

Abstract
Alzheimer's disease (AD) is a progressive neurodegenerative disorder that leads to memory deficits and death. While the number of individuals with AD is rising each year due to the longer life expectancy worldwide, current therapy can only somewhat relieve the symptoms of AD. There is no proven medication to cure or prevent the disease, possibly due to a lack of knowledge regarding the molecular mechanisms underlying disease pathogenesis. Most previous studies have accepted the “amyloid hypothesis,” in which the neuropathogenesis of AD is believed to be triggered by the accumulation of the toxic amyloid beta (Aβ) protein in the central nervous system (CNS). Lately, knowledge that may be critical to unraveling the hidden pathogenic pathway of AD has been revealed. This review concentrates on the toxicity of Aβ and the mechanism of accumulation of this toxic protein in the brain of individuals with AD and also summarizes recent advances in the study of these accumulation mechanisms together with the role of herbal medicines that could facilitate the development of more effective therapeutic and preventive strategies.
1. Introduction
Alzheimer's disease (AD) is a progressive neurodegenerative disorder named by the German physician Dr. Alois Alzheimer in 1906 [1]. This disease can occur in anyone at any age; however, it is most common among the elderly and is less prevalent in younger people. Although AD develops differently for each individual, it normally presents similar symptoms; in the early stages, the most common defect is remembering recent events or short-term memory impairment. As the disease progresses, AD patients gradually lose their ability to think and reason clearly, make judgments, solve problems, communicate, and take care of themselves. Symptoms also include confusion, irritability and aggression, mood swings, changes in personality and behavior, problems with attention and spatial orientation, trouble with language, and long-term memory loss, all of which can affect a person's daily life. AD can even lead to the death of the afflicted person in the final stages by causing malnutrition, brain death, and multiple organ failure due to the number of nerve cells that have died. At present, AD afflicts more than 26.6 million worldwide, and its prevalence is rising dramatically each year. By 2050, the number of AD patients is expected to quadruple to more than 106 million globally, and it is estimated that 1 in 85 persons will be living with the disease [2]. After several decades of study, AD is now considered as a complex disease that results from both genetic and environmental factors, such as age, gender, family history of AD, Down syndrome (DS), and the apolipoprotein E (apoE) gene. However, the actual causes of AD are still unknown. Additionally, the biochemistry of AD is not yet fully understood, even though its histopathological features in the brain are well characterized. So far, there have been a vast number of studies that have hypothesized disease mechanisms for AD, the majority of which support the amyloid hypothesis. It is believed that the neuropathogenesis of this disease may be triggered by the accumulation of toxic amyloid in the central nervous system (CNS). Therefore, a clearer understanding of how these toxic proteins accumulate in the brain of AD patients is significant for the development of more effective therapeutic and preventive strategies. Potential mechanisms related to overproduction or impaired clearance of these amyloids that may lead to its abnormal deposition in the brain as well as some possible molecular targets for AD treatment will be the focus of this review.
2. Amyloidosis
Amyloidosis is a large group of pathologic conditions in which a particular type of protein, called amyloid, is abnormally deposited in various tissues or organs. Generally, amyloid refers to misfolded peptides or proteins that demonstrate a stable, cross-beta super-secondary structure that renders it insoluble, fibrous-like, and resistant to proteolysis. Thus, amyloid may alter the normal function of tissues and cause serious changes in tissues and organs of the body [3, 4]. Amyloidosis can be inherited or acquired. In addition, the deposition of amyloid fibrils may occur in specific areas of a single tissue (localized amyloidosis) or throughout the body (systemic amyloidosis). Each type of amyloidosis is classified according to clinical signs and the main peptide or protein that constitutes the amyloid fibrils. Amyloidosis depots contain not only the major fibrillar component but also minor nonfibrillar components such as glycosaminoglycans (GAGs), apolipoprotein E (apoE), and serum amyloid P (SAP) components [3]. Despite the differences between amyloid proteins, all forms in different diseases share some common features; amyloid deposits exhibit an apple-green birefringence under a polarized light microscope after staining with the dye Congo red and appear as rigid, nonbranching fibrils 7.5 to 10 nm in diameter under extremely high magnification using an electron microscope [5]. To date, at least 28 different proteins have been identified as amyloids in humans [3, 5–7]. Several well-known examples of human amyloid-related diseases and the official nomenclature and classification of their causative agents are shown in Table 1. Alzheimer's disease (AD) and the toxicity and mechanisms of amyloid protein aggregation will be emphasized in this review.
Table 1.
Some examples of amyloid proteins, their functional precursors, and related diseases.
| Amyloid protein (abbreviation) | Precursor protein | Disease | Distribution/type | Reference |
|---|---|---|---|---|
| Aβ | Aβ precursor protein | Alzheimer's disease | Localized/hereditary or Acquired | [57] |
| Aβ | Aβ precursor protein | Cerebral amyloid angiopathy | Localized/hereditary | [58] |
| AIAPP | Pro-IAPP | Diabetes mellitus type II | Localized/??? | [59] |
| AL | Immunoglobulin light chain | Primary systemic amyloidosis | Systemic/acquired | [60] |
| AA | Serum amyloid A | Rheumatoid arthritis | Systemic/acquired | [61] |
| ATTR | Wild-type transthyretin | Senile systemic amyloidosis | Systemic/acquired | [62] |
| ATTR | Transthyretin variant | Familial amyloid polyneuropathy | Systemic/hereditary | [63] |
| AFib | Fibrinogen α-chain variant | Familial amyloidosis | Systemic/hereditary | [64] |
| Aβ2M | Beta 2 microglobulin | Hemodialysis-associated amyloidosis | Systemic/acquired | [65] |
| APrPSC | Prion protein | Creutzfeldt-Jakob disease | Localized/hereditary | [66] |
| AANF | Atrial natriuretic factor | Isolated atrial amyloidosis | Localized/acquired | [67] |
3. Toxicity of Amyloids in Alzheimer's Disease
The amyloid beta (Aβ) peptide was initially identified and biochemically characterized in 1984 [8] as a peptide that aggregated and was deposited outside neurons in the brain tissue of Alzheimer's patients, leading to the formation of neuritic plaques (also called senile or amyloid plaques) in the AD brain. The presence of these neuritic plaques is the major pathological hallmark of AD. The Aβ peptide, a principal component of these plaques, is thought to play a central role in AD and is regarded as the causative agent in development of the disease. This hypothesis has emerged from the fact that nearly all individuals with Down syndrome (DS), or trisomy 21, carry an extra copy of the amyloid precursor gene on chromosome 21. Therefore, their Aβ levels are high, and DS patients exhibit the clinical symptoms of AD around the age of 40 years [9, 10]. Aβ is a 4.2 kDa short peptide of 40–42 amino acids, generated from the intracellular cleavage of the amyloid precursor protein (APP) by the sequential action of two proteolytic enzymes, beta- (β-) secretase and gamma- (γ-) secretase. A schematic of the normal proteolytic processing of APP is shown in Figure 1.
Figure 1.
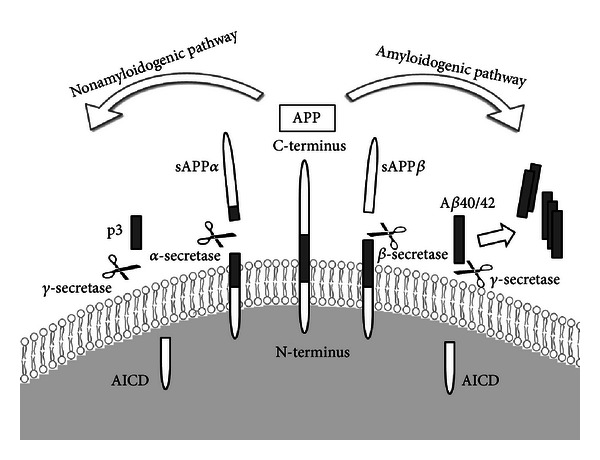
Proteolytic processing of amyloid precursor protein (APP). Amyloid precursor protein (APP) is a ubiquitously expressed integral membrane protein that can be processed in two distinct pathways. In the nonamyloidogenic pathway, APP is cleaved within the Aβ domain by the α-secretase enzyme. However, in the amyloidogenic pathway, APP is first cleaved by β-secretase (BACE1), instead of α-secretase, at the N-terminus of the Aβ domain, and this is followed by γ-secretase cleavage at the C-terminus. This sequence of events generates the Aβ amylogenic peptides, which can aggregate into oligomers and form extracellular neurotoxic plaques in the brain. Both pathways release identical APP intracellular C-terminal domain (AICD). This figure was adapted from Thinakaran and Koo (2008) [11].
APP is a single-pass transmembrane protein that is highly expressed in the brain and is concentrated at neuronal synapses [11, 12]. APP has been implicated in neuroprotection and as a regulator of neuronal cell growth, cell-cell, or cell-matrix interactions and synaptic plasticity [13]. However, its cleavage products from the amyloidogenic pathway can contribute to neurotoxicity. Indeed, soluble monomeric Aβ fragments are normally produced in the human body, but they can aggregate into various sized oligomers and insoluble fibrils, which subsequently form neuritic plaques. Aβ monomers are generated in most of the body's cells, including vascular endothelial cells [14], thyroid epithelial cells [15], and neuronal and nonneuronal cultured cells [16, 17]. However, neuronal cells seem to generate greater amounts of Aβ than other cell types [16], indicating that the Aβ peptide might play an important role in the normal physiology of the CNS. There is a notion that Aβ might serve as an essential synaptic protein in synaptic structural-functional plasticity underlying learning and memory, an idea supported by the increased long-term potentiation mediated by Aβ 40 (LTP) [18]. Therefore, the neuropathological events occurring in individuals with AD likely result from the toxicity of amyloid oligomers and fibrils, which are the aggregated forms of Aβ, rather than from its monomeric form.
As Aβ accumulates, our bodies control the amyloid level through various mechanisms. In the normal brain, the concentration of the Aβ peptide is regulated by its production from APP and influx into the brain across the blood-brain barrier (BBB), mainly via the receptor for advanced glycation end products (RAGE), and by clearance from the brain via the low-density lipoprotein receptor-related protein-1 (LRP1) and enzymatic degradation within brain [19–21]. Thus, impairment of these regulatory mechanisms could lead to the accumulation and deposition of excessive amounts of Aβ peptide in the brain of individual with AD, the details of which are described later in this paper.
The polymeric forms of Aβ trigger changes in biochemical molecules and functions in brain cells, resulting in several neuropathological abnormalities associated with the symptoms of AD. Aβ aggregate-mediated toxicity has been documented in vitro and in vivo. Initial reports in 1994 showed that elevated oxidative stress, one of the early pathological events of AD, was mediated by hydrogen peroxide (H2O2) produced through the reduction of metal ions by Aβ peptides [22–25]. This finding is consistent with the ability of Aβ to capture the transition metal ions Cu, Fe, and Zn, which are potent catalysts of oxidation [26]. These elements, particularly Zn, have also been implicated in promoting the oligomerization of Aβ peptides [27, 28]. Furthermore, extracellular and intracellular accumulation of metal ions is found in AD brains with high concentrations of Aβ plaques [29–32]. As a result of free radical production induced by Aβ, some biomolecules in the AD brain undergo conformational and structural changes due to lipid peroxidation and oxidative modification of proteins [33], leading to the dysfunction of these molecules and thereby influencing a wide array of cellular functions. Proteomic studies have identified several oxidatively modified proteins in AD [34, 35]. For example, oxidized ubiquitin carboxy-terminal hydrolase L-1 (UCH L-1) leads to proteasomal dysfunction and the consequent accumulation of damaged, misfolded, and aggregated proteins. Oxidatively modified creatine kinase BB (CK) and glutamine synthetase (GS) severely affect ATP production and the influx of calcium ions into neurons, resulting in the loss of function of ion pumps, the dysregulation of intracellular calcium homeostasis, alterations in LTP, and mitochondrial dysfunction with the release of proapoptotic factors. All of these changes could ultimately lead to neuronal death [36]. Increased oxidative stress, in turn, also promotes APP processing through the upregulation of BACE1 gene expression, which leads to an increase in AB generation [37–39].
Although the Aβ peptide plays an essential role at the synapse, Aβ aggregate-mediated toxicity impairs synaptic function, which leads to the progressive memory loss and cognitive failure associated with AD. Synaptic dysfunction is triggered by changes in synaptic structure and neurochemicals induced by oligomerized Aβ rather than amyloid plaques. Loss of synaptic terminals and LTP deficits have been demonstrated in studies of transgenic mice overexpressing mutant APP [40, 41]. Studies in the normal rodent hippocampus also showed that soluble Aβ oligomers isolated from the cerebral cortex of AD patients or from tissue culture reduced dendritic spine density and markedly inhibited LTP, resulting in the disruption of synaptic plasticity and memory [42, 43]. However, LTP was not impaired after treatment with insoluble amyloid plaque cores, suggesting that the oligomeric form rather than the deposit form is synaptotoxic. Aβ oligomer-induced synaptic dysfunction was found to be associated with a reduction in surface expression of both NMDA-type and AMPA-type glutamate receptors [44, 45] as well as postsynaptic density-95 (PSD-95) protein levels [46, 47] in cortical neurons; all three are key proteins in the postsynaptic density (PSD) involved in the regulation of synaptic function. Both glutamate receptors and nicotinic acetylcholine receptors (nAChRs) are also considerably reduced in the AD brain, possibly through Aβ-induced receptor internalization. The binding of exogenous Aβ to nAChRs facilitates internalization and intraneuronal accumulation of these toxic peptides, which could impact neuronal cells [48, 49]. Furthermore, a recent study proposed that activation of casein kinase II (CKII) by Aβ might underlie the disruption of synaptic transmission [50].
In addition to oxidative damage and synaptic failure, Aβ aggregates can induce mitochondrial dysfunction, which is another pathological hallmark of AD. The alteration of synaptic mitochondria, as a result of the buildup of Aβ, may underlie the synaptic pathology in AD [51]. Reported mitochondrial malfunctions as a consequence of membrane-localized Aβ include the inhibition of protein transport into mitochondria, the disruption of the electron transport chain leading to impaired glucose utilization in neurons, and mitochondrial damage due to an increase in reactive oxygen species (ROS) production [52]. Aβ also contributes to mitochondrial toxicity by the induction of microtubule-associated protein tau phosphorylation through specific kinase activation [53–55], which results in dissociation of tau from microtubules, the destabilization and disintegration of microtubules in axons, leading to the collapse of the neuronal transport system, and the formation of neurofibrillary tangles (NFTs) composed of aggregated hyperphosphorylated tau inside neuronal cell bodies. Moreover, proteasome-mediated degradation of misfolded protein, including tau aggregates, is inhibited by the actions of Aβ oligomers, contributing to their enhanced accumulation [56].
4. Risk Factors and Mechanisms Underlying Amyloid Beta Amyloidosis
Because the amyloid hypothesis postulated that accumulation of Aβ is the fundamental cause of AD, factors that lead to excessive levels of Aβ have been investigated in a number of studies, especially those factors associated with the pathway for overproduction and impaired clearance of amyloids. This review summarizes the latest information on factors influencing Aβ levels, as well as the molecular mechanisms involved in the above-mentioned pathways, including the schematic diagram of Aβ accumulation mechanisms influenced by these factors in Figure 2.
Figure 2.
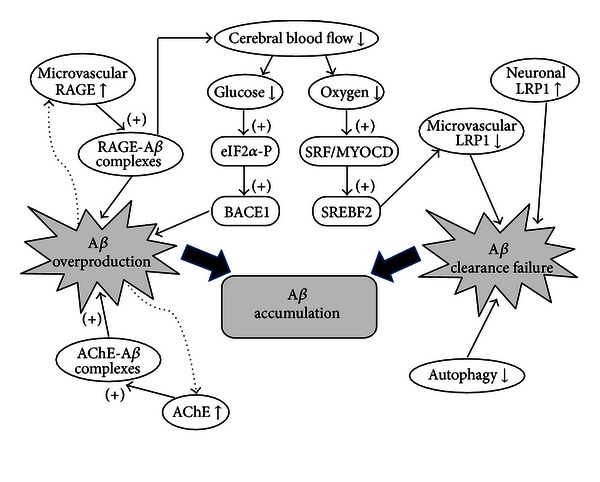
Schematic diagram of Aβ accumulation mechanism. This schematic diagram summarizes the factors influencing Aβ accumulation and the molecular mechanisms involved in the pathways for overproduction and impaired clearance of toxic Aβ peptides. Several potential targets for AD treatment, such as AChE, LRP1, and RAGE, and certain components involved in the control of cerebral blood flow and the autophagic pathway are suggested by their direct involvement in Aβ accumulation mechanism.
4.1. Cholinergic System
Cholinergic signaling was the primary factor described in the oldest AD hypothesis, which states that the progression of AD is initiated by a deficiency in the production of the vital neurotransmitter acetylcholine. Amyloid peptide was also found to be involved in this hypothesis [68] and may play a central role in producing the cholinergic deficit, as suggested by Ehrenstein et al. (1997). Amyloid peptide reduces acetylcholine (ACh) synthesis through Aβ-induced leakage of choline across cell membranes [69]. Moreover, Aβ was shown to affect nAChR levels [48]. In turn, the loss of ACh was reported to be associated with the production of Aβ. However, how the decline in ACh is linked to the increased level of Aβ has remained unclear. A known link involves acetylcholinesterase (AChE), the activity of which is increased around amyloid plaques, although it is lower in other regions of the AD brain [70, 71]. These findings were supported by later studies of the effect of Aβ on AChE expression [72, 73]. It was demonstrated that AChE could promote Aβ fibril and plaque formation both in vitro and in vivo [74, 75], which is caused by the interaction of Aβ peptide with AChE at a specific motif located close to the peripheral anionic binding site (PAS) of the enzyme [76]. Furthermore, AChE-Aβ complexes are more neurotoxic than Aβ alone, which is related to changes in components of the wingless-type MMTV integration site family (Wnt) signal transduction pathway. The level of beta-catenin, a key component of Wnt signaling, was found to be decreased through the action of AChE-Aβ complexes, whereas enhancing this pathway by lithium could block AChE-Aβ-dependent neurotoxicity [77–79]. In fact, one type of currently available drug, such as donepezil hydrochloride, a reversible competitive inhibitor of AChE, targets AChE. Unfortunately, it is not able to cure AD but reduces the symptoms for a limited period of time.
4.2. Receptor for Advanced Glycation End Products (RAGEs)
RAGE is a member of the immunoglobulin superfamily of cell surface molecules, which is able to recognize a variety of ligands such as advanced glycation end products (AGEs), HMGB1 (amphoterin), S100 protein, macrophage-1 antigen (Mac-1), and amyloid protein. The interaction between Aβ and RAGE in brain vessels mediates the transportation of circulating Aβ peptides across the BBB into the brain [20, 21, 80]. Expression of RAGE is determined by the levels of its ligands; thus, high production of Aβ results in the upregulation of RAGE, which in turn leads to the greater accumulation of toxic proteins in the brain. Thus, targeting RAGE might be beneficial for AD treatment. A study of human brains using western blotting and immunostaining analyses revealed elevated RAGE levels in the AD hippocampus [81]. In addition, a RAGE-Aβ interaction has been implicated in AD pathogenesis, and previous studies have shown that increased RAGE expression is associated with neurotoxicity. The interaction of Aβ with vascular RAGE enhanced the expression of proinflammatory cytokines and potent vasoconstrictor endothelin-1 (ET-1), which may result in decreased cerebral blood flow [20]. Insufficient blood flow was also reported to be associated with Aβ accumulation, and these details are discussed below.
4.3. Low-Density Lipoprotein Receptor-Related Protein-1 (LRP1)
LRP1 (also known as apolipoprotein E (apoE) receptor) is a member of the low-density lipoprotein (LDL) receptor family that is highly expressed in the CNS and plays a critical role in brain lipoprotein metabolism, including clearance of amyloid peptides [82]. The interaction between Aβ and LRP1 may mediate transport of Aβ out of the brain. Approximately, 70 to 90% of circulating plasma Aβ is normally controlled by the soluble form of this receptor, soluble LRP1 (sLRP1) [83]. However, whereas LRP1 along brain capillaries showed amyloid clearance capacity [84], LRP1 expressed in neurons is not likely responsible for the removal of Aβ but may instead promote neuronal uptake of Aβ via its endocytic function [85]. A study in transgenic mice overexpressing functional LRP1 receptors showed an increase in soluble brain Aβ accumulation [86]. This is consistent with the high LRP1 concentrations in neurons reported in the study of human AD hippocampi, while minimal LRP1 levels were found in microvessels of AD cases [81]. Hence, expression of capillary endothelial LRP1 and neuronal LRP1 is implicated in Aβ clearance and leads to toxic amyloid accumulation in AD brain; thus, LRP1 could be another potential target for AD treatment.
4.4. Autophagy
Autophagy is a lysosomal degradation pathway for the turnover of cytoplasmic components and aggregated proteins, including dysfunctional organelles. Autophagy is essential for maintaining cellular homeostasis. It has been suggested that alteration in autophagic processing is linked to AD pathogenesis due to its relevance in the removal of toxic Aβ aggregates as well as APP [87]. In addition, autophagy-induced Aβ production activity was implicated in a new pathway for APP processing [88]. Enhancing the autophagic degradation pathway was shown to protect neurons from Aβ-induced neurotoxicity, which was in turn increased by transcriptional silencing of the autophagic gene (Atg) [89]. Therefore, impaired autophagy could lead to Aβ accumulation. For that reason, modulation of this pathway might be a potential AD therapy. The Ser/Thr kinase mammalian target of rapamycin (mTOR), which plays a central role in autophagic regulation, or other components of the mTOR signaling pathway, could be efficient therapeutic targets for AD.
4.5. Cerebral Blood Flow
Recently, poor blood flow was suggested to be a main cause of AD. Energy and oxygen starvation in the brain resulting from insufficient blood flow potentially initiate the signaling pathways influencing Aβ biosynthesis and the brain's ability to remove this toxic protein. O'Connor et al. (2008) showed that BACE1 is regulated in response to stress from energy deprivation at the translational level [90]. An insufficient supply of glucose induces phosphorylation of the translation initiation factor eIF2alpha (eIF2α), which consequently increases BACE1 levels, resulting in overproduction of Aβ. In parallel, a study by Bell et al. (2009) reported that hypoxia also causes the impairment of brain clearance of Aβ through stimulation of serum response factor (SRF) and myocardin (MYOCD) expressions [91], which were found to be much more active in the blood vessels of brains of people with AD than in people who do not have the disease [92]. Overexpression of these two proteins in cerebrovascular smooth muscle cells (CVSMCs) negatively regulates the expression of LRP1, which is a key Aβ clearance receptor in the BBB, by stimulating the transactivation properties of the sterol regulatory element binding transcription factor 2 (SREBF2), ultimately leading to toxic Aβ accumulation. Therefore, the components of signaling pathways underlying reduced blood flow might be potential targets for AD treatment. Improving blood flow by exercise, healthy eating, or using dietary supplements may also be effective for preventing AD. Substantial evidence demonstrates an association between physical activity and improvement of cognitive decline in AD [93–95].
5. Therapeutic Perspectives of Herbal Medicine in AD
While the number of individuals with AD is rising each year due to a longer life expectancy worldwide, there is currently no drug treatment that provides a cure for AD. The currently available medications only relieve the symptoms of AD. Drugs commonly used to treat AD include AChE inhibitors, such as donepezil hydrochloride (Aricept), rivastigmine (Exelon), and galantamine (Reminyl). All three AChE inhibitors are reversible inhibitors of AChE and interact with the active site of AChE to prevent the breakdown of the vital neurotransmitter ACh, thereby allowing a higher level of ACh in the brain. Another drug, memantine (Ebixa), is an antagonist of the NMDA-type glutamate receptor. The action of memantine is quite different from that of the three AChE inhibitors. Memantine prevents neuronal cell death due to glutamate receptor overstimulation. Recently, selective monoamine oxidase B (MAO-B) inhibitors (MAOIs) were developed as agents for AD therapy [96]. MAOIs act by inhibiting the activity of the metabolizing enzyme MAO-B to prevent the breakdown of monoamine neurotransmitters, thus increasing their availability. However, these drugs are not effective for everyone with AD and can only temporarily slow down the progression of symptoms. Some users also experience adverse drug reactions or side effects. Therefore, developing alternative treatments for AD is needed.
One type of alternative treatment that could be effective in curing AD or preventing the disease is herbal medicine. Using natural compounds from plants for medication is becoming more popular because of their wide availability, low cost, and potential for fewer adverse reactions than synthetic drugs. Nevertheless, the safety and efficacy of each plant or natural product must be confirmed before human usage. Many herbs have been reported to exhibit a neuroprotective effect in AD. Herbal medications targeting the mechanisms underlying Aβ accumulation, which is now believed to be a central causative pathway in AD pathogenesis, might be the most effective approach to preventing the disease. For example, cerebral blood flow-modulating plants may be beneficial. The ethanolic extract of the Morinda citrifolia fruit, including its chloroform and ethyl acetate fractions, was recently reported to significantly improve cerebral blood flow in a mouse model, suggesting that M. citrifolia may prevent Aβ accumulation. Interestingly, increased oxidative stress and AChE activities, common problems in AD, were also attenuated by the ethanolic extract of M. citrifolia, which supports its potential to prevent AD [97, 98]. Affecting the regulation of expression of genes involved in amyloidogenesis may be another mechanism of neuronal protection by plants. The components of Caulis piperis futokadsurae were reported to selectively inhibit the expression of the APP gene [99]. The extracts from several traditional Chinese herbs such as Astragalus membranaceus [100], Paeonia suffruticosa [101], Magnolia officinalis [102], and Rhizoma anemarrhenae [103] were reported to effectively prevent memory impairment via downregulation of the expression or activity of BACE1, thereby reducing APP levels in animal models. Inhibition of BACE1 activity was also reported in an in vitro study using extracted components of Panax notoginseng [104] and Polygala tenuifolia [105]. Autophagy-regulating plants might also help to prevent AD by altering Aβ clearance through the autophagic process. There are reports, both in vivo and in vitro, that alkaloids isolated from Stephaniae tetrandrae [106] could induce the expression of microtubule-associated protein-1 light chain 3 (LC3) and autophagy-related gene 7 (Atg7), which promote autophagy and the removal of Aβ. Glycyrrhiza glabra root extract [107], including β-Elemene, an active component derived from herbs used in traditional Chinese medicine [108], induces autophagy by increasing the levels of the LC3 protein. Additionally, curcumin, a major active component of Curcuma longa (turmeric), has been proposed to be a promising candidate for treatment of AD as shown by the enhanced clearance toxic Aβ in a number of studies [109–111], although the mechanisms of action of curcumin in AD are still unclear. Our review suggests that the autophagic mechanism, which has recently been identified as a target of curcumin [112–114], may be responsible for its potent antiamyloidogenic effects. Several previous studies have supported the effectiveness of herb extracts to treat AD by influencing Aβ accumulation. Interestingly, recent studies from our laboratory also provided evidence of the medicinal uses of plants for AD protection. The ethanolic extracts of Rhinacanthus nasutus leaf and root showed a beneficial effect protecting against the neuronal cell death induced by Aβ treatment or hypoxia in a cell culture study [115, 116]. The molecular mechanisms underlying this therapeutic effect need to be investigated further.
In conclusion, AD is a progressive neurodegenerative disorder that leads to memory impairment and death. However, there is currently no proven medication to cure or stop the progression of the disease. This review focused on the “amyloid hypothesis,” which states that the neuropathogenesis of AD is triggered by the accumulation of toxic Aβ in the CNS. We highlighted the importance of medicinal plants as alternative therapeutic or preventive agents for AD in the near future.
Conflict of Interests
The authors report no conflict of interests.
Acknowledgments
We would like to thank Assoc. Prof. Dr. Rachana Santiyanont (Department of Clinical Chemistry, Faculty of Allied Health Sciences, Chulalongkorn University) for her guidance and inspiration that led us to write this review. This work was supported by the Integrated Innovation Academic Center (IIAC) Chulalongkorn University Centenary Academic Development Project (CU56-AS01) and the Higher Education Research Promotion and National Research University Project of Thailand, Office of the Higher Education Commission (AS562A). The authors would also like to thank Chulalongkorn University Graduate Scholarship to commemorate the 72nd Anniversary of his Majesty King Bhumibol Adulyadej. Finally, we are very grateful to Dr. James M. Brimson (a researcher, Department of Clinical Chemistry, Faculty of Allied Health Sciences, Chulalongkorn University) for his critical reading of the paper and for his valuable comments.
References
- 1.Alzheimer A, Stelzmann RA, Schnitzlein HN, Murtagh FR. An English translation of Alzheimer's 1907 paper, ‘uber eine eigenartige erkankung der hirnrinde’. Clinical Anatomy. 1995;8(6):429–431. doi: 10.1002/ca.980080612. [DOI] [PubMed] [Google Scholar]
- 2.Brookmeyer R, Johnson E, Ziegler-Graham K, Arrighi HM. Forecasting the global burden of Alzheimer's disease. Alzheimer's and Dementia. 2007;3(3):186–191. doi: 10.1016/j.jalz.2007.04.381. [DOI] [PubMed] [Google Scholar]
- 3.Westermark P, Benson MD, Buxbaum JN, et al. A primer of amyloid nomenclature. Amyloid. 2007;14(3):179–183. doi: 10.1080/13506120701460923. [DOI] [PubMed] [Google Scholar]
- 4.Pepys MB. Amyloidosis. Annual Review of Medicine. 2006;57:223–241. doi: 10.1146/annurev.med.57.121304.131243. [DOI] [PubMed] [Google Scholar]
- 5.Merlini G, Bellotti V. Molecular mechanisms of amyloidosis. The New England Journal of Medicine. 2003;349(6):583–596. doi: 10.1056/NEJMra023144. [DOI] [PubMed] [Google Scholar]
- 6.Rambaran RN, Serpell LC. Amyloid fibrils: abnormal protein assembly. Prion. 2008;2(3):112–117. doi: 10.4161/pri.2.3.7488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Benson MD, James S, Scott K, Liepnieks JJ, Kluve-Beckerman B. Leukocyte chemotactic factor 2: a novel renal amyloid protein. Kidney International. 2008;74(2):218–222. doi: 10.1038/ki.2008.152. [DOI] [PubMed] [Google Scholar]
- 8.Glenner GG, Wong CW. Alzheimer's disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochemical and Biophysical Research Communications. 1984;120(3):885–890. doi: 10.1016/s0006-291x(84)80190-4. [DOI] [PubMed] [Google Scholar]
- 9.Mann DMA. Alzheimer's disease and Down's syndrome. Histopathology. 1988;13(2):125–137. doi: 10.1111/j.1365-2559.1988.tb02018.x. [DOI] [PubMed] [Google Scholar]
- 10.Shamas-Ud-Din S, Holmes C. Genetics of down's syndrome and Alzheimer's disease. British Journal of Psychiatry. 2002;181:167–168. doi: 10.1017/s0007125000162014. [DOI] [PubMed] [Google Scholar]
- 11.Thinakaran G, Koo EH. Amyloid precursor protein trafficking, processing, and function. Journal of Biological Chemistry. 2008;283(44):29615–29619. doi: 10.1074/jbc.R800019200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.O'Brien RJ, Wong PC. Amyloid precursor protein processing and alzheimer's disease. Annual Review of Neuroscience. 2011;34:185–204. doi: 10.1146/annurev-neuro-061010-113613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Storey E, Cappai R. The amyloid precursor protein of Alzheimer's disease and the Aβ peptide. Neuropathology and Applied Neurobiology. 1999;25(2):81–97. doi: 10.1046/j.1365-2990.1999.00164.x. [DOI] [PubMed] [Google Scholar]
- 14.Kitazume S, Tachida Y, Kato M, et al. Brain endothelial cells produce amyloid β from amyloid precursor protein 770 and preferentially secrete the O-glycosylated form. Journal of Biological Chemistry. 2010;285(51):40097–40103. doi: 10.1074/jbc.M110.144626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schmitt TL, Steiner E, Klingler P, Lassmann H, Grubeck-Loebenstein B. Thyroid epithelial cells produce large amounts of the Alzheimer β- amyloid precursor protein (APP) and generate potentially amyloidogenic APP fragments. Journal of Clinical Endocrinology and Metabolism. 1995;80(12):3513–3519. doi: 10.1210/jcem.80.12.8530592. [DOI] [PubMed] [Google Scholar]
- 16.Fukumoto H, Tomita T, Matsunaga H, Ishibashi Y, Saido TC, Iwatsubo T. Primary cultures of neuronal and non-neuronal rat brain cells secrete similar proportions of amyloid β peptides ending at Aβ40 and Aβ42. NeuroReport. 1999;10(14):2965–2969. doi: 10.1097/00001756-199909290-00017. [DOI] [PubMed] [Google Scholar]
- 17.Hayes GM, Howlett DR, Griffin GE. Production of β-amyloid by primary human foetal mixed brain cell cultures and its modulation by exogenous soluble β-amyloid. Neuroscience. 2002;113(3):641–646. doi: 10.1016/s0306-4522(02)00191-4. [DOI] [PubMed] [Google Scholar]
- 18.Koudinov AR, Koudinova NV. Cholesterol homeostasis failure as a unifying cause of synaptic degeneration. Journal of the Neurological Sciences. 2005;229-230:233–240. doi: 10.1016/j.jns.2004.11.036. [DOI] [PubMed] [Google Scholar]
- 19.Selkoe DJ. Clearing the brain's amyloid cobwebs. Neuron. 2001;32(2):177–180. doi: 10.1016/s0896-6273(01)00475-5. [DOI] [PubMed] [Google Scholar]
- 20.Deane R, Yan SD, Submamaryan RK, et al. RAGE mediates amyloid-β peptide transport across the blood-brain barrier and accumulation in brain. Nature Medicine. 2003;9(7):907–913. doi: 10.1038/nm890. [DOI] [PubMed] [Google Scholar]
- 21.Deane R, Bell RD, Sagare A, Zlokovic BV. Clearance of amyloid-β peptide across the blood-brain barrier: Implication for therapies in Alzheimer's disease. CNS and Neurological Disorders. 2009;8(1):16–30. doi: 10.2174/187152709787601867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Behl C, Davis JB, Lesley R, Schubert D. Hydrogen peroxide mediates amyloid β protein toxicity. Cell. 1994;77(6):817–827. doi: 10.1016/0092-8674(94)90131-7. [DOI] [PubMed] [Google Scholar]
- 23.Hensley K, Carney JM, Mattson MP, et al. A model for β-amyloid aggregation and neurotoxicity based on free radical generation by the peptide: relevance to Alzheimer disease. Proceedings of the National Academy of Sciences of the United States of America. 1994;91(8):3270–3274. doi: 10.1073/pnas.91.8.3270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang X, Atwood CS, Hartshorn MA, et al. The Aβ peptide of Alzheimer's disease directly produces hydrogen peroxide through metal ion reduction. Biochemistry. 1999;38(24):7609–7616. doi: 10.1021/bi990438f. [DOI] [PubMed] [Google Scholar]
- 25.Atwood CS, Obrenovich ME, Liu T, et al. Amyloid-β: a chameleon walking in two worlds: a review of the trophic and toxic properties of amyloid-β . Brain Research Reviews. 2003;43(1):1–16. doi: 10.1016/s0165-0173(03)00174-7. [DOI] [PubMed] [Google Scholar]
- 26.Miura T, Suzuki K, Kohata N, Takeuchi H. Metal binding modes of Alzheimer's amyloid β-peptide in insoluble aggregates and soluble complexes. Biochemistry. 2000;39(23):7024–7031. doi: 10.1021/bi0002479. [DOI] [PubMed] [Google Scholar]
- 27.Huang X, Atwood CS, Moir RD, Hartshorn MA, Tanzi RE, Bush AI. Trace metal contamination initiates the apparent auto-aggregation, amyloidosis, and oligomerization of Alzheimer's Aβ peptides. Journal of Biological Inorganic Chemistry. 2004;9(8):954–960. doi: 10.1007/s00775-004-0602-8. [DOI] [PubMed] [Google Scholar]
- 28.Drago D, Bolognin S, Zatta P. Role of metal ions in the Aβ oligomerization in Alzheimer's disease and in other neurological disorders. Current Alzheimer Research. 2008;5(6):500–507. doi: 10.2174/156720508786898479. [DOI] [PubMed] [Google Scholar]
- 29.Basun H, Forssell LG, Wetterberg L, Winblad B. Metals and trace elements in plasma and cerebrospinal fluid in normal ageing and Alzheimer's disease. Journal of Neural Transmission. 1991;3(4):231–258. [PubMed] [Google Scholar]
- 30.Smith MA, Harris PLR, Sayre LM, Perry G. Iron accumulation in Alzheimer disease is a source of redox-generated free radicals. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(18):9866–9868. doi: 10.1073/pnas.94.18.9866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lovell MA, Robertson JD, Teesdale WJ, Campbell JL, Markesbery WR. Copper, iron and zinc in Alzheimer's disease senile plaques. Journal of the Neurological Sciences. 1998;158(1):47–52. doi: 10.1016/s0022-510x(98)00092-6. [DOI] [PubMed] [Google Scholar]
- 32.Religa D, Strozyk D, Cherny RA, et al. Elevated cortical zinc in Alzheimer disease. Neurology. 2006;67(1):69–75. doi: 10.1212/01.wnl.0000223644.08653.b5. [DOI] [PubMed] [Google Scholar]
- 33.Qi XL, Xiu J, Shan KR, et al. Oxidative stress induced by beta-amyloid peptide1-42 is involved in the altered composition of cellular membrane lipids and the decreased expression of nicotinic receptors in human SH-SY5Y neuroblastoma cells. Neurochemistry International. 2005;46(8):613–621. doi: 10.1016/j.neuint.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 34.Castegna A, Aksenov M, Aksenova M, et al. Proteomic identification of oxidatively modified proteins in Alzheimer's disease brain. Part I: creatine kinase BB, glutamine synthase, and ubiquitin carboxy-terminal hydrolase L-1. Free Radical Biology and Medicine. 2002;33(4):562–571. doi: 10.1016/s0891-5849(02)00914-0. [DOI] [PubMed] [Google Scholar]
- 35.Castegna A, Aksenov M, Thongboonkerd V, et al. Proteomic identification of oxidatively modified proteins in Alzheimer's disease brain. Part II: dihydropyrimidinase-related protein 2, α-enolase and heat shock cognate 71. Journal of Neurochemistry. 2002;82(6):1524–1532. doi: 10.1046/j.1471-4159.2002.01103.x. [DOI] [PubMed] [Google Scholar]
- 36.Butterfield DA, Boyd-Kimball D. Amyloid β-peptide(1-42) contributes to the oxidative stress and neurodegeneration found in Alzheimer disease brain. Brain Pathology. 2004;14(4):426–432. doi: 10.1111/j.1750-3639.2004.tb00087.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tong Y, Zhou W, Fung V, et al. Oxidative stress potentiates BACE1 gene expression and Aβ generation. Journal of Neural Transmission. 2005;112(3):455–469. doi: 10.1007/s00702-004-0255-3. [DOI] [PubMed] [Google Scholar]
- 38.Coma M, Guix FX, Ill-Raga G, et al. Oxidative stress triggers the amyloidogenic pathway in human vascular smooth muscle cells. Neurobiology of Aging. 2008;29(7):969–980. doi: 10.1016/j.neurobiolaging.2007.01.009. [DOI] [PubMed] [Google Scholar]
- 39.Quiroz-Baez R, Rojas E, Arias C. Oxidative stress promotes JNK-dependent amyloidogenic processing of normally expressed human APP by differential modification of α-, β- and γ-secretase expression. Neurochemistry International. 2009;55(7):662–670. doi: 10.1016/j.neuint.2009.06.012. [DOI] [PubMed] [Google Scholar]
- 40.Chapman PF, White GL, Jones MW, et al. Impaired synaptic plasticity and learning in aged amyloid precursor protein transgenic mice. Nature Neuroscience. 1999;2(3):271–276. doi: 10.1038/6374. [DOI] [PubMed] [Google Scholar]
- 41.Spires TL, Meyer-Luehmann M, Stern EA, et al. Dendritic spine abnormalities in amyloid precursor protein transgenic mice demonstrated by gene transfer and intravital multiphoton microscopy. Journal of Neuroscience. 2005;25(31):7278–7287. doi: 10.1523/JNEUROSCI.1879-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Walsh DM, Klyubin I, Fadeeva JV, et al. Naturally secreted oligomers of amyloid β protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416(6880):535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- 43.Shankar GM, Li S, Mehta TH, et al. Amyloid-β protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nature Medicine. 2008;14(8):837–842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Snyder EM, Nong Y, Almeida CG, et al. Regulation of NMDA receptor trafficking by amyloid-β . Nature Neuroscience. 2005;8(8):1051–1058. doi: 10.1038/nn1503. [DOI] [PubMed] [Google Scholar]
- 45.Hsieh H, Boehm J, Sato C, et al. AMPAR removal underlies Aβ-induced synaptic depression and dendritic spine loss. Neuron. 2006;52(5):831–843. doi: 10.1016/j.neuron.2006.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Almeida CG, Tampellini D, Takahashi RH, et al. Beta-amyloid accumulation in APP mutant neurons reduces PSD-95 and GluR1 in synapses. Neurobiology of Disease. 2005;20(2):187–198. doi: 10.1016/j.nbd.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 47.Roselli F, Tirard M, Lu J, et al. Soluble β-amyloid1-40 induces NMDA-Dependent Degradation of Postsynaptic Density-95 at Glutamatergic Synapses. Journal of Neuroscience. 2005;25(48):11061–11070. doi: 10.1523/JNEUROSCI.3034-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nagele RG, D'Andrea MR, Anderson WJ, Wang HY. Intracellular accumulation of β-amyloid1-42 in neurons is facilitated by the α7 nicotinic acetylcholine receptor in Alzheimer's disease. Neuroscience. 2002;110(2):199–211. doi: 10.1016/s0306-4522(01)00460-2. [DOI] [PubMed] [Google Scholar]
- 49.Bayer TA, Wirths O. Intracellular accumulation of amyloid-Beta—a predictor for synaptic dysfunction and neuron loss in Alzheimer's disease. Frontiers in Aging Neuroscience. 2010;2, article 8 doi: 10.3389/fnagi.2010.00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pigino G, Morfini G, Atagi Y, et al. Disruption of fast axonal transport is a pathogenic mechanism for intraneuronal amyloid beta. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(14):5907–5912. doi: 10.1073/pnas.0901229106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Du H, Guo L, Yan SS. Synaptic mitochondrial pathology in Alzheimer's disease. Antioxidants & Redox Signaling. 2012;16(12):1467–1475. doi: 10.1089/ars.2011.4277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Spuch C, Ortolano S, Navarro C, et al. New insights in the amyloid-Beta interaction with mitochondria. Journal of Aging Research. 2012;2012:9 pages. doi: 10.1155/2012/324968.324968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Busciglio J, Lorenzo A, Yeh J, Yankner BA. β-Amyloid fibrils induce tau phosphorylation and loss of microtubule binding. Neuron. 1995;14(4):879–888. doi: 10.1016/0896-6273(95)90232-5. [DOI] [PubMed] [Google Scholar]
- 54.Zheng WH, Bastianetto S, Mennicken F, Ma W, Kar S. Amyloid β peptide induces tau phosphorylation and loss of cholinergic neurons in rat primary septal cultures. Neuroscience. 2002;115(1):201–211. doi: 10.1016/s0306-4522(02)00404-9. [DOI] [PubMed] [Google Scholar]
- 55.Blurton-Jones M, LaFerla FM. Pathways by which Aβ facilitates tau pathology. Current Alzheimer Research. 2006;3(5):437–448. doi: 10.2174/156720506779025242. [DOI] [PubMed] [Google Scholar]
- 56.Tseng BP, Green KN, Chan JL, Blurton-Jones M, LaFerla FM. Aβ inhibits the proteasome and enhances amyloid and tau accumulation. Neurobiology of Aging. 2008;29(11):1607–1618. doi: 10.1016/j.neurobiolaging.2007.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ghiso J, Frangione B. Amyloidosis and Alzheimer's disease. Advanced Drug Delivery Reviews. 2002;54(12):1539–1551. doi: 10.1016/s0169-409x(02)00149-7. [DOI] [PubMed] [Google Scholar]
- 58.Weller RO, Massey A, Newman TA, Hutchings M, Kuo YM, Roher AE. Cerebral amyloid angiopathy: amyloid β accumulates in putative interstitial fluid drainage pathways in Alzheimer's disease. American Journal of Pathology. 1998;153(3):725–733. doi: 10.1016/s0002-9440(10)65616-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hayden MR, Tyagi SC. ‘A’ is for amylin and amyloid in type 2 diabetes mellitus. Journal of the Pancreas. 2001;2(4):124–139. [PubMed] [Google Scholar]
- 60.Sanchorawala V. Light-chain (AL) amyloidosis: diagnosis and treatment. Clinical Journal of the American Society of Nephrology. 2006;1(6):1331–1341. doi: 10.2215/CJN.02740806. [DOI] [PubMed] [Google Scholar]
- 61.Husby G. Amyloidosis and rheumatoid arthritis. Clinical and Experimental Rheumatology. 1985;3(2):173–180. [PubMed] [Google Scholar]
- 62.Westermark P, Bergström J, Solomon A, Murphy C, Sletten K. Transthyretin-derived senile systemic amyloidosis: clinicopathologic and structural considerations. Amyloid. 2003;10(supplement 1):48–54. [PubMed] [Google Scholar]
- 63.Ando Y, Nakamura M, Araki S. Transthyretin-related familial amyloidotic polyneuropathy. Archives of Neurology. 2005;62(7):1057–1062. doi: 10.1001/archneur.62.7.1057. [DOI] [PubMed] [Google Scholar]
- 64.Kyle RA. Amyloidosis: a convoluted story. British Journal of Haematology. 2001;114(3):529–538. doi: 10.1046/j.1365-2141.2001.02999.x. [DOI] [PubMed] [Google Scholar]
- 65.Argiles A, Mourad G, Axelrud-Cavadore C, Watrin A, Mion C, Cavadore JC. High-molecular-mass proteins in heamodialysis-associated amyloidosis. Clinical Science. 1989;76(5):547–552. doi: 10.1042/cs0760547. [DOI] [PubMed] [Google Scholar]
- 66.Head MW, Ironside JW. Review: creutzfeldt-Jakob disease: prion protein type, disease phenotype and agent strain. Neuropathology and applied neurobiology. 2012;38(4):296–310. doi: 10.1111/j.1365-2990.2012.01265.x. [DOI] [PubMed] [Google Scholar]
- 67.Kaye GC, Butler MG, D'Ardenne AJ. Isolated atrial amyloid contains atrial natriuretic peptide: a report of six cases. British Heart Journal. 1986;56(4):317–320. doi: 10.1136/hrt.56.4.317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pedersen WA, Kloczewiak MA, Blusztajn JK. Amyloid β-protein reduces acetylcholine synthesis in a cell line derived from cholinergic neurons of the basal forebrain. Proceedings of the National Academy of Sciences of the United States of America. 1996;93(15):8068–8071. doi: 10.1073/pnas.93.15.8068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ehrenstein G, Galdzicki Z, Lange GD. The choline-leakage hypothesis for the loss of acetylcholine in Alzheimer's disease. Biophysical Journal. 1997;73(3):1276–1280. doi: 10.1016/S0006-3495(97)78160-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ulrich J, Meier-Ruge W, Probst A, Meier E, Ipsen S. Senile plaques: staining for acetylcholinesterase and A4 protein: a comparative study in the hippocampus and entorhinal cortex. Acta Neuropathologica. 1990;80(6):624–628. doi: 10.1007/BF00307630. [DOI] [PubMed] [Google Scholar]
- 71.Moran MA, Mufson EJ, Gomez-Ramos P. Colocalization of cholinesterases with β amyloid protein in aged and Alzheimer's brains. Acta Neuropathologica. 1993;85(4):362–369. doi: 10.1007/BF00334445. [DOI] [PubMed] [Google Scholar]
- 72.Sberna G, Sáez-Valero J, Beyreuther K, Masters CL, Small DH. The amyloid β-protein of Alzheimer's disease increases acetylcholinesterase expression by increasing intracellular calcium in embryonal carcinoma P19 cells. Journal of Neurochemistry. 1997;69(3):1177–1184. doi: 10.1046/j.1471-4159.1997.69031177.x. [DOI] [PubMed] [Google Scholar]
- 73.Sberna G, Sáez-Valero J, Li QX, et al. Acetylcholinesterase is increased in the brains of transgenic mice expressing the C-terminal fragment (CT100) of the β-amyloid protein precursor of Alzheimer's disease. Journal of Neurochemistry. 1998;71(2):723–731. doi: 10.1046/j.1471-4159.1998.71020723.x. [DOI] [PubMed] [Google Scholar]
- 74.Inestrosa NC, Alvarez A, Pérez CA, et al. Acetylcholinesterase accelerates assembly of amyloid-β-peptides into Alzheimer's fibrils: possible role of the peripheral site of the enzyme. Neuron. 1996;16(4):881–891. doi: 10.1016/s0896-6273(00)80108-7. [DOI] [PubMed] [Google Scholar]
- 75.Rees T, Hammond PI, Soreq H, Younkin S, Brimijoin S. Acetylcholinesterase promotes beta-amyloid plaques in cerebral cortex. Neurobiology of Aging. 2003;24(6):777–787. doi: 10.1016/s0197-4580(02)00230-0. [DOI] [PubMed] [Google Scholar]
- 76.De Ferrari GV, Canales MA, Shin I, Weiner LM, Silman I, Inestrosa NC. A structural motif of acetylcholinesterase that promotes amyloid β-peptide fibril formation. Biochemistry. 2001;40(35):10447–10457. doi: 10.1021/bi0101392. [DOI] [PubMed] [Google Scholar]
- 77.Inestrosa NC, Alvarez A, Godoy J, Reyes A, De Ferrari GV. Acetylcholinesterase-amyloid-β-peptide interaction and Wnt signaling involvement in Aβ neurotoxicity. Acta Neurologica Scandinavica, Supplement. 2000;102(176):53–59. doi: 10.1034/j.1600-0404.2000.00308.x. [DOI] [PubMed] [Google Scholar]
- 78.Inestrosa NC, Urra S, Colombres M. Acetylcholinesterase (AChE)–amyloid-beta-peptide complexes in Alzheimer's disease. the Wnt signaling pathway. Current Alzheimer Research. 2004;1(4):249–254. doi: 10.2174/1567205043332063. [DOI] [PubMed] [Google Scholar]
- 79.Inestrosa NC, Alvarez A, Dinamarca MC, Pérez-Acle T, Colombres M. Acetylcholinesterase-amyloid-β-peptide interaction: effect of Congo Red and the role of the Wnt pathway. Current Alzheimer Research. 2005;2(3):301–306. doi: 10.2174/1567205054367928. [DOI] [PubMed] [Google Scholar]
- 80.Yan SD, Chen X, Fu J, et al. RAGE and amyloid-β peptide neurotoxicity in Alzheimer's disease. Nature. 1996;382(6593):685–691. doi: 10.1038/382685a0. [DOI] [PubMed] [Google Scholar]
- 81.Donahue JE, Flaherty SL, Johanson CE, et al. RAGE, LRP-1, and amyloid-beta protein in Alzheimer's disease. Acta Neuropathologica. 2006;112(4):405–415. doi: 10.1007/s00401-006-0115-3. [DOI] [PubMed] [Google Scholar]
- 82.Kang DE, Pietrzik CU, Baum L, et al. Modulation of amyloid β-protein clearance and Alzheimer's disease susceptibility by the LDL receptor-related protein pathway. Journal of Clinical Investigation. 2000;106(9):1159–1166. doi: 10.1172/JCI11013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sagare A, Deane R, Bell RD, et al. Clearance of amyloid-β by circulating lipoprotein receptors. Nature Medicine. 2007;13(9):1029–1031. doi: 10.1038/nm1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Shibata M, Yamada S, Ram Kumar S, et al. Clearance of Alzheimer's amyloid-β1-40 peptide from brain by LDL receptor-related protein-1 at the blood-brain barrier. Journal of Clinical Investigation. 2000;106(12):1489–1499. doi: 10.1172/JCI10498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Fuentealba RA, Liu Q, Zhang J, et al. Low-density lipoprotein receptor-related protein 1 (LRP1) mediates neuronal Aβ42 uptake and lysosomal trafficking. PLoS ONE. 2010;5(7) doi: 10.1371/journal.pone.0011884.e11884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zerbinatti CV, Wozniak DF, Cirrito J, et al. Increased soluble amyloid-β peptide and memory deficits in amyloid model mice overexpressing the low-density lipoprotein receptor-related protein. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(4):1075–1080. doi: 10.1073/pnas.0305803101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zhou F, van Laar T, Huang H, Zhang L. APP and APLP1 are degraded through autophagy in response to proteasome inhibition in neuronal cells. Protein & Cell. 2011;2(5):377–383. doi: 10.1007/s13238-011-1047-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Nixon RA. Autophagy, amyloidogenesis and Alzheimer disease. Journal of Cell Science. 2007;120(23):4081–4091. doi: 10.1242/jcs.019265. [DOI] [PubMed] [Google Scholar]
- 89.Hung SY, Huang WP, Liou HC, Fu WM. Autophagy protects neuron from Aβ-induced cytotoxicity. Autophagy. 2009;5(4):502–510. doi: 10.4161/auto.5.4.8096. [DOI] [PubMed] [Google Scholar]
- 90.O'Connor T, Sadleir KR, Maus E, et al. Phosphorylation of the translation initiation factor eIF2α increases BACE1 levels and promotes amyloidogenesis. Neuron. 2008;60(6):988–1009. doi: 10.1016/j.neuron.2008.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bell RD, Deane R, Chow N, et al. SRF and myocardin regulate LRP-mediated amyloid-β clearance in brain vascular cells. Nature Cell Biology. 2009;11(2):143–153. doi: 10.1038/ncb1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Chow N, Bell RD, Deane R, et al. Serum response factor and myocardin mediate arterial hypercontractility and cerebral blood flow dysregulation in Alzheimer's phenotype. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(3):823–828. doi: 10.1073/pnas.0608251104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Winchester J, Dick MB, Gillen D, et al. Walking stabilizes cognitive functioning in Alzheimer's disease (AD) across one year. Archives of Gerontology and Geriatrics. 2012;56(1):96–103. doi: 10.1016/j.archger.2012.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Intlekofer KA, Cotman CW. Exercise counteracts declining hippocampal function in aging and Alzheimer's disease. doi: 10.1016/j.nbd.2012.06.011. Neurobiology of Disease. In press. [DOI] [PubMed] [Google Scholar]
- 95.Maesako M, Uemura K, Kubota M, et al. Exercise is more effective than diet control in preventing high fat diet-induced beta-amyloid deposition and memory deficit in amyloid precursor protein transgenic mice. The Journal of Biological Chemistry. 2012;287(27):23024–23033. doi: 10.1074/jbc.M112.367011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Thomas T. Monoamine oxidase-B inhibitors in the treatment of Alzheimers disease. Neurobiology of Aging. 2000;21(2):343–348. doi: 10.1016/s0197-4580(00)00100-7. [DOI] [PubMed] [Google Scholar]
- 97.Muralidharan P, Ravi Kumar V, Balamurugan G. Protective effect of Morinda citrifolia fruits on β-amyloid (25–35) induced cognitive dysfunction in mice: an experimental and biochemical study. Phytotherapy Research. 2010;24(2):252–258. doi: 10.1002/ptr.2922. [DOI] [PubMed] [Google Scholar]
- 98.Pachauri SD, Tota S, Khandelwal K, et al. Protective effect of fruits of Morinda citrifolia L. on scopolamine induced memory impairment in mice: a behavioral, biochemical and cerebral blood flow study. Journal of Ethnopharmacology. 2012;139(1):34–41. doi: 10.1016/j.jep.2011.09.057. [DOI] [PubMed] [Google Scholar]
- 99.Xia W, Zeng JP, Chen LB, et al. Inhibition of β-amyloid precursor protein gene in SK-N-SH cells by piperlonguminine/dihydropiperlonguminine components separated from Chinese herbal medicine Futokadsura stem. The Chinese Journal of Physiology. 2007;50(4):157–163. [PubMed] [Google Scholar]
- 100.Li WZ, Wu WY, Huang DK, et al. Protective effects of astragalosides on dexamethasone and Abeta25-35 induced learning and memory impairments due to decrease amyloid precursor protein expression in 12-month male rats. Food and Chemical Toxicology. 2012;50(6):1883–1890. doi: 10.1016/j.fct.2012.03.064. [DOI] [PubMed] [Google Scholar]
- 101.Su SY, Cheng CY, Tsai TH, Hsieh CL. Paeonol Protects Memory after Ischemic Stroke via Inhibiting beta-secretase and Apoptosis. Evidence-based complementary and alternative medicine. eCAM. 2012;2012:11 pages. doi: 10.1155/2012/932823.932823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lee YJ, Choi DY, Han SB, et al. Inhibitory effect of ethanol extract of magnolia officinalis on memory impairment and amyloidogenesis in a transgenic mouse model of Alzheimer's disease via regulating beta-secretase activity. Phytotherapy Research. 2012;26(12):1884–1889. doi: 10.1002/ptr.4643. [DOI] [PubMed] [Google Scholar]
- 103.Huang JF, Shang L, Liu P, et al. Timosaponin-BII inhibits the up-regulation of BACE1 induced by Ferric Chloride in rat retina. BMC Complementary and Alternative Medicine. 2012;12(1):p. 189. doi: 10.1186/1472-6882-12-189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wang YH, Du GH. Ginsenoside Rg1 inhibits β-secretase activity in vitro and protects against Aβ-induced cytotoxicity in PC12 cells. Journal of Asian Natural Products Research. 2009;11(7):604–612. doi: 10.1080/10286020902843152. [DOI] [PubMed] [Google Scholar]
- 105.Jia H, Jiang Y, Ruan Y, et al. Tenuigenin treatment decreases secretion of the Alzheimer's disease amyloid β-protein in cultured cells. Neuroscience Letters. 2004;367(1):123–128. doi: 10.1016/j.neulet.2004.05.093. [DOI] [PubMed] [Google Scholar]
- 106.Gong K, Chen C, Zhan Y, Chen Y, Huang Z, Li W. Autophagy-related gene 7 (ATG7) and reactive oxygen species/extracellular signal-regulated kinase regulate tetrandrine-induced autophagy in human hepatocellular carcinoma. The Journal of Biological Chemistry. 2012;287(42):35576–35588. doi: 10.1074/jbc.M112.370585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Yo YY, Shieh GS, Hsu KF, Chao-Liang W, Shiau AL. Licorice and licochalcone-a induce autophagy in lncap prostate cancer cells by suppression of bcl-2 expression and the mtor pathway. Journal of Agricultural and Food Chemistry. 2009;57(18):8266–8273. doi: 10.1021/jf901054c. [DOI] [PubMed] [Google Scholar]
- 108.Liu J, Zhang Y, Qu J, et al. β-Elemene-induced autophagy protects human gastric cancer cells from undergoing apoptosis. BMC Cancer. 2011;11, article 183 doi: 10.1186/1471-2407-11-183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Yang F, Lim GP, Begum AN, et al. Curcumin inhibits formation of amyloid β oligomers and fibrils, binds plaques, and reduces amyloid in vivo. Journal of Biological Chemistry. 2005;280(7):5892–5901. doi: 10.1074/jbc.M404751200. [DOI] [PubMed] [Google Scholar]
- 110.Mishra S, Palanivelu K. The effect of curcumin (turmeric) on Alzheimer's disease: an overview. Annals of Indian Academy of Neurology. 2008;11(1):13–19. doi: 10.4103/0972-2327.40220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Zhang C, Browne A, Child D, Tanzi RE. Curcumin decreases amyloid-β peptide levels by attenuating the maturation of amyloid-β precursor protein. Journal of Biological Chemistry. 2010;285(37):28472–28480. doi: 10.1074/jbc.M110.133520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Jia YL, Li J, Qin ZH, Liang ZQ. Autophagic and apoptotic mechanisms of curcumin-induced death in K562 cells. Journal of Asian Natural Products Research. 2009;11(11):918–928. doi: 10.1080/10286020903264077. [DOI] [PubMed] [Google Scholar]
- 113.Han J, Pan XY, Xu Y, et al. Curcumin induces autophagy to protect vascular endothelial cell survival from oxidative stress damage. Autophagy. 2012;8(5):812–825. doi: 10.4161/auto.19471. [DOI] [PubMed] [Google Scholar]
- 114.Kim JY, Cho TJ, Woo BH, et al. Curcumin-induced autophagy contributes to the decreased survival of oral cancer cells. Archives of Oral Biology. 2012;57(8):1018–1025. doi: 10.1016/j.archoralbio.2012.04.005. [DOI] [PubMed] [Google Scholar]
- 115.Brimson JM, Brimson SJ, Brimson CA, Rakkhitawatthana V, Tencomnao T. Rhinacanthus nasutus extracts prevent glutamate and amyloid-beta neurotoxicity in HT-22 mouse hippocampal cells: possible active compounds include lupeol, stigmasterol and beta-sitosterol. International Journal of Molecular Sciences. 2012;13(4):5074–5097. doi: 10.3390/ijms13045074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Brimson JM, Tencomnao T. Rhinacanthus nasutus protects cultured neuronal cells against hypoxia induced cell death. Molecules. 2011;16(8):6322–6338. doi: 10.3390/molecules16086322. [DOI] [PMC free article] [PubMed] [Google Scholar]