

Abstract
Using large-scale small molecule screening techniques, Li et al. have identified a compound that inhibits the UT-B urea channel. They propose that this or similar compounds could be used as aquaretic agents to increase water excretion without changes in electrolyte excretion. Such compounds would potentially be useful in treatment of hyponatremic disorders. Here we review the physiological basis for the action of urea channel inhibitors in the kidney and assess their clinical potential.
A paper by Li and colleagues in this month’s edition of Kidney International,[1] together with two recent papers from the laboratory of Alan Verkman [2][3], introduce two new chemical families of drugs that increase water excretion by inhibiting urea channels expressed in the kidney. The agent studied by Li et al., PU-14, was shown to increase water excretion in rodents with relatively little effect on the excretion of electrolytes, thereby putting it into a functional class of drugs (along with the vaptans) termed “aquaretic” agents. The authors project possible roles for these urea channel inhibitors (UCIs) in the treatment of hyponatremic states associated with volume expansion, e.g. congestive heart failure, hepatic cirrhosis, nephrotic syndrome, and the syndrome of inappropriate antidiuresis. Based on current knowledge about the physiological roles of renal urea channels, potential advantages and disadvantages of such agents for human use can be posited.
Renal urea channels are termed “UT-B” (coded by the SLC14A1 gene) and “UT-A” (coded by the SLC14A2 gene). The latter gene produces several protein products as a result of alternative splicing and use of alternative promoters. Two of these, UT-A isoform 1 and UT-A isoform 3, are expressed in the inner medullary collecting ducts (IMCDs), while a third, UTA isoform 2, is expressed in the thin descending limbs of Henle. Originally, these proteins were thought to mediate carrier-mediated transport, but recent x-ray crystallography studies have revealed that the UT-B protein is channel-like in character [4]. The agents described by Li and colleagues in this issue of KI and by Verkman’s group were identified by small molecule screening of drug libraries using an assay based on the fact that endogenous UT-B in erythrocyte membranes protects against osmotic lysis when exposed to gradients of a chemical analog of urea, acetamide. Candidate UCIs were therefore identified by their abilities to sensitize red blood cells to osmotic lysis. Consequently, these drugs are UT-B inhibitors, although Li et al showed that their agent, PU-14, can weakly inhibit UT-A isoform 1. How do UCIs increase water excretion? In brief, in the absence of urea channel activity, endogenous urea becomes a powerful de facto osmotic diuretic. Understanding why this is true requires a deeper discussion of the physiology of urea transport in the context of the urinary concentrating mechanism.
The role of urea transport in renal water conservation is widely misunderstood. The old notion that urea gradients in the inner medulla are involved in generating an axial sodium chloride gradient (based largely on the Kokko-Rector passive countercurrent model [5]) has been dispelled based on findings in urea channel knockout mice (UT-A1 and 3) demonstrating that the absence of urea channels in the inner medullary collecting duct does not alter sodium chloride concentrations in inner medullary tissue [6]. Rather, our understanding of the role of urea in the medullary concentrating mechanism has returned to a view originally expressed by Berliner and colleagues in 1959 [7], which is summarized as follows:
In mammals including humans, urea is the prime molecular vehicle for excretion of excess nitrogen when dietary protein intake exceeds that required for growth and repair (plus the small amounts of arginine utilized for production of nitric oxide and the small amount of glutamine used for the production of ammonium in the kidney). Consequently, when protein intake is high, the rate of urea excretion is high. From an osmotic perspective, the amount of urea excreted is substantial, creating a conundrum. The large amount of urea excreted creates an osmotic load in the renal tubule lumens. If the same amounts of any other solute (e.g. mannitol) were introduced into the tubule lumens, a massive osmotic diuresis would occur. Thus, the requirement to excrete large amounts of urea potentially conflicts with the need to conserve water. Nature’s solution to this problem is urea channel-mediated accumulation of urea in the renal medullary interstitium, which osmotically balances the urea in the collecting duct lumen, thereby preventing urea-dependent osmotic diuresis that would otherwise occur.
How does urea accumulate in the inner medullary interstitium? All of the known urea channel isoforms are involved (Figure 1). The combination of UT-A isoform 1 and isoform 3 in the collecting duct provides a source of urea delivered to the inner medullary interstitium from the collecting duct lumen. However, a source of urea is not enough because blood flow to the inner medulla would tend to dissipate whatever urea gradients are generated. Dissipation however is prevented by countercurrent exchange of urea which occurs in specialized structures called “vascular bundles” located in the inner part of the inner stripe of the outer medulla. These vascular bundles utilize the urea channel UT-B of the descending vasa recta and UT-A isoform 2 of the descending limb of Henle to rapidly return urea that effluxes from the fenestrated ascending vasa recta. A failure of this countercurrent exchange process would rapidly deplete urea from the inner medulla. An additional feature of this mechanism is the UT-B urea channels present in erythrocyte membranes which allows urea inside of red cells to exit rapidly enough to be included in this recycling process. The ability of PU-14 to block UT-B in erythrocytes and descending vasa recta, therefore subverts the countercurrent exchange process necessary for retention of urea in the inner medulla. Indeed, direct measurements of solute concentrations in the inner medullary tissue by Li and colleagues confirmed that PU-14 dissipates urea from the inner medulla, while leaving sodium concentrations unaffected. Consequently, as discussed above, there will not be enough urea in the inner medulla to osmotically balance the urea in the inner medullary collecting duct lumen, causing the luminal urea to behave as an osmotic diuretic.
Figure 1. Diagram illustrating roles of urea channels in the process responsible for accumulation of urea in renal inner medulla.
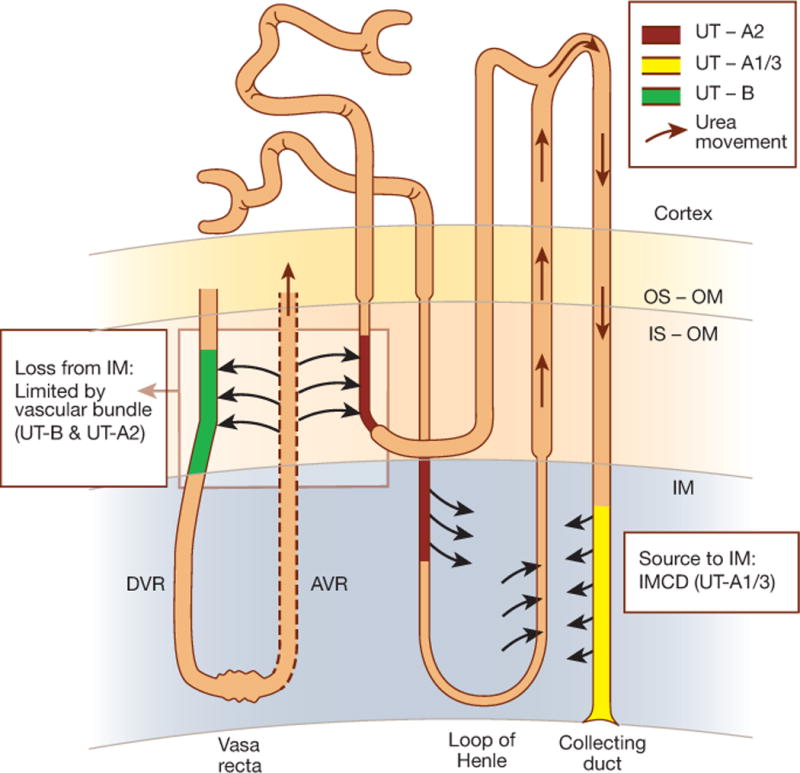
See text for explanation. DVR, descending vasa recta; AVR, ascending vasa recta; OS-OM, outer stripe of outer medulla; IS-OM, inner stripe of outer medulla; IM, inner medulla; IMCD, inner medullary collecting duct. UT-B, UT-A1, UT-A2, and UT-A3 are urea channel isoforms described in the text.
Do UCIs have clinical potential? Li et al have proposed use as aquaretic agents for treatment of hyponatremic disorders. Obviously, developing a drug for clinical use is a long and expensive process. We would argue that further groundwork is required to target UT-A rather than UT-B. The authors have targeted UT-B primarily because of ready availability of an assay using red blood cells. However, an argument could be made that a UT-A inhibitor would be a better target based on the physiology reviewed above. First, UT-A inhibitors may be more effective as aquaretics than UT-B selective drugs because they would inhibit both the source of urea to the inner medulla (UT-A 1 and 3 in the IMCD) and the countercurrent exchanger in the vascular bundles (UT-A2). By inhibiting urea absorption in the inner medullary collecting duct, UT-A inhibitors (but not UT-B inhibitors) would also increase fractional excretion of urea. This could be of value in patients with hyponatremia due to heart failure in whom azotemia is common. In addition, UT-A inhibitors would potentially have fewer adverse effects since UT-A’s only known physiological function is in the kidney whereas UT-B inhibitors could have effects elsewhere, e.g. causing hemolysis as illustrated by the screening assay. Development of UT-A selective UCIs could be accomplished either by additional large-scale small-molecule screens using an appropriate assay for transport via UT-A, or by designed modifications of UT-B selective agents. The latter option would be benefited by further X-ray crystallography work to discover the structure of UT-A. Also, before the development of a therapeutic UCI, attention should be paid to questions of oral availability and potency, which can in principle be optimized through systematic modifications of the hypothetical UT-A selective lead compound in the manner followed by Anderson et al. for UT-B inhibitors [3].
Could UCIs compete with vaptans (vasopressin V2 receptor blockers) for use in hyponatremic disorders? Given the entirely different mechanism of action, we believe that UCIs could provide a potentially useful option for clinicians, with a different spectrum of potential adverse effects. Indeed, UCIs are predicted to be more selective for water versus Na transport compared with vaptans since the transport via the epithelial sodium channel is strongly regulated by vasopressin (via the V2 receptor) in the renal collecting duct [8][9]. In contrast, UCIs would be of little utility in the treatment of autosomal dominant polycystic kidney disease, where vaptans show great promise as a result of their abilities to block vasopressin-dependent signaling [10]. An additional caveat is that the efficacy of UCIs would be predicted to be dependent on the rate of urea excretion and consequently on the level of protein intake and nitrogen balance in the patient. As demonstrated by Yao et al. [2], when rats were given a low protein diet, a UT-B inhibitor was without demonstrable effect.
Overall, the paper by Li and colleagues provides us with a nice ‘proof of principle’ study introducing a new class of drugs potentially useful in the treatment of hyponatremic disorders. It will be interesting to follow future developments with this class of drugs.
Acknowledgments
M.A.K.’s laboratory is supported by the Intramural Budget of the National Heart, Lung and Blood Institute (Project ZO1-HL001285 to M. A. Knepper). C.A.M is a medical student from New York University in the National Institutes of Health (NIH) Medical Research Scholars Program, a public-private partnership supported jointly by the NIH and the Foundation for the NIH (http://www.cc.nih.gov/training/mrsp/index.html).
Footnotes
Disclosure
Private donors to the Foundation for the NIH include Pfizer Inc., The Leona M. and Harry B. Helmsley Charitable Trust, and the Howard Hughes Medical Institute.
References
- 1.Li F, Lei T, Zhu J, et al. A Novel Small-molecule Urea Transporter Inhibitor Acts as a Potential Diuretic. Kidney Int. doi: 10.1038/ki.2013.62. This issue. [DOI] [PubMed] [Google Scholar]
- 2.Yao C, Anderson MO, Zhang J, et al. Triazolothienopyrimidine inhibitors of urea transporter UT-B reduce urine concentration. J Am Soc Nephrol. 2012;23:1210–1220. doi: 10.1681/ASN.2011070751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Anderson MO, Zhang J, Liu Y, et al. Nanomolar potency and metabolically stable inhibitors of kidney urea transporter UT-B. J Med Chem. 2012;55:5942–5950. doi: 10.1021/jm300491y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Levin EJ, Cao Y, Enkavi G, et al. Structure and permeation mechanism of a mammalian urea transporter. Proc Natl Acad Sci USA. 2012;109:11194–11199. doi: 10.1073/pnas.1207362109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kokko JP, Rector FC., Jr Countercurrent multiplication system without active transport in inner medulla. Kidney International. 1972;2:214–223. doi: 10.1038/ki.1972.97. [DOI] [PubMed] [Google Scholar]
- 6.Fenton RA, Chou CL, Stewart GS, et al. Urinary concentrating defect in mice with selective deletion of phloretin-sensitive urea transporters in the renal collecting duct. Proc Natl Acad Sci USA. 2004;101:7469–7474. doi: 10.1073/pnas.0401704101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Berliner RW, Levinsky NG, Davidson DG, Eden M. Dilution and concentration of the urine and the action of antidiuretic hormone. Am J Med. 1958;27:730–744. doi: 10.1016/0002-9343(58)90377-2. [DOI] [PubMed] [Google Scholar]
- 8.Tomita K, Pisano JJ, Knepper MA. Control of sodium and potassium transport in the cortical collecting duct of the rat. Effects of bradykinin, vasopressin, and deoxycorticosterone. Journal of Clinical Investigation. 1985;76:132–136. doi: 10.1172/JCI111935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ecelbarger CA, Kim GH, Terris J, et al. Vasopressin-mediated regulation of ENaC abundance in rat kidney. American Journal of Physiology: Renal Physiology. 2000;279:F46–F53. doi: 10.1152/ajprenal.2000.279.1.F46. [DOI] [PubMed] [Google Scholar]
- 10.Torres VE, Chapman AB, Devuyst O, et al. Tolvaptan in patients with autosomal dominant polycystic kidney disease. N Engl J Med. 2012;367:2407–2418. doi: 10.1056/NEJMoa1205511. [DOI] [PMC free article] [PubMed] [Google Scholar]