

Summary
Axon degeneration is an evolutionarily conserved process that drives the loss of damaged axons and is an early event in many neurological disorders, and so it is important to identify the molecular constituents of this poorly understood mechanism. Here we demonstrate that the Phr1 E3 ubiquitin ligase is a central component of this axon degeneration program. Loss of Phr1 results in prolonged survival of severed axons both in the peripheral and central nervous systems, as well as preservation of motor and sensory nerve terminals. Phr1 depletion increases the axonal level of the axon survival molecule nicotinamide mononucleotide adenyltransferase 2 (NMNAT2), and NMNAT2 is necessary to mediate Phr1-dependent axon stability. The profound long-term protection of peripheral and central mammalian axons following Phr1 deletion suggests that pharmacological inhibition of Phr1 function may be an attractive therapeutic candidate for ameliorating axon loss in neurological disease.
Introduction
Axon degeneration is a hallmark of many neurological disorders including neuropathies and neurodegenerative diseases. An active and evolutionarily conserved mechanism drives axon loss (Wang and Barres, 2012), so inhibiting this process is an attractive therapeutic target for treating a wide-range of neurological disorders. Acute severing of axons leads to a process commonly referred to as Wallerian degeneration, consisting of axon breakdown, glial and macrophage responses, and clearance of myelin debris (George and Griffin, 1994). Wallerian degeneration is delayed for at least a week by the ectopic expression of the Wallerian degeneration slow (WldS) protein, which acts via a gain-of-function mechanism requiring the axonal relocalization of its active component nicotinamide mononucleotide adenylyltransferase, NMNAT1 (Babetto et al., 2010; Sasaki et al., 2009a). Axon degeneration occurs not only in response to trauma, but also occurs in metabolic, inflammatory, and hereditary neuropathies as well as neurodegenerative diseases (Vargas and Barres, 2007). Remarkably, expression of WldS or axonal NMNAT can delay axon loss in many of these disease models suggesting the existence of a general axon loss program (Wang and Barres, 2012).
While ectopic expression of NMNAT orthologs can delay axon loss, the endogenous mechanisms that promote axon degeneration are largely unknown. Others and we have recently identified neuron intrinsic pathways that promote axon death (Miller et al., 2009; Osterloh et al., 2012; Wakatsuki et al., 2012; Wishart et al., 2012), and have designed screens to identify additional components (Bhattacharya et al., 2012; Fang et al., 2012; Gerdts et al., 2012). In a hypomorphic mouse mutant of dual-leucine zipper kinase (DLK), we demonstrated axon protection for at least 52h after axotomy, and identified JNK and SCG10 as downstream signaling effectors (Miller et al., 2009; Shin et al., 2012). However, only deletion of the sterile alpha- and armadillo-motif-containing protein (SARM) protein confers WldS-comparable axon protection in mice for days after peripheral nerve transection, yet its mechanism of action remains unknown (Osterloh et al., 2012). To advance our understanding of the endogenous processes orchestrating axon death it is thus necessary to identify essential components of the degeneration program and to define the underlying mechanisms.
PHR (PAM-Highwire-Rpm-1) ligases are an evolutionarily conserved family of large E3 ubiquitin ligases that are central regulators of multiple aspects of axonal biology. Mutations of PHR orthologs from worms to mice lead to dramatic defects in the development of synapses and axons (Bloom et al., 2007; Culican et al., 2009; D’Souza et al., 2005; Lewcock et al., 2007; Schaefer et al., 2000; Wan et al., 2000; Zhen et al., 2000). Recent work in invertebrates demonstrates that PHR proteins also accelerate the regenerative response following nerve injury (Nix et al., 2011; Xiong et al., 2010), and delay synapse loss in genetic models of cytoskeletal instability (Massaro et al., 2009). The involvement of Phr1 (also know as Mycbp2) in fundamental aspects of axon biology prompted us to test the role of Phr1 in axon degeneration in mice. Here we demonstrate that Phr1 is a central component of the axonal degeneration program. Excision of Phr1 in adult mice leads to long-term preservation of PNS and CNS axons, and Phr1 promotes the self-destruction of injured axons by limiting the availability of the essential axonal maintenance factor NMNAT2. This identifies Phr1 as a candidate therapeutic target for inhibiting axonal loss due to trauma and disease.
Results
Phr1 deletion protects axons and nerve terminals in vivo
Loss of Phr1 during development in mice impairs axon guidance and leads to perinatal lethality (Bloom et al., 2007; Lewcock et al., 2007), so we disrupted Phr1 in young adults using a previously characterized conditional Phr1 allele (Bloom et al., 2007) and a Tamoxifen-inducible Cre recombinase under the control of the β-actin promoter (Hayashi and McMahon, 2002). Tamoxifen administration induced the excision of the floxed region within the Phr1 gene (Method section), and we confirmed in vitro that Cre-dependent excision leads to a decrease in Phr1 protein levels (Figure S2A). Phr1 adult conditional knock out (Phr1 aKO) mice appeared healthy for up to six weeks after tamoxifen administration.
In Phr1 aKO mice we observed profound axon preservation following injury, comparable to that described in WldS (Mack et al., 2001) and SARM KO mutants (Osterloh et al., 2012) (Figure 1A–D). Axons degenerate about two days after sciatic nerve axotomy in wild-type (WT) mice (Beirowski et al., 2004), with axons of smaller caliber degenerating faster than large caliber axons (Vargas and Barres, 2007). In Phr1 aKO mutants, there is a dramatic delay in loss of both small and large axons, with ~ 75% of myelinated fibers structurally intact five days after axotomy, and ~25% of myelinated fibers persisting for at least ten days (Figure 1A, B). Electron microscopy demonstrates intact axonal organelles and cytoskeleton 10 days after injury (Figure 1C), as well as persistence of unmyelinated sensory axons with their associated glia for at least three days (Figure 1C). Biochemical analysis confirms maintenance of cytoskeletal elements; neurofilament subunits are degraded within three days of injury in WT tibial nerves, but are preserved for at least 10 days in axotomized Phr1 aKO nerves (Figure 1D). Taken together, these results show that Phr1 genetic ablation confers a remarkable delay of axon degeneration in adult mice, protecting small, large and unmyelinated sensory axons.
Figure 1.

Phr1 adult conditional KO (Phr1 aKO) delays axon degeneration in the CNS and PNS. (a) Toluidine-blue stained cross-sections of axotomized sciatic nerve distal to the cut site, showing delayed axon degeneration in Phr1 aKO compared to littermate controls (WT). (b) Quantification of intact myelinated axons of cut and unlesioned contralateral sciatic nerves. (c) Electron micrographs of the nerves shown in (a). Black arrowheads point to preserved small unmyelinated sensory fibers (Remak bundles) in Phr1 aKO nerves. White arrows point to the preservation of large caliber axons, whose cytoskeleton and organelles remain intact for at least 10 days (inset). (d) Representative Western blot of tibial nerves at the indicated time points after transection, showing preservation of neurofilaments (heavy, NF-H; medium, NF-M; light, NF-L chains) in Phr1 aKO mice (n=3). (e) Toluidine-blue stained cross-sections of optic nerve distal to the cut site demonstrate almost complete preservation of axons in Phr1 aKO mice five days after eye enucleation, a time-point when WT optic nerves are extensively degenerated, as quantified in (g). (f) Electron micrographs of the same optic nerves shown in (e), demonstrating axon death in WT samples, and morphological axon preservation in Phr1 aKO samples (insets) five days after lesion.
Many debilitating neurodegenerative conditions affect central axons, so we tested if Phr1 also regulates axon degeneration in the CNS. We axotomized retinal ganglion neurons, and assayed axon survival quantitatively in the optic nerve distal to the transection site. Five days after lesion WT nerves display extensive axon degeneration with gliosis and axonal replacement by fibrotic tissue. In contrast, lesioned optic nerves from Phr1 aKO mice appear similar to unlesioned contralateral nerves, with preservation of the vast majority of axons (Figure 1E, F, G).
We next explored whether Phr1 regulates the stability of terminal specializations such as motor and sensory endings, which are particularly vulnerable in a variety of neuropathies. We first assessed the integrity of neuromuscular junctions (NMJs). In wild type mice, tibialis anterioris muscles are completely denervated by three days after sciatic nerve axotomy. In axotomized Phr1 aKO mice, motor nerve terminal degeneration is delayed, with significant numbers of NMJs remaining occupied (Figure 2A, B). We next examined skin sensory endings, finding an ~3-fold increase in the number of intraepidermal fibers (IEPFs) remaining in the Phr1 aKO compared to WT three days after injury (p < 0.05), (Figure S1). Hence, loss of Phr1 preserves both axons and their specialized motor and sensory arbors following injury.
Figure 2.

Loss of Phr1 slows neuromuscular junction degeneration. (a) Presynaptic motoneuron terminals (anti-synaptophysin and anti-neurofilament M, green) and postsynaptic endplates (bungarotoxin, red) from the tibialis anterioris muscle remain apposed in Phr1 aKO mice three days after sciatic nerve trasection, whereas complete denervation is observed in WT samples, as quantified in (b).
Phr1 loss of function protects neurites cell-autonomously after disparate insults
To investigate whether Phr1 KO promotes axon survival in a neuron-autonomous manner, we cultured embryonic dorsal root ganglia neurons (DRGs) of Phr1 floxed mice in the absence of other cell types. We confirmed loss of the neuronal Phr1 protein after lentivirus-transduction of Cre recombinase (Figure S2A). Distal WT neurites degenerate within 6h after transection displaying features such as swelling and fragmentation (Figure 3A, B). Axon degeneration is delayed for at least 24h following axotomy in Phr1 KO neurons (Figure 3A, B), demonstrating that Phr1 can act autonomously in the neuron to control axonal stability. Using this culture model we next demonstrated that axonal degeneration induced by the neurotoxin vincristine, a chemotherapeutic agent that causes peripheral neuropathy, is also delayed in Phr1 KO neurons (Figure 3A, B). Thus, Phr1 functions downstream of distinct insults. Finally we decreased Phr1 levels using shRNA to exclude the possibility that axon preservation is an allele specific phenotype. Three independent shRNAs for Phr1 decrease Phr1 protein levels and delay axon degeneration following axotomy (Figure S2B, C). Taken together, these in vitro studies demonstrate that Phr1 can function cell autonomously within neurons to promote axon degeneration induced by both traumatic and neurotoxic insults. While this in vitro protection is clear, it is less robust than that conferred by WldS in vitro even though the in vivo protection in these two mutants is similar (Babetto et al., 2010). This difference is consistent with Phr1 having additional axoprotective functions in non-neuronal cells, and highlights the importance of in vivo analysis to identify the most significant axoprotective pathways.
Figure 3.

Phr1 functions within the neuron to promote degeneration in response to both traumatic and neurotoxic insults. (a) Phase-contrast images of transected or vincristine-treated neurites are shown from Phr1 KO or control (WT) embryonic dorsal root ganglia (DRG) cultures at the indicated time points after injury. The time-course of axonal fragmentation, quantified as degeneration index (see Methods) is shown in (b). Values indicated are the mean ± standard error, and are representative of at least 3 repeats of each analysis (axotomy and vincristine-treatment).
Phr1 regulates DLK levels in the mouse
To investigate the mechanism of action of Phr1, we initially focused on the MAP3K DLK, which is the best-characterized target of Phr1-family ubiquitin ligases in invertebrates (Collins et al., 2006; Nakata et al., 2005; Xiong et al., 2010). While we previously found no alteration of DLK levels in embryonic Phr1 KO whole-head lysates (Bloom et al., 2007), here we measured DLK levels in Phr1 KO DRG cultures and found a 3.9 ± 0.2 -fold increase compared to controls (p<0.005) (Figure S3A). The difference between our current findings in DRGs and prior findings in embryonic head may reflect a cell type difference or we may have missed a change in DLK levels in embryonic CNS neurons because they were masked by expression in non-neuronal cells. The increase in DLK abundance in the DRG demonstrates that regulation of DLK levels by PHR ligases is evolutionarily conserved in invertebrates and vertebrates.
While DLK levels are increased in the Phr1 KO cells, this increase is unlikely to mediate the axoprotective Phr1 KO phenotype because loss of DLK, rather than its activation, promotes axon survival (Miller et al., 2009). To test directly whether elevated levels of DLK can protect axons, we overexpressed DLK in cultured DRG neurons. DLK overexpression does not delay neurite degeneration after axotomy (9h after cut, WT degeneration index = 0.51±0.06 SEM; overexpressed DLK degeneration index = 0.59±0.04 SEM, p>0.1, n=3). These results indicate that Phr1 has a different functionally relevant target in axons that mediates the change in axon stability.
Phr1 protects axons via increased levels of the axonal survival factor NMNAT2
The Phr1 axon protection phenotype in vivo is similar to that afforded by WldS (Mack et al., 2001), whose active component is NMNAT1. In mice and humans there are three isoforms of NMNATs, and each promotes the survival of axons when overexpressed (Gilley and Coleman, 2010; Sasaki et al., 2009a; Yahata et al., 2009). Of these, NMNAT2 is necessary for nerve development (Hicks et al., 2012) and behaves as an endogenous axon survival factor. NMNAT2 is enriched in neurons, is axonally transported, and has a much shorter half-life than the other NMNAT isoforms. In vitro loss of NMNAT2 triggers axonal degeneration and its overexpression delays axon degeneration (Gilley and Coleman, 2010). Therefore, we tested the hypothesis that Phr1 may limit neuronal NMNAT2 abundance.
The lack of both reliable and sensitive antibodies to NMNAT2 precluded an assessment of endogenous NMNAT2 levels. Instead, we overexpressed a functional hisMyc-tagged NMNAT2 in DRG cultures, and found that NMNAT2 levels in Phr1 KO cultures are 2.0-fold higher (±0.2 SEM, p<0.05) than in control cultures (Figure 4A). Analogous experiments with tagged NMNAT1 and NMNAT3 found no difference in the levels of these proteins in Phr1 KO neurons compared to control (Figure S3B). Hence, Phr1-dependent regulation is selective for the NMNAT isoform with a short half-life, and is not a general response to viral-mediated expression (Figure S3B). Moreover, endogenous NMNAT2 mRNA levels are not significantly different in Phr1 KO and control neurons (qPCR, p > 0.1), consistent with post-transcriptional regulation of NMNAT2 by Phr1. To investigate the subcellular localization of this excess NMNAT2, we stained Phr1 KO and control cultures expressing hisMyc-tagged NMNAT2. NMNAT2 is increased in Phr1 KO neurites (Figure 4B, C) but not cell bodies (Figure S3C). Following injury NMNAT2 protein is rapidly lost from distal axons as previously described (Gilley and Coleman, 2010), reaching background levels by 2.5h after injury in controls axons. In contrast, Phr1 KO axons maintain higher levels of NMNAT2 after injury, falling to uncut control levels 5h after axotomy (Figure 4B, C). Hence, NMNAT2 levels are elevated in Phr1 KO axons during the commitment phase of axonal degeneration, the period during which exogenous NMNAT must be present to block axonal degeneration (Sasaki and Milbrandt, 2010).
Figure 4.
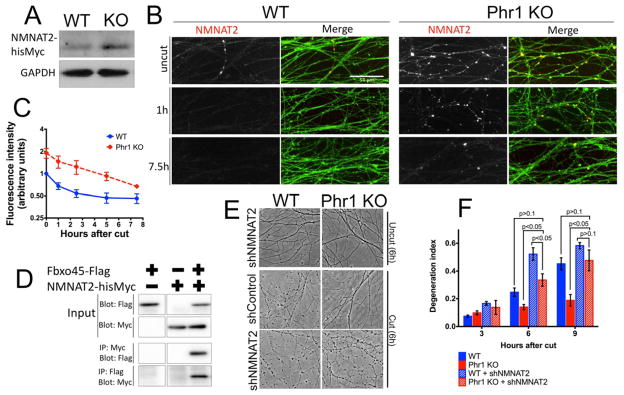
Increased levels of NMNAT2 mediates Phr1 KO protection. (a) Western blot of embryonic DRGs transduced with NMNAT2-hisMyc, showing ~2-fold increase of NMNAT2 protein in the Phr1 KO (n=3). (b) Representative anti-Myc immunostaining (NMNAT2), counterstained for axons (anti-βIII-tubulin, green), shows NMNAT2 in axons at the indicated time points after axotomy. NMNAT2 levels decrease after injury in both WT and Phr KO samples, but are consistently higher in Phr1 KO samples, as quantified in (c) (n=3). (d) Co-immunoprecipitation studies from cultured cells demonstrate binding between Fbxo45-Flag, the substrate recognition domain of the Phr1 ligase, and NMNAT2-hisMyc. (e) Phase contrast images of uncut and cut DRGs (6h after axotomy) showing reversion of the Phr1 KO protection when NMNAT2 is knocked-down via shRNA (shNMNAT2), as quantified in the time-course shown in (f).
The increase in NMNAT2 protein levels in Phr1 KO neurons is consistent with the Phr1 E3 ubiquitin ligase directly targeting NMNAT2 for degradation. To test this hypothesis, we assayed for direct binding between NMNAT2 and Fbxo45, the substrate-recognition subunit of the Phr1 ubiquitin ligase complex (Wu et al., 2007; Saiga et al., 2009). When co-expressed in cultured cells, reciprocal co-immunoprecipitation experiments demonstrate binding between Fbxo45 and NMNAT2 (Figure 4D). While this finding is consistent with Phr1 acting as an ubiquitin ligase to regulate NMNAT2 protein levels, the eventual loss of NMNAT2 protein from injured Phr1 mutant axons indicates that additional mechanisms must also promote NMNAT2 degradation.
Taken together, these data suggest that loss of Phr1 delays axonal degeneration by elevating the levels of the axon survival factor NMNAT2. This model predicts that reducing the levels of NMNAT2 should revert the axon protection phenotype in the Phr1 mutant. To test this model, we used lentiviral shRNA expression to reduce neuronal NMNAT2 transcript levels by 75% as measured by quantitative RT-PCR (not shown). This partial knockdown of endogenous NMNAT2 does not induce spontaneous axonal degeneration, and only slightly accelerates axotomy-induced axonal degeneration in control cultures (Figure 4 E, F). In contrast, this reduction in NMNAT2 potently reverts the delay of axonal degeneration following axotomy of Phr1 KO axons (Figure 4E, F). Degeneration is comparable to control preparations 6h after axotomy, and no Phr1-dependent protection is evident 9h after axotomy (Figure 4F). These findings support the model that elevated levels of endogenous NMNAT2 mediate the axon protection in the Phr1 KO.
Discussion
This study demonstrates a dramatic delay of Wallerian degeneration in Phr1 mutant mice. The axon protection is evident in several neuronal types including myelinated and unmyelinated axons, central and peripheral nerves, and sensory and motor terminals. Although there may be differences in the mechanisms of degeneration in these different neurons (Ebenezer et al., 2007; Gillingwater and Ribchester, 2001), the broad protection afforded by the Phr1 KO strongly suggests that Phr1 is a core regulatory component of a fundamental axon degeneration mechanism in mammals. Indeed, the axoprotective phenotype of the Phr1 KO is similar to that of the recently described SARM1 mutant mice, with preservation of axotomized peripheral axons for more than a week and persistence of NMJs for at least three days (Osterloh et al., 2012). For Phr1, we have identified the axon survival protein NMNAT2 as a functional downstream target. Importantly, this is the first report in mammals of a regulatory factor for NMNAT2. Modulating the levels of NMNAT2 alters the rate of Wallerian degeneration not only after axotomy in vitro, but also in mouse models of human tauopathy (Ljungberg et al., 2012). Hence, strategies to manipulate NMNAT2 are particularly attractive therapeutic candidates for treating axonopathies in a broad range of conditions.
Phr1 plays an evolutionarily conserved role in various aspects of axon biology (Po et al., 2010). Our findings now demonstrate that PHR proteins play an evolutionarily conserved role in the control of axonal degeneration. In mutants of the Phr1 ortholog highwire in Drosophila, motor nerve terminals are protected from genetic disruption of the cytoskeleton (Massaro et al., 2009). In addition, a recent study in Drosophila showed that Highwire promotes axon degeneration by limiting the levels of the Drosophila NMNAT enzyme (Xiong et al., 2012). Hence, PHR regulation of axonal degeneration via control of NMNAT isoform levels is an evolutionary conserved component of a program that likely balances axonal maintenance and degeneration. How does Phr1/Highwire regulate NMNAT2 and Drosophila NMNAT? Phr1 is an E3 ubiquitin ligase that can ubiquitinate and degrade targets (Han et al., 2008) and we show here that NMNAT2 binds to Fbxo45, the substrate recognition domain of the Phr1 ubiquitin ligase complex. In addition, NMNAT2 and Drosophila NMNAT accumulate after MG132-mediated inhibition of degradative pathways, so NMNAT2 is likely regulated via the ubiquitin proteasome system (Gilley and Coleman, 2010; Xiong et al., 2012). However others and we have yet to detect ubiquitination of NMNAT2 (Xiong et al., 2012), raising the possibility of additional regulatory mechanisms.
The extent of Phr1 aKO protection is similar to that of the gain-of-function WldS mutant (Mack et al., 2001). In the Wlds mutant the axoprotective NMNAT1 enzyme is mistargeted to axons and functionally compensates for the loss of NMNAT2 after injury. However in contrast to the chimeric WldS protein, which is a gain-of-function mutation, Phr1 is an endogenous protein that limits the levels of NMNAT2 and and promotes axonal degeneration. Two interesting questions arise from this finding. First, how do increased levels of NMNAT2 protect axons when the protein is still rapidly lost within hours of injury? This is an issue both in the Phr1 mutant and when NMNAT2 is overexpressed in wild type neurons. Indeed, overexpression of NMNAT2 in wild type neurons leads to long-term protection but the protein in undetectable within a few hours after injury (Figure 4 and Gilley and Coleman, 2010). Elevated levels of NMNAT2 may have a unique function in the first hours after injury, or low levels residual protein may still function long after the injury. In the Phr1 KO, additional targets may also contribute to axonal protection. Second, why do neurons have a mechanism to restrict the levels of an axoprotective molecule such as NMNAT2? Short-lived axoprotective molecules are maintained in the axon by constant replenishment via anterograde axonal transport, and so their presence in the axon can serve as a signal of a healthy connection with the cell body (Gilley and Coleman, 2010; Shin et al., 2012). Axons that are not well connected can therefore be rapidly and efficiently eliminated, reducing the possibility of damaging adjacent axons, and clearing the way for regeneration. Such a mechanism would require molecules such as Phr1 to regulate the levels of axoprotective factors such as NMNAT2.
In conclusion, we have identified Phr1 as a key regulator of a core axon degeneration program that balances axon survival and loss in various neuronal cell types. The profound long-term protection of injured mammalian axons following deletion of Phr1 suggests that pharmacological inhibition of Phr1 function could be an attractive therapeutic strategy for ameliorating axon loss in neurological diseases in which axonopathy is a major component, such as peripheral neuropathies, glaucoma, multiple sclerosis, and some neurodegenerative diseases (Vargas and Barres, 2007). The great challenge will be in designing specific inhibitors of the Phr1 ubiquitin ligase, as efforts to target ubiquitin ligases are in their infancy (Zhao and Sun, 2012).
Methods
Mouse dorsal root ganglia (DRG) cultures
Spot DRG cultures were described previously (Shin et al., 2012). E13.5 Phr1 floxed-allele (Bloom et al., 2007) or CD1 (Charles River) DRGs were cultured with 1 μM 5-fluoro-2′-deoxyuridine and 1 μM uridine (Sigma) to block nonneuronal cell division. To avoid confounding effects from somatic death, at DIV 2 DRGs were transduced with Bcl-XL lentivirus, which blocks apoptosis but not injury-induced degeneration (Vohra et al., 2010). Cre recombinase (KO) or FCIV (WT) were introduced via lentiviral transduction in Phr1 floxed DRGs from the same preparation at DIV 2-4. ShNMNAT2, or shControl, was applied at DIV 4 to avoid complete NMNAT2 knock-down and subsequent spontaneous axon degeneration. ShPhr1, or DLK overexpressing lentiviruses were applied to CD1 DRGs at DIV 2. See Supplemental material for details of constructs. At DIV 8-9, after complete neurite development, cultures were lysed, axotomized, or treated with vincristine as per the specific experiment, and neurite degeneration analyzed as described (Sasaki et al., 2009b).
Ultrastructural studies
For assessing optic axon survival mice were perfused transcardially (ice-cold 3% glutaraldehyde (Polysciences) in 0.1M phosphate buffer. Sciatic nerves and optic nerve segments distal to the axotomy site were harvested, the first mm was discarded. See Supplementary material for production of semi-thin sections and ultrathin sections. From these sections a blinded investigator quantified intact axons 5 mm distally from the axotomy site by established methods (Beirowski et al., 2009; Beirowski et al., 2012; Marina et al., 2012).
Western blot
Immunoprecipitated material (obtained as described in the Supplementary material) and DRGs were lysed in Laemmli buffer. Snap-frozen tibial nerves were homogenized in RIPA buffer with protease inhibitor tablet (cOmplete Mini EDTA-free, Roche). Western blot was performed with standard protocols. Antibodies are listed in the Supplemental material. Intensities of protein bands were normalized by comparison with the loading control (βIII-tubulin or GAPDH).
Immunocytochemistry
Immunostaining was carried out on glass slides (BD Falcon) as described (Shin et al., 2012). Neurons were stained for NMNAT2-hisMyc using anti-Myc antibody (9B11, Cell Signaling, 1:500) and counterstained for βIII-tubulin (TUJ, Aves lab, Inc. 1:500), with the appropriate Alexa-conjugated secondary antibodies (Invitrogen). Confocal stacks were obtained with a Nikon D-Eclipse C1 confocal microscope and z-projected. The axonal NMNAT2 level was assessed with ImageJ. Images were thresholded for βIII-tubulin immunoreactivity to define the axonal area, and the average anti-Myc immunofluorescence intensity was measured within the defined area and background was subtracted.
Mouse husbandry and surgery
Phr1 floxed mice (Bloom et al., 2007) were bred to B6. Cg-Tg(CAG-cre/Esr1*)5Amc/J mice (Jackson lab) that express a Tamoxifen-inducible Cre-recombinase. Tamoxifen (Sigma) was injected intraperitoneally (0.1 mg/g) to all littermates, including those without the Cre allele (wild-type, WT), for 2 consecutive days for 3 weeks starting at 60–80 days of age. Surgical axotomy is described in the Supplementary Material. At the indicated times mice were euthanized, or intracardially perfused, and tissue dissected. To test for Cre-recombination, a small portion of the tail-tip was collected before tamoxifen-injection, and DRGs were dissected at the end-point of the experiment. PCR analysis (Bloom et al., 2007) verified the appearance of a recombination-specific band in DRGs from mice expressing Cre. Mouse handling and surgery was supervised by the Division of Comparative Medicine of the Washington University in Saint Louis.
Neuromuscular junction (NMJ) staining and quantification
Surgery was performed 2 weeks after the first tamoxifen injection. Tibialis anterior muscles were collected from mice intracardially perfused (4% PFA in 0.1 M PBS) and postfixed for 2h at RT. Cryopreservation, cryosectioning of 60 μm-thick sections, and staining procedure were performed as described (Babetto et al., 2010, and Supplemental material). For morphological NMJ quantification, endplate occupancy was assessed by a masked investigator. Full and partial occupancy was defined as a state when the postsynaptic bungarotoxin labeling was completely or partially apposed by the axon terminal. 50–100 NMJs were quantified for each muscle preparation, from 3 mice per genotype.
Statistical Analysis
A Student T-test or ANOVA, with Tukey post-hoc, was used to test statistical significance. P values >0.05 were considered not significant. Data are presented as mean ± SEM.
Supplementary Material
Highlights.
Loss of the E3 ligase Phr1 strongly protects injured mammalian PNS and CNS axons.
Phr1 functions within neurons to promote axon loss induced by diverse insults.
Phr1 promotes axon degeneration by limiting the amount of the survival factor NMNAT2.
Acknowledgments
We are grateful to Dr. L.B. Holzman and Prof. G. Melino for sharing reagents. We thank the members of the DiAntonio and Milbrandt laboratories and Catherine Collins for helpful discussions. We thank Dr. Susan Culican for experimental advice and Kelli Simburger and Sylvia Johnson for their technical assistance. The work was supported by the American-Italian Cancer Foundation (AICF) Post-doctoral research fellowship (E.B.), the European Molecular Biology Organization (EMBO) long-term fellowship (B.B.), the Muscular Dystrophy Association (MDA) Development Award (B.B.), and the NIH: DA020812 to A.D., and NS065053 and NS078007 to J.M. and A.D.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Babetto E, Beirowski B, Janeckova L, Brown R, Gilley J, Thomson D, Ribchester RR, Coleman MP. Targeting NMNAT1 to axons and synapses transforms its neuroprotective potency in vivo. J Neurosci. 2010;30:13291–13304. doi: 10.1523/JNEUROSCI.1189-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beirowski B, Babetto E, Gilley J, Mazzola F, Conforti L, Janeckova L, Magni G, Ribchester RR, Coleman MP. Non-nuclear Wld(S) determines its neuroprotective efficacy for axons and synapses in vivo. J Neurosci. 2009;29:653–668. doi: 10.1523/JNEUROSCI.3814-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beirowski B, Berek L, Adalbert R, Wagner D, Grumme DS, Addicks K, Ribchester RR, Coleman MP. Quantitative and qualitative analysis of Wallerian degeneration using restricted axonal labelling in YFP-H mice. J Neurosci Methods. 2004;134:23–35. doi: 10.1016/j.jneumeth.2003.10.016. [DOI] [PubMed] [Google Scholar]
- Beirowski B, Gustin J, Armour SM, Yamamoto H, Viader A, North BJ, Michan S, Baloh RH, Golden JP, Schmidt RE, et al. Sir-two-homolog 2 (Sirt2) modulates peripheral myelination through polarity protein Par-3/atypical protein kinase C (aPKC) signaling. Proc Natl Acad Sci U S A. 2012;108:E952–961. doi: 10.1073/pnas.1104969108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya MR, Gerdts J, Naylor SA, Royse EX, Ebstein SY, Sasaki Y, Milbrandt J, DiAntonio A. A model of toxic neuropathy in Drosophila reveals a role for MORN4 in promoting axonal degeneration. J Neurosci. 2012;32:5054–5061. doi: 10.1523/JNEUROSCI.4951-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloom AJ, Miller BR, Sanes JR, DiAntonio A. The requirement for Phr1 in CNS axon tract formation reveals the corticostriatal boundary as a choice point for cortical axons. Genes Dev. 2007;21:2593–2606. doi: 10.1101/gad.1592107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins CA, Wairkar YP, Johnson SL, DiAntonio A. Highwire restrains synaptic growth by attenuating a MAP kinase signal. Neuron. 2006;51:57–69. doi: 10.1016/j.neuron.2006.05.026. [DOI] [PubMed] [Google Scholar]
- Culican SM, Bloom AJ, Weiner JA, DiAntonio A. Phr1 regulates retinogeniculate targeting independent of activity and ephrin-A signalling. Mol Cell Neurosci. 2009;41:304–312. doi: 10.1016/j.mcn.2009.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Souza J, Hendricks M, Le Guyader S, Subburaju S, Grunewald B, Scholich K, Jesuthasan S. Formation of the retinotectal projection requires Esrom, an ortholog of PAM (protein associated with Myc) Development. 2005;132:247–256. doi: 10.1242/dev.01578. [DOI] [PubMed] [Google Scholar]
- Ebenezer GJ, McArthur JC, Thomas D, Murinson B, Hauer P, Polydefkis M, Griffin JW. Denervation of skin in neuropathies: the sequence of axonal and Schwann cell changes in skin biopsies. Brain. 2007;130:2703–2714. doi: 10.1093/brain/awm199. [DOI] [PubMed] [Google Scholar]
- Fang Y, Soares L, Teng X, Geary M, Bonini NM. A novel Drosophila model of nerve injury reveals an essential role of Nmnat in maintaining axonal integrity. Curr Biol. 2012;22:590–595. doi: 10.1016/j.cub.2012.01.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George R, Griffin JW. Delayed macrophage responses and myelin clearance during Wallerian degeneration in the central nervous system: the dorsal radiculotomy model. Exp Neurol. 1994;129:225–236. doi: 10.1006/exnr.1994.1164. [DOI] [PubMed] [Google Scholar]
- Gerdts J, Sasaki Y, Vohra B, Marasa J, Milbrandt J. Image-based screening identifies novel roles for IkappaB kinase and glycogen synthase kinase 3 in axonal degeneration. J Biol Chem. 2012;286:28011–28018. doi: 10.1074/jbc.M111.250472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilley J, Coleman MP. Endogenous Nmnat2 is an essential survival factor for maintenance of healthy axons. PLoS Biol. 2010;8:e1000300. doi: 10.1371/journal.pbio.1000300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillingwater TH, Ribchester RR. Compartmental neurodegeneration and synaptic plasticity in the Wld(s) mutant mouse. J Physiol. 2001;534:627–639. doi: 10.1111/j.1469-7793.2001.00627.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han S, Witt RM, Santos TM, Polizzano C, Sabatini BL, Ramesh V. Pam (Protein associated with Myc) functions as an E3 ubiquitin ligase and regulates TSC/mTOR signaling. Cell Signal. 2008;20:1084–1091. doi: 10.1016/j.cellsig.2008.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi S, McMahon AP. Efficient recombination in diverse tissues by a tamoxifen-inducible form of Cre: a tool for temporally regulated gene activation/inactivation in the mouse. Dev Biol. 2002;244:305–318. doi: 10.1006/dbio.2002.0597. [DOI] [PubMed] [Google Scholar]
- Hicks AN, Lorenzetti D, Gilley J, Lu B, Andersson KE, Miligan C, Overbeek PA, Oppenheim R, Bishop CE. Nicotinamide mononucleotide adenylyltransferase 2 (Nmnat2) regulates axon integrity in the mouse embryo. PLoS ONE. 2012;7:e47869. doi: 10.1371/journal.pone.0047869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewcock JW, Genoud N, Lettieri K, Pfaff SL. The ubiquitin ligase Phr1 regulates axon outgrowth through modulation of microtubule dynamics. Neuron. 2007;56:604–620. doi: 10.1016/j.neuron.2007.09.009. [DOI] [PubMed] [Google Scholar]
- Ljungberg MC, Ali YO, Zhu J, Wu CS, Oka K, Zhai RG, Lu HC. CREB-activity and nmnat2 transcription are down-regulated prior to neurodegeneration, while NMNAT2 over-expression is neuroprotective, in a mouse model of human tauopathy. Hum Mol Genet. 2012;21:251–267. doi: 10.1093/hmg/ddr492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mack TG, Reiner M, Beirowski B, Mi W, Emanuelli M, Wagner D, Thomson D, Gillingwater T, Court F, Conforti L, et al. Wallerian degeneration of injured axons and synapses is delayed by a Ube4b/Nmnat chimeric gene. Nat Neurosci. 2001;4:1199–1206. doi: 10.1038/nn770. [DOI] [PubMed] [Google Scholar]
- Marina N, Bull ND, Martin KR. A semiautomated targeted sampling method to assess optic nerve axonal loss in a rat model of glaucoma. Nat Protoc. 2012;5:1642–1651. doi: 10.1038/nprot.2010.128. [DOI] [PubMed] [Google Scholar]
- Massaro CM, Pielage J, Davis GW. Molecular mechanisms that enhance synapse stability despite persistent disruption of the spectrin/ankyrin/microtubule cytoskeleton. J Cell Biol. 2009;187:101–117. doi: 10.1083/jcb.200903166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller BR, Press C, Daniels RW, Sasaki Y, Milbrandt J, DiAntonio A. A dual leucine kinase-dependent axon self-destruction program promotes Wallerian degeneration. Nat Neurosci. 2009;12:387–389. doi: 10.1038/nn.2290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakata K, Abrams B, Grill B, Goncharov A, Huang X, Chisholm AD, Jin Y. Regulation of a DLK-1 and p38 MAP kinase pathway by the ubiquitin ligase RPM-1 is required for presynaptic development. Cell. 2005;120:407–420. doi: 10.1016/j.cell.2004.12.017. [DOI] [PubMed] [Google Scholar]
- Nix P, Hisamoto N, Matsumoto K, Bastiani M. Axon regeneration requires coordinate activation of p38 and JNK MAPK pathways. Proc Natl Acad Sci U S A. 2011;108:10738–10743. doi: 10.1073/pnas.1104830108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterloh JM, Yang J, Rooney TM, Fox AN, Adalbert R, Powell EH, Sheehan AE, Avery MA, Hackett R, Logan MA, et al. dSarm/Sarm1 is required for activation of an injury-induced axon death pathway. Science. 2012;337:481–484. doi: 10.1126/science.1223899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Po MD, Hwang C, Zhen M. PHRs: bridging axon guidance, outgrowth and synapse development. Curr Opin Neurobiol. 2010;20:100–107. doi: 10.1016/j.conb.2009.12.007. [DOI] [PubMed] [Google Scholar]
- Saiga T, Fukuda T, Matsumoto M, Tada H, Okano HJ, Okano H, Nakayama KI. Fbxo45 forms a novel ubiquitin ligase complex and is required for neuronal development. Mol Cell Biol. 2009;29:3529–3543. doi: 10.1128/MCB.00364-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki Y, Milbrandt J. Axonal degeneration is blocked by nicotinamide mononucleotide adenylyltransferase (Nmnat) protein transduction into transected axons. J Biol Chem. 2010;285:41211–41215. doi: 10.1074/jbc.C110.193904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki Y, Vohra BP, Baloh RH, Milbrandt J. Transgenic mice expressing the Nmnat1 protein manifest robust delay in axonal degeneration in vivo. J Neurosci. 2009a;29:6526–6534. doi: 10.1523/JNEUROSCI.1429-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki Y, Vohra BP, Lund FE, Milbrandt J. Nicotinamide mononucleotide adenylyl transferase-mediated axonal protection requires enzymatic activity but not increased levels of neuronal nicotinamide adenine dinucleotide. J Neurosci. 2009b;29:5525–5535. doi: 10.1523/JNEUROSCI.5469-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer AM, Hadwiger GD, Nonet ML. rpm-1, a conserved neuronal gene that regulates targeting and synaptogenesis in C. elegans. Neuron. 2000;26:345–356. doi: 10.1016/s0896-6273(00)81168-x. [DOI] [PubMed] [Google Scholar]
- Shin JE, Miller BR, Babetto E, Cho Y, Sasaki Y, Qayum S, Russler EV, Cavalli V, Milbrandt J, Diantonio A. SCG10 is a JNK target in the axonal degeneration pathway. Proc Natl Acad Sci U S A. 2012;109:E3696–3705. doi: 10.1073/pnas.1216204109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas ME, Barres BA. Why is Wallerian degeneration in the CNS so slow? Annu Rev Neurosci. 2007;30:153–179. doi: 10.1146/annurev.neuro.30.051606.094354. [DOI] [PubMed] [Google Scholar]
- Vohra BP, Sasaki Y, Miller BR, Chang J, DiAntonio A, Milbrandt J. Amyloid precursor protein cleavage-dependent and -independent axonal degeneration programs share a common nicotinamide mononucleotide adenylyltransferase 1-sensitive pathway. J Neurosci. 2010;30:13729–13738. doi: 10.1523/JNEUROSCI.2939-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakatsuki S, Saitoh F, Araki T. ZNRF1 promotes Wallerian degeneration by degrading AKT to induce GSK3B-dependent CRMP2 phosphorylation. Nat Cell Biol. 2012;13:1415–1423. doi: 10.1038/ncb2373. [DOI] [PubMed] [Google Scholar]
- Wan HI, DiAntonio A, Fetter RD, Bergstrom K, Strauss R, Goodman CS. Highwire regulates synaptic growth in Drosophila. Neuron. 2000;26:313–329. doi: 10.1016/s0896-6273(00)81166-6. [DOI] [PubMed] [Google Scholar]
- Wang JT, Barres BA. Axon degeneration: where the Wlds things are. Curr Biol. 2012;22:R221–223. doi: 10.1016/j.cub.2012.02.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart TM, Rooney TM, Lamont DJ, Wright AK, Morton AJ, Jackson M, Freeman MR, Gillingwater TH. Combining comparative proteomics and molecular genetics uncovers regulators of synaptic and axonal stability and degeneration in vivo. PLoS Genet. 2012;8:e1002936. doi: 10.1371/journal.pgen.1002936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C, Daniels RW, DiAntonio A. DFsn collaborates with Highwire to down-regulate the Wallenda/DLK kinase and restrain synaptic terminal growth. Neural Dev. 2007;2:16. doi: 10.1186/1749-8104-2-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong X, Hao Y, Sun K, Li J, Li X, Mishra B, Soppina P, Wu C, Hume RI, Collins CA. The Highwire ubiquitin ligase promotes axonal degeneration by tuning levels of Nmnat protein. PLoS Biol. 2012;10:e1001440. doi: 10.1371/journal.pbio.1001440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong X, Wang X, Ewanek R, Bhat P, Diantonio A, Collins CA. Protein turnover of the Wallenda/DLK kinase regulates a retrograde response to axonal injury. J Cell Biol. 2010;191:211–223. doi: 10.1083/jcb.201006039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yahata N, Yuasa S, Araki T. Nicotinamide mononucleotide adenylyltransferase expression in mitochondrial matrix delays Wallerian degeneration. J Neurosci. 2009;29:6276–6284. doi: 10.1523/JNEUROSCI.4304-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Sun Y. Cullin-RING ligases (CRLs) as attractive anti-cancer targets. Curr Pharm Des. 2012 doi: 10.2174/13816128113199990300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhen M, Huang X, Bamber B, Jin Y. Regulation of presynaptic terminal organization by C. elegans RPM-1, a putative guanine nucleotide exchanger with a RING-H2 finger domain. Neuron. 2000;26:331–343. doi: 10.1016/s0896-6273(00)81167-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.