

Abstract
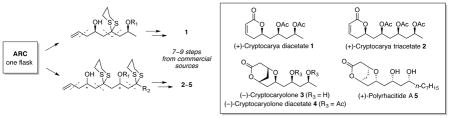
A unified synthetic strategy to the Cryptocarya family of natural products has been achieved employing four-component fragment unions in a “single flask” exploiting Anion Relay Chemistry (ARC). Functionalization of the ARC adducts permits rapid construction of five polyhydroxylated di- and tetrahydropyrone natural products of the Cryptocarya class (1–5), in a total of 7–9 steps from commercially available materials.
Recently, polyhydroxylated dihydropyrone natural products and derivatives have drawn significant attention from the biochemical and synthetic communities.1 Cryptocarya diacetate 1, cryptocarya triacetate 2, cryptocaryolone 3 and the diacetate 4, as well as polyrhacitide A 5, all belong to this general class. The natural sources – the bark of South African Cryptocarya latifolia (1–4)2 and the Chinese ant Polyrhacis lamellidens (5)3 – are used in folk medicine as analgesics. Structural analogs of 1 have also been associated with α-tubulin binding activity, based on the premise that the common α,β-unsaturated lactone moiety may operate as a Michael acceptor for a specific lysine α-tubulin residue (Lys352), in conjunction with stabilization of the adduct via hydrogen-bonding.1b
Numerous strategically diverse syntheses have been reported leading to the construction of different subsets of these natural products.4 In particular, She et al. utilized Anion Relay Chemistry (ARC) in a concise route to 2–4.4e In this context, we recently developed an augmented ARC-based strategy to access members of the Cryptocarya class in a more efficient manner.
Anion Relay Chemistry (ARC) comprises a powerful tactic to assemble complex fragments in natural product total synthesis.5 The utility of ARC also resides in application to diversity-oriented syntheses (DOS); that is, the potential to introduce rapidly structural complexity from readily available fragments.6 In Type I ARC, two electrophiles are tethered to an anionic linchpin (6) via [1,4]-Brook rearrangement (Scheme 1a), whereas in Type II ARC, the use of a bifunctional linchpin (8) permits the relay of the negative charge of an initiating nucleophile to a distal carbon atom prior to electrophilic capture (Scheme 1b). The latter version of ARC in particular holds the promise for iterative additions of diverse linchpins, by means of repeated charge relay in a way not dissimilar to “living polymerization.”7
Scheme 1.

Type I and Type II Anion Relay Chemistry
Based on this paradigm, we envisioned that ARC could be extended vis-à-vis the work of She to construct the advanced carbon skeletons required for 1–5 via a four-component “single flask” union. Specifically, we reasoned that access to targets 1–5 (Scheme 2) could be achieved via acetylation or intramolecular oxa-Michael addition of polyol derivatives (15), which in turn would arise via syn-diastereoselective reduction of a suitable hydroxy-ketone (16). In all cases, formation of the dihydropyrone motif would arise from ring-closing metathesis of the corresponding polyketide precursor (17), the latter constructed utilizing appropriate ARC tactics.
Scheme 2.

Comparison of synthetic strategies
Cryptocarya diacetate (1) comprised a useful first model to fuel the development of reaction conditions both for the key four-component ARC step (Scheme 3) and for the subsequent transformations. Towards this end, application of Type I ARC involving nucleophilic attack of (S)-methyl oxirane by 2-lithio-2-TBS-1,3-dithiane (Li-22), followed by a solvent-triggered Brook rearrangement to regenerate a carbon nucleophile at the 2-position of the dithiane (24), would permit capture of a second electrophile. The significant basicity of 24 however would preclude use of allyl-oxiranes as the terminating electrophile to access directly 27, given the propensity of allylic epoxides for elimination. We turned instead to a more general synthetic approach, importantly involving minimal protecting-group manipulations, employing (S)-epichlorohydrin as the second electrophile.8 As previously reported, lithio-dithianes are known under our conditions to attack exclusively at the 3-position (in particular avoiding direct displacement of the chloride),9 with the resulting chlorohydrin anion (25) being stable at −40 °C. Formation of a new electrophilic terminal epoxide (26) can then be achieved simply by warming the reaction mixture to room temperature once consumption of 24 is complete (TLC). We also discovered that introduction of epichlorohydrin prior to triggering the Brook rearrangement, orchestrated by addition of hexamethyl phosphoramide (HMPA), results in a 1.6-fold increase in union efficiency; carbanion 24 thus reacts with the excess electrophile as soon as generated. Addition of vinylmagnesium bromide under copper-catalyzed conditions to epoxide 26 in the same flask completes construction of the advanced homoallylic alcohol skeleton 27, obtained in 75% yield in a “single flask” from ethylene oxide. Pleasingly, the average yield for each of the three carbon-carbon bond forming steps nears 90% (Scheme 3).
Scheme 3.
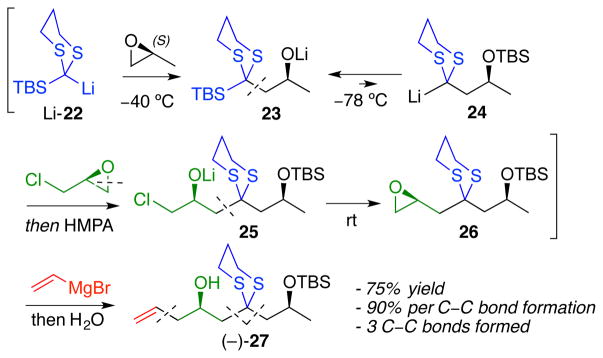
Type I ARC in the synthesis of 1
Dithiane 27 was next subjected to mercury(II)-mediated hydrolysis,10 which proved to be superior to oxidative dithiane removal protocols.11 Subsequent treatment with acryloyl chloride in the presence of Hünig’s base12 furnishes acrylate 28 in 97% yield over 2 steps. Next, ring-closing metathesis (RCM), carried out under microwave irradiation,13 employing the Hoveyda-Grubbs 2nd generation catalyst, completed construction of the dihydropyrone moiety in 87% yield. For the RCM conversion, revealing the carbonyl moiety and maintaining TBS protection on the adjacent hydroxyl group in 28 proved to be beneficial. Following RCM, the silyl protecting group was removed in 84% yield employing trifluoroacetic acid (TFA) and thioanisole,14 thus setting the stage for a syn-diastereoselective reduction of the β-hydroxyketone in 29.15 Use of diethyl methoxyborane and sodium borohydride proceeded smoothly, however, a boronate ester was initially isolated instead of the expected diol. Methanolysis released the desired diol (30) in 70% overall yield. Bis-acetylation then completed construction of cryptocarya diacetate 1. The synthetic sequence proceeded in 7 steps and 25% overall yield (Scheme 4).
Scheme 4.

Synthesis of cryptocarya diacetate (1)
The syntheses of 2–5, in turn, relied on Type II ARC, which permits introduction of the requisite additional carbinol motif in the polyol skeleton (Scheme 5). To this end, ARC union of 2-methyl-1,3-dithiane 31 with bifunctional linchpin (R)-20 and epichlorohydrin,16 followed by copper(I)-mediated addition of vinylmagnesium bromide, provided again in a “single flask” advanced homoallylic alcohol 32 in 52% yield. Application of the dithiane hydrolysis, acylation and ring-closing metathesis protocols developed for 1 permitted isolation of dihydropyrone 34 in 51% yield for the 3 steps from 32. Deprotection and directed reduction with sodium borohydride next furnished syn-triol 36, which without purification was either per-acetylated to yield cryptocarya triacetate 2 [65% over 2 steps, 17% overall, for 7 steps from (R)-20] or subjected to catalytic acid to trigger an intramolecular oxa-Michael cyclization to furnish cryptocaryolone 3 in 60% for the final 2 steps [16% overall also from (R)-20]. Cryptocaryolone diacetate (4) was then obtained in near quantitative yield upon treatment with acetic anhydride.4c
Scheme 5.

Synthesis of targets 2–4
To access polyrhacitide A (5), we exploited the previously developed ARC Type II protocol, now employing n-heptyldithiane 37, prepared by condensation of 1,3-propanedithiol and n-octanal,17 as the initiating nucleophile (Scheme 6). The change in nature of the dithiane alkyl chain, however, had a detrimental impact on the ARC reaction efficiency (36%), probably due to the increased steric hindrance at the dithiane nucleophilic carbon. Notwithstanding this shortcoming, the reaction was carried out on gram-scale, thus readily furnishing material to complete the synthesis following a similar route designed for 2–4. Specifically, dithiane removal, acylation of the homoallylic alcohol and RCM led to dihydropyrone 39 (59% over 3 steps). Removal of the TBS protecting group (95%), syn-diastereoselective reduction (83%) and oxa-Michael cyclization (98%) then completed construction of polyrhacitide A (5), in a total of 7 steps and 16% overall yield from (S)-20 (Scheme 6).
Scheme 6.

Synthesis of polyrhacitide A (5)
In summary, Anion Relay Chemistry (ARC) has been exploited for the rapid construction of a small library of polyhydroxylated pyrone natural products, which could be accessed in 7–9 steps from commercially available materials. Initial studies, utilizing the Type I ARC tactic, led to the synthesis of (+)-cryptocarya diacetate 1, which was extended to the Type II ARC protocol to furnish (+)-cryptocarya triacetate 2, (−)-cryptocaryolone 3, (−)-cryptocaryolone diacetate 4 and (+)-polyrhacitide A 5. Application of ARC tactics for the construction of more complex congeners of this class of bioactive natural products continues in our laboratory.
Supplementary Material
Acknowledgments
Financial support was provided by NIH through grant CA-19033. We thank Drs. G. Furst and R. Kohli at the University of Pennsylvania for assistance in obtaining NMR and high-resolution mass spectra, respectively.
Footnotes
Supporting Information Available: Experimental details and spectral data for all new compounds.
References
- 1.For a review of recent syntheses of dihydropyrone-containing natural products see Marco JA, Carda M, Murga J, Falomir E. Tetrahedron. 2007;63:2929–2958.for biological background information including structure-activity relationship (SAR) studies see Carda M, Murga J, Díaz-Oltra S, García-Pla J, Paños J, Falomir E, Trigili C, Díaz JF, Barasoain I, Marco JA. Eur J Org Chem. 2013:1116–1123.
- 2.(a) Drewes SE, Sehlapelo BM, Horn MM, Scott-Shaw R, Sandor P. Phytochemistry. 1995;38:1427–1430. [Google Scholar]; (b) Zschocke S, van Staden J. J Ethnopharmacol. 2000;71:473–478. doi: 10.1016/s0378-8741(00)00183-5. [DOI] [PubMed] [Google Scholar]
- 3.Jiang ZH, Yang QX, Tanaka T, Kouno I. J Nat Prod. 2008;71:724–727. doi: 10.1021/np070558l. [DOI] [PubMed] [Google Scholar]
- 4.(a) Jørgensen KB, Suenaga T, Nakata T. Tetrahedron Lett. 1999;40:8855–8858. [Google Scholar]; (b) Hunter TJ, O’Doherty GA. Org Lett. 2001;3:2777–2780. doi: 10.1021/ol016399t. [DOI] [PubMed] [Google Scholar]; (c) Smith CM, O’Doherty GA. Org Lett. 2003;5:1959–1962. doi: 10.1021/ol0345529. [DOI] [PubMed] [Google Scholar]; (d) Umarye JD, Lessmann T, García AB, Mamane V, Sommer S, Waldmann H. Chem Eur J. 2007;13:3305–3319. doi: 10.1002/chem.200601698. [DOI] [PubMed] [Google Scholar]; (e) Wang X, Wang W, Zheng H, Su Y, Jiang T, He Y, She X. Org Lett. 2009;11:3136–3138. doi: 10.1021/ol901024t. [DOI] [PubMed] [Google Scholar]; (f) Mohapatra DK, Bhimireddy E, Sivarama Krishnarao P, Das PP, Yadav JS. Org Lett. 2011;13:744–747. doi: 10.1021/ol1029854. [DOI] [PubMed] [Google Scholar]; (g) Albury AMM, Jennings MP. J Org Chem. 2012;77:6929–6936. doi: 10.1021/jo301122b. [DOI] [PubMed] [Google Scholar]
- 5.Smith AB, III, Adams CM. Acc Chem Res. 2004;37:365–377. doi: 10.1021/ar030245r. [DOI] [PubMed] [Google Scholar]
- 6.Schreiber SL. Science. 2000;287:1964–1969. doi: 10.1126/science.287.5460.1964.see: Smith AB, III, Kim WS. Proc Natl Acad Sci U S A. 2011;108:6787–6892. doi: 10.1073/pnas.1015265108.Smith AB, III, Han H, Kim WS. Org Lett. 2011;13:3328–3331. doi: 10.1021/ol2010598.
- 7.Hirao A, Nakahama S. Acta Polymer. 1998;49:133–144. [Google Scholar]
- 8.Inverting the order of electrophile addition resolved this problem in the (+)-rimocidin synthetic studies: Smith AB, III, Foley M, Dong S, Orbin A. J Org Chem. 2009;74:5987–6001. doi: 10.1021/jo900765p.
- 9.Smith AB, III, Pitram SM, Boldi AM, Gaunt MJ, Sfouggatakis C, Moser WH. J Am Chem Soc. 2003;125:14435–14445. doi: 10.1021/ja0376238. and references within. [DOI] [PubMed] [Google Scholar]
- 10.Wang H, Shuhler BJ, Xian M. J Org Chem. 2007;72:4280–4283. doi: 10.1021/jo070346t. [DOI] [PubMed] [Google Scholar]
- 11.Examples in Chen MJ, Tsai YM. Tetrahedron. 2011;67:1564–1574.Spangenberg T, Aubry S, Kishi Y. Tetrahedron Lett. 2010;51:1782–1785. doi: 10.1016/j.tetlet.2010.01.105.
- 12.Trost BM, Yeh VSC. Org Lett. 2002;4:3513–3516. doi: 10.1021/ol026665i. [DOI] [PubMed] [Google Scholar]
- 13.Varray S, Gauzy C, Lamaty F, Lazaro R, Martinez J. J Org Chem. 2000;65:6787–6790. doi: 10.1021/jo000898a. [DOI] [PubMed] [Google Scholar]
- 14.Sunazuka T, Hirose T, Chikaraishi N, Harigaya Y, Hayashi M, Komiyama K, Sprengeler PA, Smith AB, III, Ōmura S. Tetrahedron. 2005;61:3789–3803. [Google Scholar]
- 15.Munakata R, Katakai H, Ueki T, Kurosaka J, Takao KI, Tadano K. J Am Chem Soc. 2003;125:14722–14723. doi: 10.1021/ja038732p. [DOI] [PubMed] [Google Scholar]
- 16.For the preparation of linchpin 20 (one step from commercially available 22) and examples of Type II ARC see Smith AB, III, Xian M. J Am Chem Soc. 2006;128:66–67. doi: 10.1021/ja057059w.
- 17.Morokuma K, Takahashi K, Ishihara J, Hatakeyama S. Chem Commun. 2005:2265–2267. doi: 10.1039/b500660k. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.