX− fibers contain expanded deletions rather than point mutations. There is also evidence that NRTIs can induce point mutations in mtDNA (A. Osborne and D. Wangh, personal communication). Nevertheless, accelerated expansion of preexisting mutations does appear to be the best model for NRTI toxicity. In support of early life mutations, previous work4,5 (discussed in ref. 3) and a very recent detailed study6 reported that a sizable proportion of mtDNA mutation load indeed originates in early development or even in the germline. Although this seems to confirm that mutations in mtDNA arise early in life, the actual pathogenic effect of these early mutations is uncertain. A disproportionate majority of mutations found in these studies4,5,6 are non-pathogenic. As a note of caution, the scenarios drawn here are necessarily a simplification. The two mechanisms are not mutually exclusive; the rate of expansion may change with age and cell type. In conclusion, the jury is still out on the case of early life pathogenic mtDNA mutations, but the importance of solving the case is clear.
Acknowledgments
K.K. is supported by the Ellison Foundation and US National Institutes of Health grants AG19787, NS058988 and AG035223.
Footnotes
COMPETING FINANCIAL INTERESTS
The author declares no competing financial interests.
References
- 1.Payne BAI, et al. Nat Genet. 2011;43:xxx–yyy. [Google Scholar]
- 2.Elson JL, Samuels D, Turnbull D, Chinnery P. Am J Hum Genet. 2001;68:802–806. doi: 10.1086/318801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Khrapko K, Nekhaeva E, Kraytsberg Y, Kunz W. Mutat Res. 2003;522:13–19. doi: 10.1016/s0027-5107(02)00306-8. [DOI] [PubMed] [Google Scholar]
- 4.Michikawa Y, Mazzucchelli F, Bresolin N, Scarlato G, Attardi G. Science. 1999;286:774–779. doi: 10.1126/science.286.5440.774. [DOI] [PubMed] [Google Scholar]
- 5.Wang Y, et al. Proc Natl Acad Sci USA. 2001;98:4022–4027. doi: 10.1073/pnas.061013598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.He Y, et al. Nature. 2010;464:610–614. doi: 10.1038/nature08802. [DOI] [PMC free article] [PubMed] [Google Scholar]