

Abstract
Aims/hypothesis
Beta cells situated in the islet of Langerhans respond more vigorously to glucose than do dissociated beta cells. Mechanisms for this discrepancy were studied by comparing insulin-producing MIN6 cells aggregated into pseudoislets with MIN6 monolayer cells and mouse and human islets.
Methods
MIN6 monolayers, pseudoislets and mouse and human islets were exposed to glucose, α-ketoisocaproic acid (KIC), pyruvate, KIC plus glutamine and the phosphatidylinositol 3-kinase (PI3K) inhibitors LY294002 or wortmannin. Insulin secretion (ELISA), cytoplasmic Ca2+ concentration ([Ca2+]c; microfluorometry), glucose oxidation (radiolabelling), the expression of genes involved in mitochondrial metabolism (PCR) and the phosphorylation of insulin receptor signalling proteins (western blotting) were measured.
Results
Insulin secretory responses to glucose, pyruvate, KIC and glutamine were higher in pseudoislets than monolayers and comparable to those of human islets. Glucose oxidation and genes for mitochondrial metabolism were upregulated in pseudoislets compared with single cells and monolayers, respectively. Phosphorylation at the inhibitory S636/639 site of IRS-1 was significantly higher in monolayers and dispersed human and mouse cells than pseudoislets and intact human and mouse islets. PI3K inhibition only slightly attenuated glucose-stimulated insulin secretion from monolayers, but substantially reduced that from pseudoislets and human and mouse islets without suppressing the glucose-induced [Ca2+]c response.
Conclusions/interpretation
We propose that islet architecture is critical for proper beta cell mitochondrial metabolism and IRS-1 signalling, and that PI3K regulates insulin secretion at a step distal to the elevation of [Ca2+]c.
Electronic supplementary material
The online version of this article (doi:10.1007/s00125-013-2903-3) contains peer-reviewed but unedited supplementary material, which is available to authorised users.
Keywords: Beta cell, Ca2+, Insulin secretion, IRS-1, Islets, Mitochondrial metabolism, PI3-kinase
Introduction
Insulin secreted from the pancreatic beta cell is the main glucose-lowering hormone. The loss of beta cell function plays a key role in the development of both type 1 and 2 diabetes mellitus. The beta cell is located in the islets of Langerhans, which are scattered in the pancreas and comprise approximately 1% of the volume of the gland [1]. In addition to insulin-secreting beta cells, the islets also contain other hormone-producing cell types, including glucagon-, somatostatin- and pancreatic polypeptide-secreting cells, as well as non-endocrine cells [1]. This makes it important to consider the contribution of non-beta cells in beta cell islet experimentation.
The relative difficulties of isolating large numbers of primary islets, and their mixed cell populations, have made insulin-producing cell lines an important tool in beta cell research. The mouse-derived insulinoma MIN6 cell line is glucose-responsive [2], although the response is modest, with a typically twofold to fourfold rise in glucose-stimulated insulin secretion (GSIS) [3]. A larger secretory response to a glucose challenge depends on the location of the beta cell in the islet [4]. When challenged with high glucose levels, dispersed islet cells secrete less insulin than beta cells within the intact islet [5]. Interestingly, when dispersed islet cells were allowed to reaggregate into islet-like structures, so-called pseudoislets, much of the secretory capacity was regained [6]. The strategy of allowing cells to aggregate was also attempted with MIN6 cells, which form cell clusters the size of primary islets [7]. Despite containing only insulin-producing cells, these MIN6 pseudoislets showed enhanced glucose responsiveness compared with MIN6 cells cultured in monolayers [7].
In the present study, we characterised the detailed dynamic insulin secretory response of MIN6 pseudoislets to different metabolisable and non-metabolisable secretagogues. The secretory phenotype was compared not only with that of MIN6 cells grown in monolayers, but also with that of human and mouse islets. We demonstrate that the superior secretory characteristics of MIN6 cells aggregated to form pseudoislets compared with MIN6 monolayers depended on mitochondrial metabolism and was related to differences in IRS-1 phosphorylation.
Methods
Cell culture
Mouse insulinoma MIN6 monolayer cells were cultured in 250 ml tissue culture flasks (Becton Dickson Labware, Franklin Lakes, NJ, USA) at 37°C (95% O2 and 5% CO2) in DMEM (Invitrogen, Paisley, UK) as previously described [3]. MIN6 pseudoislets were prepared by aggregating 3 × 106 dispersed cells cultured in Petri dishes made of non-adherent plastic (Becton Dickson Labware) for 3–5 days using the same culture condition as for monolayers [7]. All experiments were performed between passages 20 and 30. Human islets were cultured for 4–8 days before experiments in CMRL medium containing 5.5 mmol/l glucose and supplemented with 10% FBS, 1% l-glutamine, 100 U/ml penicillin and 100 μg/ml streptomycin. Islets from the pancreases of C57Bl/6 mice were isolated using collagenase. The mouse islets obtained were cultured for 2 days in RPMI 1640 (Invitrogen) medium containing 11.1 mmol/l glucose supplemented with 10% FBS, 100 units/ml penicillin and 100 μg/ml streptomycin.
To prepare the dispersed cells, groups of 100 human and mouse islets were dispersed in 0.5% trypsin for 10–12 min and 3–5 min, respectively, and then treated with DNase I (Qiagen GmbH, Hilden, Germany) for 2 min. The resulting cell suspensions were placed in poly-l-lysine-coated plates and cultured for 48 h in the respective culture medium. The use of human and mouse islets was approved by the local ethical committees (Dnr 2010/006 for human islets and Dnr C106/11 for mouse islets).
Insulin secretion
Insulin secretion was measured from MIN6 monolayers and MIN6 pseudoislets as well as mouse and human islets. For static incubation experiments, 105 MIN6 cells were seeded into 12-well tissue culture plates (Becton Dickson Labware) and cultured for 3 days. The cultured monolayer cells or groups of 20 pseudoislets or human islets were preincubated for 60 min at 37°C in 1 ml KRB HEPES buffer consisting of 130 mmol/l NaCl, 4.8 mmol/l KCl, 1.2 mmol/l MgSO4, 1.2 mmol/l KH2PO4, 1.2 mmol/l CaCl2, 5 mmol/l NaHCO3 and 5 mmol/l HEPES, titrated to pH 7.4 with NaOH and supplemented with 1 mg/ml BSA and 2 mmol/l glucose. Subsequently, the cells were incubated in 1 ml KRB HEPES buffer containing either 2 or 20 mmol/l glucose for 30 min at 37°C. Aliquots (200 μl) of medium were collected for determination of insulin secretion. Total protein was measured as previously described [3].
Insulin secretion was also measured dynamically from monolayer cells, groups of 20 pseudoislets and human and mouse islets by perifusing in the presence of 2 and 20 mmol/l glucose as previously described [8]. Individual pseudoislets and human islets were also perifused in 2 and 20 mmol/l glucose. For the dynamic insulin measurement, 5 × 104 MIN6 cells were attached to the central part of poly-l-lysine-coated coverslips and cultured for 3 days. The samples were collected at 2 mmol/l glucose during either 10 min for individual islets or 20 min for other preparations. Subsequently, the medium was exchanged to either 20 mmol/l glucose, 2 mmol/l pyruvate, 20 mmol/l alpha-ketoisocaproic acid (KIC), 10 mmol/l KIC plus 10 mmol/l glutamine, or 30 mmol/l KCl, and sample collection was continued for another 20 min. In some experiments, LY294002 (50 μmol/l) or wortmannin (100 nmol/l for monolayers, pseudoislets and mouse islets; 1 μmol/l for human islets) was introduced into the perifusion medium 30 min prior to sampling and was present throughout the experiment. Insulin was measured by ELISA as previously described [8].
Simultaneous measurements of insulin and cytoplasmic Ca2+ concentration
For simultaneous measurements of insulin and cytoplasmic Ca2+ concentration ([Ca2+]c), pseudoislets were loaded with 1 μmol/l Fura-2 LR acetoxymethyl ester (TEFLabs, Inc., Austin, TX, USA) by incubating them for 60 min at 37°C in KRB HEPES buffer containing 2 mmol/l glucose and 1 mg/ml BSA. After rinsing, groups of islets were allowed to attach to the central part of poly-l-lysine-coated coverslips. The chamber was placed on the stage of an inverted microscope (Eclipse TE2000U; Nikon, Tokyo, Japan). The pseudoislets were perifused for 60 min with 2 mmol/l glucose in KRB HEPES buffer supplemented with 1 mg/ml BSA at 37°C at a rate of 160 μl/min. [Ca2+]c was recorded by dual wavelength fluorometry as previously described [9]. During the [Ca2+]c recordings, the perifusate was collected in 1 min intervals for subsequent insulin measurements.
To measure the effect of phosphatidylinositol 3-kinase (PI3K) inhibition, the attached pseudoislets were perifused for 30 min in 2 mmol/l glucose. Wortmannin (100 nmol/l) or LY294002 (50 μmol/l) was subsequently added and perifusion continued for another 30 min before [Ca2+]c was measured and fractions of perifusate were collected for insulin measurement in the continued presence of the inhibitors.
Western blotting
To determine the levels of specific proteins, western blotting was performed on MIN6 monolayers, MIN6 pseudoislets and human and mouse islets with some modifications as previously described [3]. Immunoblotting was conducted with antibodies against IRS-1, IRS-2, phosphorylated (p)-IRS-1 S636/639, p-IRS-1 S612, p-IRS-1 S307, p-Akt (S473), p-Akt (T308), p-S6 ribosomal protein (S235/236), total Akt, total S6 ribosomal protein, pancreatic and duodenal homeobox 1 (PDX1; Cell Signaling, Danvers, MA, USA), glucokinase, GLUT2 and actin (Santa Cruz Biotechnology, Santa Cruz, CA, USA). The immunoreactive bands were imaged with the Fluor-S MultiImager MAX (Bio-Rad, Hercules, CA, USA) and quantified with Quantity One software (Bio-Rad).
mRNA expression by real-time PCR
Total RNA was extracted from MIN6 monolayers and pseudoislets using the RNeasy Mini Kit (Qiagen GmbH). The reverse transcription reaction was performed with 1 μg total RNA using RT2 First-Strand Kit (Qiagen GmbH). The cDNA obtained was processed by quantitative real-time reverse transcriptase PCR of 84 genes involved in the mitochondrial electron transport chain and oxidative phosphorylation complexes, and 12 housekeeping genes including internal controls using the RT2 Profiler PCR Array kit (RT2 Profiler PCR Array Mouse Mitochondrial Energy Metabolism, PAMM-008; Qiagen GmbH) and a Stratagene Mx3000p real-time PCR system (Stratagene, La Jolla, CA, USA). The ΔΔCt-based fold change in pseudoislets compared with monolayers was obtained by uploading the raw threshold cycle data to an integrated web-based software package (RT2 Profiler PCR Array Data Analysis version 3.5; Qiagen GmbH) for the PCR array system, using the following formula: fold 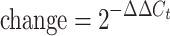 , where ΔΔCt = ΔCt (pseudoislets) − ΔCt (monolayers), and ΔCt = Ct (gene of interest) − Ct (average of housekeeping genes). Based on PCR array results, some genes were validated by real-time PCR. The real-time PCR product was quantified by measuring SYBR Green (Agilent Technologies, Santa Clara, CA, USA) fluorescent dye incorporation with ROX dye reference and normalised to the housekeeping genes β-actin (Actb), glyceraldehyde-3-phosphate dehydrogenase (GAPDH), hypoxanthine guanine phosphoribosyl transferase (Hprt) and heat shock protein 90 kDa alpha (cytosolic), class B member 1 (Hsp90ab1). The primers used are listed in electronic supplementary material (ESM) Table 1.
, where ΔΔCt = ΔCt (pseudoislets) − ΔCt (monolayers), and ΔCt = Ct (gene of interest) − Ct (average of housekeeping genes). Based on PCR array results, some genes were validated by real-time PCR. The real-time PCR product was quantified by measuring SYBR Green (Agilent Technologies, Santa Clara, CA, USA) fluorescent dye incorporation with ROX dye reference and normalised to the housekeeping genes β-actin (Actb), glyceraldehyde-3-phosphate dehydrogenase (GAPDH), hypoxanthine guanine phosphoribosyl transferase (Hprt) and heat shock protein 90 kDa alpha (cytosolic), class B member 1 (Hsp90ab1). The primers used are listed in electronic supplementary material (ESM) Table 1.
Glucose oxidation rate
MIN6 monolayer cells and pseudoislets were harvested, in triplicate per observation, and transferred to incubation vials containing KRB HEPES buffer, supplemented with 20 mmol/l glucose containing (15.5 GBq/mol) d-[U-14C]glucose. The vials were incubated for 90 min at 37°C under an atmosphere of 95/5% O2/CO2, with slow shaking. Metabolism was arrested by adding 17 μmol/l antimycin A. Subsequently, the released labelled 14CO2 was trapped in 250 μl hyamine hydroxide during incubation at 37°C for 2 h, and the radioactivity was measured by liquid scintillation counting. Cell numbers in aliquots of the harvested MIN6 monolayers were counted in a Becton Dickinson FACSCalibur flow cytometer (Becton Dickinson, San Jose, CA, USA). As for the pseudoislets, triplicate groups of 50 islets harvested in parallel to those incubated with labelled glucose were dispersed as described above, and the cell number was counted in the flow cytometer. The glucose oxidation rates were subsequently expressed per number of cells.
Data analysis
Results are presented as means ± SEM. Statistical significance for the difference between two conditions was analysed using the Student’s t test. Multiple comparisons between different groups were assessed using ANOVA followed by Bonferroni’s post hoc test. A value p < 0.05 was considered statistically significant.
Results
GSIS
In static incubation, pseudoislet insulin release increased approximately sixfold when the glucose concentration was raised from 2 to 20 mmol/l glucose, whereas for the monolayers it doubled (Fig. 1). The magnitude of the secretory response of human islets was similar to that of the MIN6 pseudoislets.
Fig. 1.

Insulin secretion from MIN6 monolayers (white bars), MIN6 pseudoislets (grey bars) and human islets (black bars) exposed to 2 or 20 mmol/l glucose for 30 min. The level of insulin released to the medium was measured and normalised to protein level. Results are means ± SEM of five separate experiments. *p < 0.05 compared with 2 mmol/l glucose; † p < 0.05 compared with pseudoislets
Perifusion of individual MIN6 pseudoislets in the presence of 2 mmol/l glucose showed insulin oscillations with a periodicity of 4.3 ± 0.3 min, which were similar to those observed for human islets (4.1 ± 0.4 min) (ESM Fig. 1). Augmentation of the glucose concentration to 20 mmol/l increased the amplitude of the insulin pulses without altering the periodicity of the pseudoislets and human islets (ESM Fig. 1).
Insulin secretion dynamics in response to nutrient and depolarising stimuli
Dynamic insulin secretion from pseudoislets in response to 20 mmol/l glucose, pyruvate, KIC, KIC in combination with glutamine, and high KCl was analysed and compared with that generated from monolayer cells and human islets (Fig. 2, Table 1). Secreted insulin level was determined in the presence of 2 mmol/l glucose for 10 min prior to the elevation of glucose or the introduction of other secretagogues (basal), during the first 10 min after stimulation, corresponding to first-phase insulin secretion and for ten subsequent minutes corresponding to second-phase insulin secretion. A rise in the glucose concentration to 20 mmol/l caused a significant increase in both the first and second phases of insulin secretion in the pseudoislets, monolayers and human islets (Fig. 2a, Table 1). The augmentation of insulin secretion from the pseudoislets was significantly higher than that from the monolayers and similar to that observed from human islets.
Fig. 2.

Insulin secretion from monolayers (dotted line) and groups of pseudoislets (solid line) and human islets (dashed line) in the presence of glucose (Glu) (a), pyruvate (Pyr) (b), KIC (c), KIC plus glutamine (d), or a high concentration of KCl (e). Insulin released to the media was measured and normalised to protein level. Results are representative of five separate experiments
Table 1.
Insulin secretion from MIN6 monolayer cells, pseudoislets and human islets in the presence of different secretagogues
| Secretagogue | Insulin secretion [fmol (μg protein)−1] | ||
|---|---|---|---|
| Monolayer cells | Pseudoislets | Human islets | |
| Glu 2 mmol/l | 5.1 ± 0.8 | 6.4 ± 0.1 | 8.4 ± 0.6 |
| Glu 20 mmol/l, first phase | 7.5 ± 0.8*† | 36.1 ± 0.7* | 54.3 ± 8.6* |
| Glu 20 mmol/l, second phase | 6.9 ± 0.8*† | 45.1 ± 12.3* | 43.6 ± 7.3* |
| Glu 2 mmol/l | 6.8 ± 0.2 | 6.3 ± 0.8 | 11.5 ± 3.2 |
| Pyr first phase | 8.4 ± 0.2 | 11.3 ± 3.7* | 14.7 ± 1.3† |
| Pyr second phase | 8.0 ± 0.9† | 15.7 ± 1.7* | 10.7 ± 2.9 |
| Glu 2 mmol/l | 7.1 ± 0.4 | 8.7 ± 1.6 | 16.7 ± 6.2 |
| KIC first phase | 7.9 ± 0.3† | 21.5 ± 2.5* | 42.9 ± 10.2*† |
| KIC second phase | 11.9 ± 0.6* | 15.3 ± 2.3* | 39.3 ± 14.5*† |
| Glu 2 mmol/l | 7.9 ± 0.2 | 7.1 ± 0.16 | 14.4 ± 4.6 |
| KIC + Gln first phase | 13.6 ± 0.5* | 17.2 ± 2.3* | 59.0 ± 3.5*† |
| KIC + Gln second phase | 11.9 ± 0.5*† | 20.5 ± 2.5* | 43.2 ± 13.8*† |
| Glu 2 mmol/l | 4.4 ± 0.1 | 8.5 ± 0.61 | 13.0 ± 4.5 |
| KCl first phase | 10.1 ± 0.8* | 15.2 ± 0.3* | 37.8 ± 8.9*† |
| KCl second phase | 12.1 ± 0.3* | 7.2 ± 2.7 | 6.1 ± 0.6* |
Results are means ± SEM of between five and eight separate experiments, which were obtained by calculating the area under the curve from each group of the experiments in Fig. 3
Cells and islets were perifused in the presence of 2 or 20 mmol/l glucose (Glu), 2 mmol/l pyruvate (Pyr), 20 mmol/l KIC, and 10 mmol/l KIC plus 10 mmol/l glutamine (Gln) or 30 mmol/l KCl. Secreted insulin was measured for 10 min at 2 mmol/l glucose and in the initial (first phase) and subsequent (second phase) 10 min after the introduction of secretagogues
*p < 0.05 compared with 2 mmol/l glucose; † p < 0.05 compared with pseudoislets
When 2 mmol/l pyruvate was introduced, a small, initial and transient increase in insulin secretion was observed for all preparations (Fig. 2b). In the pseudoislets, but not in the monolayer cells and human islets, pyruvate induced a significant rise in second-phase insulin secretion (Table 1).
KIC (20 mmol/l) induced significant first- and second-phase insulin release from the pseudoislets but increased only the second phase of insulin secretion from the monolayers. In the human islets, KIC elicited a pronounced rise in both first- and second-phase insulin secretion. When the pseudoislets were exposed to 10 mmol/l KIC plus 10 mmol/l glutamine, first- and second-phase insulin secretion was somewhat augmented compared with secretion observed in the presence of KIC alone (Fig. 2d, Table 1). In monolayer cells, the combination caused a small but significant first-phase insulin secretion. In human islets, the combination elicited an accentuated first-phase and second-phase insulin secretion. In the pseudoislets, depolarisation by KCl caused a prompt transient response involving only first-phase secretion (Fig. 2e, Table 1). A similar but more pronounced first-phase secretory response was observed in human islets. In contrast, monolayer cells showed a significant increase in the second-phase insulin secretory response as well (Table 1).
Expression of genes related to mitochondrial respiration and electron transport
To investigate how genes encoding components of the mitochondrial electron transport and oxidative phosphorylation complex were affected in the pseudoislets and MIN6 monolayers, the transcript levels of 84 genes were determined where 77 genes were expressed in both pseudoislets and monolayer cells; a majority (76%) of the genes showed a 1.4-fold or more increase in the pseudoislets compared with the monolayers (Table 2). Genes that showed at least a twofold up- or downregulation in the array were also validated by quantitative RT-PCR (Table 2).
Table 2.
Expression of mitochondrial respiration and electron transport genes in MIN6 monolayer cells and MIN6 pseudoislets
| Gene symbol | Fold regulation (PCR array) | Validation (qRT-PCR) | Gene symbol | Fold regulation (PCR array) | Validation (qRT-PCR) |
|---|---|---|---|---|---|
| Lhpp | 1.4658 | Ndufa7 | 2.3978 | 3.10 ± 0.42 | |
| Atp4a | −1.1394 | Ndufa8 | 2.0163 | 1.85 ± 0.13* | |
| Atp5a1 | 1.8554 | Ndufab1 | −2.2788 | 1.92 ± 0.34* | |
| Atp5b | 1.7553 | Ndufb10 | 2.073 | 1.96 ± 0.35* | |
| Atp5c1 | 1.7432 | Ndufb2 | 1.2939 | ||
| Atp5d | 1.6955 | Ndufb3 | 1.2156 | ||
| Atp5f1 | 1.8046 | Ndufb4 | 1.5175 | ||
| Atp5g1 | 1.2498 | Ndufb5 | 1.4557 | ||
| Atp5g2 | 1.8046 | Ndufb6 | 1.6264 | ||
| Atp5g3 | 2.0874 | 1.61 ± 0.08* | Ndufb7 | 1.4658 | |
| Atp5h | 1.3395 | Ndufb8 | 1.6955 | ||
| Atp5j | 1.7798 | Ndufb9 | 1.9208 | ||
| Atp5j2 | 1.6378 | Ndufc1 | 2.0303 | 1.88 ± 0.34* | |
| Atp5o | 1.7073 | Ndufc2 | 1.7675 | ||
| Atp6v0a2 | 1.5602 | Ndufs1 | −1.1878 | ||
| Atp6v1c2 | 3.4623 | 2.48 ± 0.59* | Ndufs2 | 1.1032 | |
| Bcs1l | 1.7675 | Ndufs3 | 1.7922 | ||
| Cox11 | 2.2372 | 1.73 ± 0.18* | Ndufs4 | −1.0485 | |
| Cox4i1 | 2.6239 | 2.50 ± 0.69* | Ndufs5 | 1.1742 | |
| Cox5a | 1.1421 | Ndufs6 | 2.0445 | 3.58 ± 0.83* | |
| Cox5b | 1.6264 | Ndufs7 | 2.2528 | 2.54 ± 0.47* | |
| Cox6a1 | 1.5281 | Ndufs8 | 1.9885 | ||
| Cox6a2 | –3.383 | −2.37 ± 0.28* | Ndufv1 | 2.0874 | 3.17 ± 0.20* |
| Cox6b1 | 1.4257 | Ndufv2 | 2.3648 | 2.42 ± 0.35* | |
| Cox6b2 | 1.9885 | Ndufv3 | 2.0587 | 4.41 ± 0.56* | |
| Cox6c | 1.8172 | Oxa1l | 2.2842 | 2.14 ± 0.38* | |
| Cox7a2 | 1.1824 | Ppa1 | −1.8382 | ||
| Cox7a2l | 2.9317 | 3.17 ± 0.70* | Ppa2 | 1.285 | |
| Cox7b | 1.571 | Sdha | 2.073 | 1.56 ± 0.17* | |
| Cox8a | 2.1019 | 1.82 ± 0.31* | Sdhb | 1.4061 | |
| Cox8c | 1.6955 | Sdhc | 1.4159 | ||
| Cyc1 | 1.285 | Sdhd | 1.8298 | ||
| Ndufa1 | 1.9476 | Uqcr11 | 1.2326 | ||
| Ndufa10 | 1.507 | Uqcrc1 | 1.9612 | ||
| Ndufa2 | 1.7073 | Uqcrc2 | 1.9612 | ||
| Ndufa3 | 1.604 | Uqcrfs1 | 1.8683 | ||
| Ndufa4 | 1.2326 | Uqcrh | 2.1166 | 3.5 ± 0.6* | |
| Ndufa5 | 1.8298 | Uqcrq | 1.8426 | ||
| Ndufa6 | 2.3161 | 2.77 ± 0.52* |
Results are means ± SEM of four separate experiments
Differential mRNA expression levels of genes encoding the components of mitochondrial electron transport and oxidative phosphorylation in MIN6 pseudoislets compared with monolayers was measured by PCR array and validated by quantitative RT-PCR (qRT-PCR)
*p < 0.05 compared with MIN6 monolayer cells
Glucose oxidation
MIN6 pseudoislets had a markedly higher glucose oxidation rate at 20 mmol/l glucose (3.57 ± 0.63 [pmol glucose/cell × 90 min]) compared with cells growing in monolayers (1.09 ± 0.34 [pmol glucose/cell × 90 min]) (p < 0.006 using an unpaired Student’s t test; n = 6 in both groups). The technique did not allow a direct comparison between pseudoislets and adherent monolayer cells, and we cannot exclude the possibility that the suspended cells would have shown a different rate of oxidation compared with the monolayers.
Expression of beta cell proteins
Levels of proteins connected with beta cell differentiation and function were measured in the pseudoislets, monolayers and human islets. In agreement with previous reports [7, 10] a similar insulin content was observed in the pseudoislets and monolayer cells (ESM Fig. 2). These levels were, however, only half those obtained for the human islets. Levels of PDX1, glucokinase and GLUT2 did not differ between the monolayers, pseudoislets and human islets (ESM Fig. 3). PDX-1 protein and glucokinase mRNA levels have been demonstrated to be similar in MIN6 cells and mouse islets [11, 12] In addition, both MIN6 cells and mouse islets express high levels of GLUT2 mRNA [13].
IRS-1 phosphorylation and PI3K activity
The total levels of IRS-1 in the pseudoislets, as well as the total levels of IRS-2 (data not shown), were no different from those obtained for the monolayer cells (Fig. 3a). Levels of the inhibitory phosphorylation at S636/639 of IRS-1 were lower in pseudoislets than monolayer cells (Fig. 3b), but no differences were observed for inhibitory sites S307 and S612 (data not shown). We also measured whether S636/639 phosphorylation affected PI3K activity by measuring the level of Akt phosphorylation at T308 as this site was reported to correlate with PI3K activity in other cell types [14, 15]. In MIN6 monolayers, phosphorylation of Akt at site T308 was significantly reduced compared with that seen in the pseudoislets (Fig. 4). When phosphorylation of IRS-1 at S636/639 was measured in intact and dispersed human and mouse islets, levels were lower in intact islets than dispersed cells (Fig. 3d,f).
Fig. 3.

Levels of total IRS-1 and IRS-1 phosphorylated at S636/639 (p-IRS-1) were measured by western blotting in MIN6 pseudoislets (PI) and monolayers (MO) (a,b), human islets (HU-I) and dispersed human islets (HU-D) (c,d), and mouse islets (MOU-I) and dispersed mouse islets (MOU-D) (e,f). Protein levels were normalised to actin for IRS-1, and total IRS-1 for p-IRS1 (S636/639). Results are means ± SEM of four or five separate experiments. *p < 0.05 compared with pseudoislets or intact islets
Fig. 4.
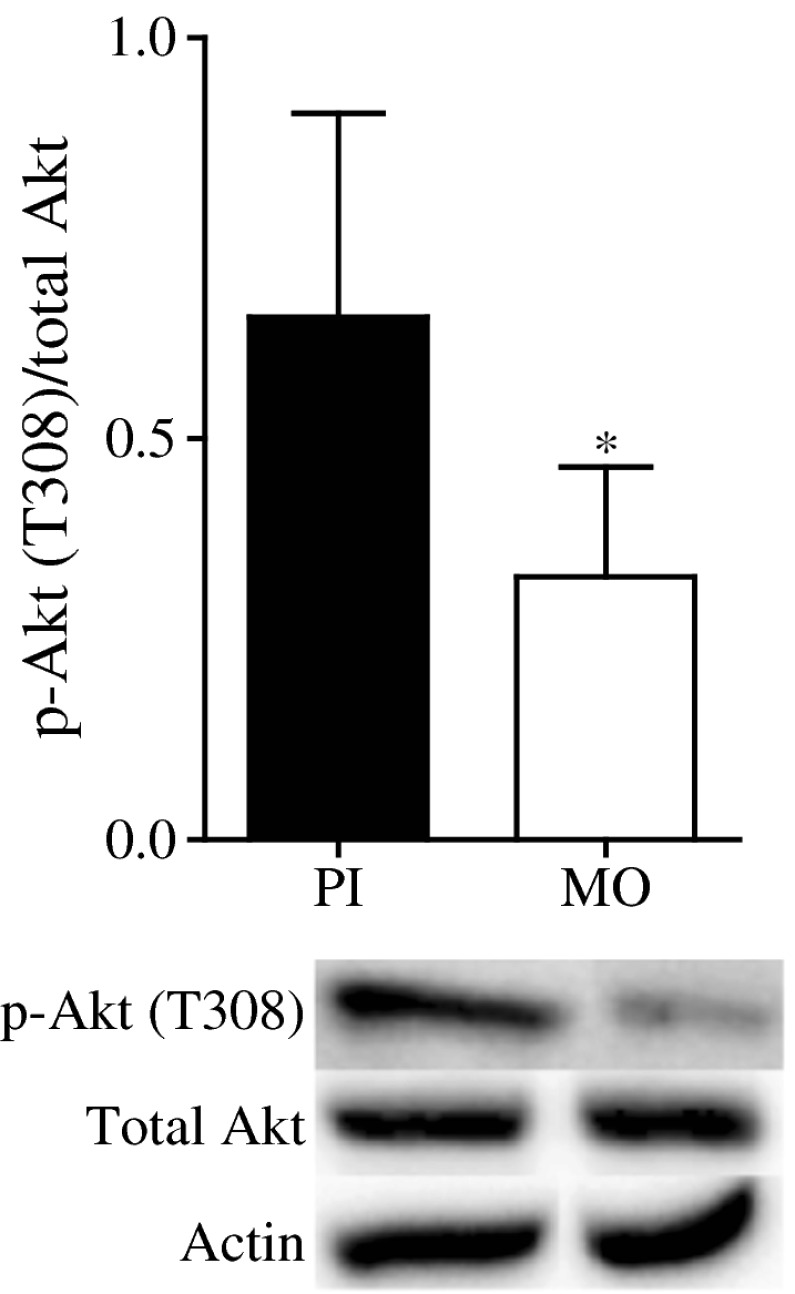
Levels of Akt phosphorylated at T308 were measured by western blotting in MIN6 pseudoislets (PI) and monolayers (MO). Protein levels were normalised to actin for Akt, and total Akt for p-Akt (T308). Results are means ± SEM of four separate experiments. *p < 0.05 compared with pseudoislets
To determine whether the differences in p-IRS-1 at S636/639 were contributing to the observed differences in secretory patterns via PI3K, we measured insulin secretion in the presence of the PI3K inhibitors LY294002 and wortmannin. PI3K inhibition was further studied by measuring the phosphorylation of Akt. LY294002 (50 μmol/l) blocked Akt phosphorylation in all preparations. Similar results were obtained with 50 nmol/l wortmannin in MIN6 cells, pseudoislets and mouse islets, whereas 1 μmol/l wortmannin was required to obtain inhibition in human islets (data not shown). The inhibitor LY294002, which was introduced 30 min prior to glucose elevation, decreased GSIS from the pseudoislets and human islets but did not affect the already low secretion from the monolayer cells during static incubation (ESM Fig. 4). In dynamic measurements, basal insulin secretion from pseudoislets, monolayer cells and human and mouse islets perifused in the presence of 2 mmol/l glucose was not affected by either LY294002 or wortmannin (Fig. 5, Table 3). When the glucose concentration was increased to 20 mmol/l, insulin secretion from the pseudoislets and human and mouse islets exposed to either of the inhibitors was significantly reduced in both the first and second phases (Fig. 5b–d, Table 3). In contrast, a reduction in the first but not the second, phase of GSIS was observed in monolayer cells (Fig. 5a, Table 3). Total levels of phosphorylation of Akt and S6 ribosomal protein, which were located downstream of PI3K, did not differ between monolayers and pseudoislets (data not shown). When MIN6 monolayers and pseudoislets were treated with LY294002 for 60 min, an increased level of IRS-1 phosphorylation at S636/639 was observed in pseudoislets compared with monolayer cells (Fig. 6), but no differences were observed at S307 and S612 (data not shown).
Fig. 5.

Glucose (Glu)-stimulated insulin secretion from MIN6 monolayers (a), pseudoislets (b), human islets (c) and mouse islets (d) in the absence (solid line) or presence of the PI3K inhibitors wortmannin (grey dotted line) or LY294002 (black dashed line). Insulin released to the medium was measured and normalised to protein. Results are representative of four or five separate experiments
Table 3.
Effects of PI3K inhibition on GSIS from MIN6 monolayer cells, pseudoislets and human and mouse islets
| PI3K inhibitor | Insulin secretion [fmol (μg protein)−1] | |||
|---|---|---|---|---|
| Monolayer | Pseudoislets | Human islets | Mouse islets | |
| Glu 2 mmol/l | 7.4 ± 2.6 | 6.2 ± 0.8 | 8.6 ± 0.3 | 5.3 ± 0.9 |
| Glu 20 mmol/l first phase | 18.2 ± 1.5* | 40.1 ± 4.5* | 65.1 ± 6.4* | 31.5 ± 10.2* |
| Glu 20 mmol/l second phase | 12.7 ± 1.8* | 55.5 ± 11.9* | 51.7 ± 5.5* | 43.5 ± 11.3* |
| Glu 2 mmol/l + W | 5.9 ± 0.6 | 5.8 ± 0.4 | 9.7 ± 0.4 | 9.6 ± 0.9 |
| Glu 20 mmol/l + W first phase | 9.4 ± 1.3*† | 12.9 ± 0.6*† | 20.5 ± 0.5*† | 19.3 ± 7.3*† |
| Glu 20 mmol/l + W second phase | 10.8 ± 1.8* | 13.2 ± 0.8*† | 17.6 ± 0.5*† | 22.7 ± 5.2*† |
| Glu 2 mmol/l + LY | 4.4 ± 1.0 | 8.3 ± 0.3 | 11.9 ± 2.3 | 8.3 ± 1.2 |
| Glu 20 mmol/l + LY first phase | 11.6 ± 1.9*† | 12.6 ± 0.3*† | 22.7 ± 3.1*† | 23.9 ± 2.7*† |
| Glu 20 mmol/l + LY second phase | 11.4 ± 2.5* | 11.5 ± 0.2*† | 19.1 ± 2.9*† | 27.0 ± 5.2*† |
Results are means ± SEM of four or five separate experiments, which were obtained by calculating the area under the curve from Fig. 7
Cells and islets, exposed or not to the PI3K inhibitor wortmannin (W) or LY294002 (LY), were perifused. Secreted insulin was measured for 10 min at 2 mmol/l glucose (Glu) and in the initial (first phase) and subsequent (second phase) 10 min after raising the glucose concentration to 20 mmol/l
*p < 0.05 compared with 2 mmol/l glucose; † p < 0.05 compared with control
Fig. 6.

Levels of IRS-1 phosphorylated at S636/639 (p-IRS-1) were measured by western blotting and normalised to total IRS-1 in MIN6 monolayers and pseudoislets with or without the PI3K inhibitor LY294002 (LY). Results are representative of five separate experiments. *p < 0.05 compared with control. MO, monolayers; PI pseudoislets
Next, we investigated the extent to which the reduction in GSIS observed in the pseudoislets exposed to PI3K inhibitors involved alterations in [Ca2+]c. In control pseudoislets, a rise in the glucose concentration from 2 to 20 mmol/l elicited a transient decrease in [Ca2+]c followed by a marked increase that peaked within 2 min after the elevation of glucose (Fig. 7a, Table 4). After the initial peak, [Ca2+]c decreased to a plateau from which distinct oscillations (∼2/min) appeared. The [Ca2+]c response was paralleled by a pronounced initial insulin secretory peak followed by oscillatory insulin levels (Fig. 7a). In pseudoislets pretreated with LY294002, a blunted GSIS was paralleled by a [Ca2+]c response that was similar to control but with lower basal [Ca2+]c and a higher frequency of the glucose-induced oscillations (Fig. 7b, Table 4). [Ca2+]c was also recorded in pseudoislets pretreated with wortmannin. Although this drug markedly reduced insulin secretion (Fig. 5b), it had no effect on [Ca2+]c (Table 4). The effects of acute inhibition of PI3K on [Ca2+]c were also investigated in the pseudoislets. While LY294002 markedly accelerated the [Ca2+]c oscillations (Fig. 7c), there was no effect of wortmannin (Fig. 7d).
Fig. 7.
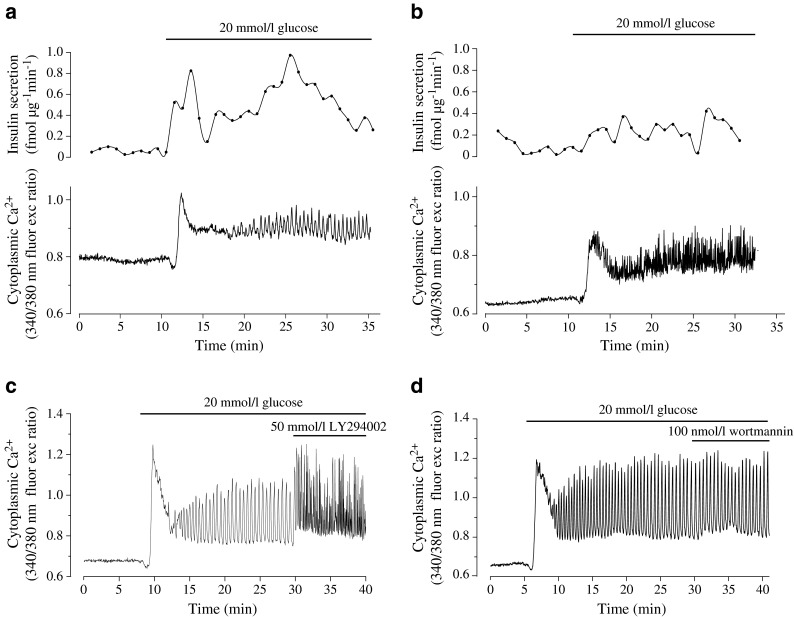
Glucose-induced changes in [Ca2+]c and insulin secretion in MIN6 pseudoislets exposed to PI3K inhibitors. Simultaneous recordings of [Ca2+]c and insulin release in control (a) or LY294002-treated (prior to and during perifusion) pseudoislets (b). Changes in [Ca2+]c in the pseudoislets after acute exposure to LY94002 (c) or wortmannin (d). Whereas the parallel recordings of insulin and [Ca2+]c are representative of three independent experiments, the [Ca2+]c recordings are representative of 26 (a), 13 (b), 7 (c) and 8 (d) pseudoislets, respectively, from between two and eight independent experiments; fluor exc, fluorescence excitation
Table 4.
Effects of PI3K inhibition on glucose-induced [Ca2+]c responses in MIN6 pseudoislets
| Measurement | Control (n = 26) | LY pretreatment (n = 13) | W pretreatment (n = 6) |
|---|---|---|---|
| Basal level (nmol/l) | 118 ± 5 | 88 ± 9* | 102 ± 5 |
| Initial lowering (nmol/l) | 16 ± 4 | 7 ± 1 | 15 ± 1 |
| Response delay (min) | 1.37 ± 0.03 | 1.52 ± 0.09 | 1.34 ± 0.06 |
| Glucose-induced peak amplitude (nmol/l) | 133 ± 11 | 134 ± 25 | 176 ± 12 |
| Average stimulated level (nmol/l) | 180 ± 4 | 148 ± 18 | 199 ± 7 |
| Oscillation frequency (min−1) | 2.18 ± 0.09 | 8.68 ± 0.56* | 2.74 ± 0.06 |
Results are means ± SEM for the number of pseudoislets indicated
[Ca2+]c was measured in pseudoislets during an increase in the glucose concentration from 2 (basal) to 20 (stimulated) mmol/l in the absence (control) or presence of LY294002 (LY) or wortmannin (W). Basal [Ca2+]c, the magnitude of the glucose-induced initial lowering of [Ca2+]c, the time between the elevation of the glucose concentration and the increase in [Ca2+]c, the amplitude of the initial glucose-induced increase in [Ca2+]c, the average [Ca2+]c under stimulated conditions as well as the frequency of glucose-induced [Ca2+]c oscillations were quantified for each pseudoislet
*p < 0.05 compared with control
Discussion
The question of why beta cells situated in the islet of Langerhans perform better than separated beta cells was addressed using insulin-secreting MIN6 cells arranged as pseudoislets or monolayers [7, 10]. A number of mechanisms, including intercellular communication by direct cell–cell contact and an exchange of small molecules through gap junctions and through paracrine effects have been suggested to explain the phenomenon [7, 10, 16–18]. Indeed, in MIN6 pseudoislets, the calcium-dependent cell adhesion molecule E-cadherin and the gap junction protein connexin 36, which are required for proper insulin secretion, were upregulated compared with monolayer cells [7, 18]. In addition, increases in ins1 mRNA levels were higher in the pseudoislets than the monolayers [19]. In the present study, MIN6 cells grown as pseudoislets showed a higher secretory capacity in response to glucose, in agreement with previous reports [7, 16]. The notion of the insulin secretory characteristics of pseudoislets being similar to those of primary islets was further strengthened when the detailed secretory dynamics were investigated in the pseudoislets. Pulsatile insulin secretion was observed from pseudoislets, with pulse durations and amplitude regulation by glucose similar to those observed for primary islets [20–22]. The enhanced secretory characteristics of MIN6 pseudoislets in the present study involved both first- and second-phase insulin secretion, which were equally increased compared with the monolayer cells, in agreement with a previous report [10].
Impaired glucokinase activity and a lowered expression of GLUT2 have been connected with impaired insulin secretion [23, 24]. The similar levels of these two proteins observed in the present and a previous study [25] suggest that the causes for the reduced secretion observed in monolayer cells lie distal to the initial steps of glucose metabolism. When pyruvate was administered extracellularly, the tricarboxylic acid induced insulin secretion from pseudoislets. The inability of pyruvate to elicit insulin secretion has been connected with low levels of the monocarboxylate transporter as well as a low expression of lactate dehydrogenase in the beta cell [26, 27]. KIC is a mitochondrial substrate and acts as a potent insulin secretagogue [28]. The keto acid induced both first- and second-phase of insulin secretion from MIN6 pseudoislets [7]. When glutamine was also included, insulin secretion was enhanced from MIN6 pseudoislets but not to the same degree as was observed for human islets.
In this context, species differences cannot be ruled out. Direct comparisons of secretory responses between pseudoislets and mouse islets for some of the secretagogues used here and in a previous study [16] showed that the secretory responses were equivalent. For the remaining secretagogues, mouse islet secretory characteristics have been studied [29, 30] but not directly compared with those of MIN6 pseudoislets. Rather than performing a direct comparison between MIN6 pseudoislets and mouse islets for the latter secretagogues, we conducted these experiments with human islets. This choice was based on the fact that there are no studies in which the secretory characteristics of MIN6 pseudoislets and human islets have been directly compared.
In this comparison, we could not exclude the influence of non-beta cells in the human islets as such an influence is present [31] and is exemplified by studies showing that glutamine can stimulate glucagon secretion [32] and that pyruvate is preferentially oxidised in non-beta cells in both mouse [33] and human [34] islets. In addition, the extent to which alpha cells influence insulin secretion from pseudoislets has been addressed [35]. Based on these results and the observation that genes encoding the mitochondrial electron transport and oxidative phosphorylation complex as well as glucose oxidation were upregulated in pseudoislets compared with monolayers, we propose that enhanced mitochondrial metabolism contributed to the improved secretory capacity of cells in the pseudoislets.
In addition to enhanced metabolism, cell aggregation seems to be associated with alterations in IRS phosphorylation. Paracrine insulin signalling can be envisaged to be more pronounced when cells are aggregated in the islet-like structures, and we therefore wanted to investigate the insulin signalling pathway. Insulin secreted from beta cells binds to the insulin receptor, from which the signal is transduced via IRSs. The IRS proteins have multiple effects on the beta cell, including on survival via the PI3K/Akt pathway and on insulin gene transcription via PI3K/mTOR/p70s6 kinase [36–38]. It has been reported that manipulation of IRS-1 levels affects both the secretion and synthesis of insulin [39, 40]. Although we observed no significant differences in the total levels of the IRS proteins in the present study, levels of the inhibitory serine phosphorylation site S636/S639 of IRS-1 were higher in monolayers than pseudoislets. The differential IRS-1 phosphorylation at site S636/639 is also proposed to contribute to the increased insulin secretion observed with pseudoislets and monolayer cells. Such serine/threonine phosphorylations uncouple the IRS proteins from their upstream and downstream effectors [41].
In human skeletal muscle cells, increased S636 phosphorylation of IRS-1 led to a reduced association of IRS-1 with PI3K [42]. A similar mechanism may be present in beta cells, since inhibitors of PI3K significantly reduced first-phase insulin secretion from pseudoislets and human islets but had only minor effects on the secretory patterns of monolayer cells. In accordance with this finding, PI3K inhibition has previously been reported to decrease the first phase of insulin secretion from MIN6 monolayers and mouse islets [43]. In contrast, other studies have suggested that PI3K inhibition results in increased GSIS [44–47]. Differences in experimental design may account for some of the discrepancy. In support for a role of PI3K as a positive regulator of insulin secretion, mouse models lacking expression of PI3K regulatory subunits [48] or of the catalytic subunit of the type 1B PI3K isoform [49] show reduced GSIS. PI3K inhibition does not, however, affect glucose metabolism [44, 50]. Our study showed that the suppression of insulin secretion by PI3K inhibitors could not be explained by a reduction in [Ca2+]c, and PI3K seems to act at a distal step of the secretory process. Accordingly, wortmannin had no influence on the [Ca2+]c responses, and the increased frequency of [Ca2+]c oscillations caused by LY294002 is probably related to the ability of this compound to block voltage-gated K+ channels (Kv channels) in β-cells [44].
Acute inhibition of PI3K by wortmannin has previously been demonstrated not to have any effect on glucose metabolism or ATP content [44, 50]. Thus, there is no evidence for a direct causal link between the changes in metabolism and IRS-1 phosphorylation observed between MIN6 pseudoislets and monolayer cells. In conclusion, the present study indicates that enhanced mitochondrial metabolism and the differential phosphorylation of IRS-1 contribute towards the enhanced secretory capacity of beta cells situated in islets compared with dissociated beta cells.
Electronic supplementary material
Below is the link to the electronic supplementary material.
(PDF 56 kb)
(PDF 42 kb)
(PDF 44 kb)
(PDF 57 kb)
(PDF 34 kb)
Acknowledgements
We would like to thank U. Landegren and M. Kamali-Moghaddam (Department of Immunology, Genetics and Pathology, Uppsala University, Uppsala, Sweden) for assistance with the PCR array.
Funding
Grants from the Swedish Medical Research Council (72X-14019, 67X-14643), Swedish Diabetes Association, Family Ernfors Foundation, EFSD/MSD, Novo-Nordisk Foundation and Uppsala University supported the study.
Duality of interest
The authors declare there is no duality of interest associated with this manuscript.
Contribution statement
AC performed secretory, PCR and western blot experiments and participated in study design, data analysis, interpretation and writing of the manuscript. OD designed the [Ca2+]c experiment and participated in the acquisition of data, data analysis and writing of the manuscript. AT participated in [Ca2+]c data analysis and manuscript writing. SS participated in the design of the glucose oxidation experiment, acquisition of data, data analysis and manuscript writing. PB participated in the study design, data interpretation, manuscript writing and overall supervision of the study. All authors critically revised and approved the final version of the manuscript.
Abbreviations
- [Ca2+]c
Cytoplasmic Ca2+ concentration
- GSIS
Glucose-stimulated insulin secretion
- KIC
Alpha-ketoisocaproic acid
- PDX1
Pancreatic and duodenal homeobox 1
- PI3K
Phosphotidylinositol 3-kinase
References
- 1.Orci L, Unger RH. Functional subdivision of islets of Langerhans and possible role of D cells. Lancet. 1975;2:1243–1244. doi: 10.1016/S0140-6736(75)92078-4. [DOI] [PubMed] [Google Scholar]
- 2.Miyazaki JI, Araki K, Yamato E, et al. Establishment of a pancreatic beta-cell line that retains glucose-inducible insulin-secretion: special reference to expression of glucose transporter isoforms. Endocrinology. 1990;127:126–132. doi: 10.1210/endo-127-1-126. [DOI] [PubMed] [Google Scholar]
- 3.Thörn K, Bergsten P. Fatty acid-induced oxidation and triglyceride formation is higher in insulin-producing MIN6 cells exposed to oleate compared to palmitate. J Cell Biochem. 2010;111:497–507. doi: 10.1002/jcb.22734. [DOI] [PubMed] [Google Scholar]
- 4.Pipeleers D, Intveld P, Maes E, Vandewinkel M. Glucose-induced insulin release depends on functional cooperation between islet cells. Proc Natl Acad Sci USA Biol Sci. 1982;79:7322–7325. doi: 10.1073/pnas.79.23.7322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Salomon D, Meda P. Heterogeneity and contact-dependent regulation of hormone secretion by individual B cells. Exp Cell Res. 1986;162:507–520. doi: 10.1016/0014-4827(86)90354-X. [DOI] [PubMed] [Google Scholar]
- 6.Hopcroft DW, Mason DR, Scott RS. Insulin secretion from perifused rat pancreatic pseudoislets. In Vitro Cell Dev Biol. 1985;21:421–427. doi: 10.1007/BF02620828. [DOI] [PubMed] [Google Scholar]
- 7.Hauge-Evans AC, Squires PE, Persaud SJ, Jones PM. Pancreatic beta-cell-to-beta-cell interactions are required for integrated responses to nutrient stimuli: enhanced Ca2+ and insulin secretory responses of MIN6 pseudoislets. Diabetes. 1999;48:1402–1408. doi: 10.2337/diabetes.48.7.1402. [DOI] [PubMed] [Google Scholar]
- 8.Bergsten P, Hellman B. Glucose-induced amplitude regulation of pulsatile insulin secretion from individual pancreatic islets. Diabetes. 1993;42:670–674. doi: 10.2337/diabetes.42.5.670. [DOI] [PubMed] [Google Scholar]
- 9.Dyachok O, Tufveson G, Gylfe E. Ca2+-induced Ca2+ release by activation of inositol 1,4,5-trisphosphate receptors in primary pancreatic β-cells. Cell Calcium. 2004;36:1–9. doi: 10.1016/j.ceca.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 10.Luther MJ, Hauge-Evans A, Souza KL, et al. MIN6 beta-cell-beta-cell interactions influence insulin secretory responses to nutrients and non-nutrients. Biochem Biophys Res Commun. 2006;343:99–104. doi: 10.1016/j.bbrc.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 11.Alejandro EU, Lim GE, Mehran AE, et al. Pancreatic beta-cell Raf-1 is required for glucose tolerance, insulin secretion, and insulin 2 transcription. FASEB J. 2011;25:3884–3895. doi: 10.1096/fj.10-180349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Quintens R, Hendrickx N, Lemaire K, Schuit F. Why expression of some genes is disallowed in beta-cells. Biochem Soc Trans. 2008;36:300–305. doi: 10.1042/BST0360300. [DOI] [PubMed] [Google Scholar]
- 13.Ishihara H, Asano T, Tsukuda K, et al. Pancreatic beta cell line MIN6 exhibits characteristics of glucose metabolism and glucose-stimulated insulin secretion similar to those of normal islets. Diabetologia. 1993;36:1139–1145. doi: 10.1007/BF00401058. [DOI] [PubMed] [Google Scholar]
- 14.Alessi DR, James SR, Downes CP, et al. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr Biol. 1997;7:261–269. doi: 10.1016/S0960-9822(06)00122-9. [DOI] [PubMed] [Google Scholar]
- 15.Stokoe D, Stephens LR, Copeland T, et al. Dual role of phosphatidylinositol-3,4,5-trisphosphate in the activation of protein kinase B. Science. 1997;277:567–570. doi: 10.1126/science.277.5325.567. [DOI] [PubMed] [Google Scholar]
- 16.Kelly C, Guo H, McCluskey JT, Flatt PR, McClenaghan NH. Comparison of insulin release from MIN6 pseudoislets and pancreatic islets of Langerhans reveals importance of homotypic cell interactions. Pancreas. 2010;39:1016–1023. doi: 10.1097/MPA.0b013e3181dafaa2. [DOI] [PubMed] [Google Scholar]
- 17.Dahl U, Sjodin A, Semb H. Cadherins regulate aggregation of pancreatic β-cells in vivo. Development. 1996;122:2895–2902. doi: 10.1242/dev.122.9.2895. [DOI] [PubMed] [Google Scholar]
- 18.Rogers GJ, Hodgkin MN, Squires PE. E-cadherin and cell adhesion: a role in architecture and function in the pancreatic islet. Cell Physiol Biochem. 2007;20:987–994. doi: 10.1159/000110459. [DOI] [PubMed] [Google Scholar]
- 19.Roderigo-Milne H, Hauge-Evans AC, Persaud SJ, Jones PM. Differential expression of insulin genes 1 and 2 in MIN6 cells and pseudoislets. Biochem Biophys Res Commun. 2002;296:589–595. doi: 10.1016/S0006-291X(02)00913-0. [DOI] [PubMed] [Google Scholar]
- 20.Bergsten P, Hellman B. Glucose-induced cycles of insulin release can be resolved into distinct periods of secretory activity. Biochem Biophys Res Commun. 1993;192:1182–1188. doi: 10.1006/bbrc.1993.1541. [DOI] [PubMed] [Google Scholar]
- 21.Lin JM, Fabregat ME, Gomis R, Bergsten P. Pulsatile insulin release from islets isolated from three subjects with type 2 diabetes. Diabetes. 2002;51:988–993. doi: 10.2337/diabetes.51.4.988. [DOI] [PubMed] [Google Scholar]
- 22.Song SH, Kjems L, Ritzel R, et al. Pulsatile insulin secretion by human pancreatic islets. J Clin Endocrinol Metab. 2002;87:213–221. doi: 10.1210/jc.87.1.213. [DOI] [PubMed] [Google Scholar]
- 23.Tinto N, Zagari A, Capuano M, et al. Glucokinase gene mutations: structural and genotype-phenotype analyses in MODY children from South Italy. PLoS One. 2008;3:e1870. doi: 10.1371/journal.pone.0001870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ban N, Yamada Y, Someya Y, et al. Hepatocyte nuclear factor-1α recruits the transcriptional co-activator p300 on the GLUT2 gene promoter. Diabetes. 2002;51:1409–1418. doi: 10.2337/diabetes.51.5.1409. [DOI] [PubMed] [Google Scholar]
- 25.Hauge-Evans AC, Squires PE, Belin VD, et al. Role of adenine nucleotides in insulin secretion from MIN6 pseudoislets. Mol Cell Endocrinol. 2002;191:167–176. doi: 10.1016/S0303-7207(02)00051-5. [DOI] [PubMed] [Google Scholar]
- 26.Zhao C, Wilson MC, Schuit F, Halestrap AP, Rutter GA. Expression and distribution of lactate/monocarboxylate transporter isoforms in pancreatic islets and the exocrine pancreas. Diabetes. 2001;50:361–366. doi: 10.2337/diabetes.50.2.361. [DOI] [PubMed] [Google Scholar]
- 27.Sekine N, Cirulli V, Regazzi R, et al. Low lactate-dehydrogenase and high mitochondrial glycerol phosphate dehydrogenase in pancreatic β-cells: potential role in nutrient sensing. J Biol Chem. 1994;269:4895–4902. [PubMed] [Google Scholar]
- 28.Mohammed JS, Wang Y, Harvat TA, Oberholzer J, Eddington DT. Microfluidic device for multimodal characterization of pancreatic islets. Lab Chip. 2009;9:97–106. doi: 10.1039/b809590f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.MacDonald MJ. Differences between mouse and rat pancreatic islets: succinate responsiveness, malic enzyme, and anaplerosis. Am J Physiol Endocrinol Metab. 2002;283:E302–E310. doi: 10.1152/ajpendo.00041.2002. [DOI] [PubMed] [Google Scholar]
- 30.Welsh M, Hellerstrom C, Andersson A. Respiration and insulin release in mouse pancreatic islets. Effects of L-leucine and 2-ketoisocaproate in combination with D-glucose and L-glutamine. Biochim Biophys Acta. 1982;721:178–184. doi: 10.1016/0167-4889(82)90066-0. [DOI] [PubMed] [Google Scholar]
- 31.Pipeleers D, Kiekens R, Ling Z, Wilikens A, Schuit F. Physiologic relevance of heterogeneity in the pancreatic beta-cell population. Diabetologia. 1994;37(Suppl 2):S57–S64. doi: 10.1007/BF00400827. [DOI] [PubMed] [Google Scholar]
- 32.Kuhara T, Ikeda S, Ohneda A, Sasaki Y. Effects of intravenous infusion of 17 amino acids on the secretion of GH, glucagon, and insulin in sheep. Am J Physiol. 1991;260:E21–E26. doi: 10.1152/ajpendo.1991.260.1.E21. [DOI] [PubMed] [Google Scholar]
- 33.Hellman B, Idahl LA, Lernmark A, Sehlin J, Taljedal IB. The pancreatic beta-cell recognition of insulin secretagogues. Comparisons of glucose with glyceraldehyde isomers and dihydroxyacetone. Arch Biochem Biophys. 1974;162:448–457. doi: 10.1016/0003-9861(74)90204-5. [DOI] [PubMed] [Google Scholar]
- 34.Henquin JC, Dufrane D, Nenquin M. Nutrient control of insulin secretion in isolated normal human islets. Diabetes. 2006;55:3470–3477. doi: 10.2337/db06-0868. [DOI] [PubMed] [Google Scholar]
- 35.Brereton H, Carvell MJ, Persaud SJ, Jones PM. Islet alpha-cells do not influence insulin secretion through cell-cell contact. Diabet Med. 2007;24:30–30. doi: 10.1111/j.1464-5491.2007.02126.x. [DOI] [PubMed] [Google Scholar]
- 36.Leibiger IB, Brismar K, Berggren PO. Novel aspects on pancreatic beta-cell signal-transduction. Biochem Biophys Res Commun. 2010;396:111–115. doi: 10.1016/j.bbrc.2010.02.174. [DOI] [PubMed] [Google Scholar]
- 37.Burks DJ, White MF. IRS proteins and beta-cell function. Diabetes. 2001;50(Suppl 1):S140–S145. doi: 10.2337/diabetes.50.2007.S140. [DOI] [PubMed] [Google Scholar]
- 38.Boura-Halfon S, Zick Y. Phosphorylation of IRS proteins, insulin action, and insulin resistance. Am J Physiol Endocrinol Metab. 2009;296:E581–E591. doi: 10.1152/ajpendo.90437.2008. [DOI] [PubMed] [Google Scholar]
- 39.Kulkarni RN, Winnay JN, Daniels M, et al. Altered function of insulin receptor substrate-1-deficient mouse islets and cultured beta-cell lines. J Clin Invest. 1999;104:R69–R75. doi: 10.1172/JCI8339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Aspinwall CA, Qian WJ, Roper MG, Kulkarni RN, Kahn CR, Kennedy RT. Roles of insulin receptor substrate-1, phosphatidylinositol 3-kinase, and release of intracellular Ca2+ stores in insulin-stimulated insulin secretion in beta-cells. J Biol Chem. 2000;275:22331–22338. doi: 10.1074/jbc.M909647199. [DOI] [PubMed] [Google Scholar]
- 41.Zick Y. Insulin resistance: a phosphorylation-based uncoupling of insulin signaling. Trends Cell Biol. 2001;11:437–441. doi: 10.1016/s0962-8924(01)02129-8. [DOI] [PubMed] [Google Scholar]
- 42.Bouzakri K, Roques M, Gual P, et al. Reduced activation of phosphatidylinositol-3 kinase and increased serine 636 phosphorylation of insulin receptor substrate-1 in primary culture of skeletal muscle cells from patients with type 2 diabetes. Diabetes. 2003;52:1319–1325. doi: 10.2337/diabetes.52.6.1319. [DOI] [PubMed] [Google Scholar]
- 43.Aoyagi K, Ohara-Imaizumi M, Nishiwaki C, Nakamichi Y, Nagamatsu S. Insulin/phosphoinositide 3-kinase pathway accelerates the glucose-induced first-phase insulin secretion through TrpV2 recruitment in pancreatic β-cells. Biochem J. 2010;432:375–386. doi: 10.1042/BJ20100864. [DOI] [PubMed] [Google Scholar]
- 44.El-Kholy W, MacDonald P, Lin JH, et al. The phosphatidylinositol 3-kinase inhibitor LY294002 potently blocks Kv currents in beta cells via a direct mechanism. Diabetes. 2003;52:A368–A368. doi: 10.1096/fj.02-0802fje. [DOI] [PubMed] [Google Scholar]
- 45.Hagiwara S, Sakurai T, Tashiro F, et al. An inhibitory role for phosphatidylinositol 3-kinase in insulin secretion from pancreatic B cell line MIN6. Biochem Biophys Res Commun. 1995;214:51–59. doi: 10.1006/bbrc.1995.2255. [DOI] [PubMed] [Google Scholar]
- 46.Eto K, Yamashita T, Tsubamoto Y, et al. Phosphatidylinositol 3-kinase suppresses glucose-stimulated insulin secretion by affecting post-cytosolic [Ca2+] elevation signals. Diabetes. 2002;51:87–97. doi: 10.2337/diabetes.51.1.87. [DOI] [PubMed] [Google Scholar]
- 47.Zawalich WS, Zawalich KC. A link between insulin resistance and hyperinsulinemia: inhibitors of phosphatidylinositol 3-kinase augment glucose-induced insulin secretion from islets of lean, but not obese, rats. Endocrinology. 2000;141:3287–3295. doi: 10.1210/en.141.9.3287. [DOI] [PubMed] [Google Scholar]
- 48.Kaneko K, Ueki K, Takahashi N, et al. Class IA phosphatidylinositol 3-kinase in pancreatic β cells controls insulin secretion by multiple mechanisms. Cell Metab. 2010;12:619–632. doi: 10.1016/j.cmet.2010.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.MacDonald PE, Joseph JW, Yau D, et al. Impaired glucose-stimulated insulin secretion, enhanced intraperitoneal insulin tolerance, and increased β-cell mass in mice lacking the p110gamma isoform of phosphoinositide 3-kinase. Endocrinology. 2004;145:4078–4083. doi: 10.1210/en.2004-0028. [DOI] [PubMed] [Google Scholar]
- 50.Gao Z, Konrad RJ, Collins H, Matschinsky FM, Rothenberg PL, Wolf BA. Wortmannin inhibits insulin secretion in pancreatic islets and beta-TC3 cells independent of its inhibition of phosphatidylinositol 3-kinase. Diabetes. 1996;45:854–862. doi: 10.2337/diabetes.45.7.854. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PDF 56 kb)
(PDF 42 kb)
(PDF 44 kb)
(PDF 57 kb)
(PDF 34 kb)