

Abstract
Coordinated growth of the skull and brain are vital to normal human development. Craniosynostosis, the premature fusion of the calvarial bones of the skull, is a relatively common pediatric disease, occurring in 1 in 2500 births, and requires significant surgical management, especially in syndromic cases. Syndromic craniosynostosis is caused by a variety of genetic lesions, most commonly by activating mutations of FGFRs 1–3, and inactivating mutations of TWIST1. In a mouse model of TWIST1 haploinsufficiency, cell mixing between the neural crest-derived frontal bone and mesoderm-derived parietal bone accompanies coronal suture fusion during embryonic development. However, the relevance of lineage mixing in craniosynostosis induced by activating FGFR mutations is unknown. Here, we demonstrate a novel mechanism of suture fusion in the Apert Fgfr2S252W mouse model. Using Cre/lox recombination we simultaneously induce expression of Fgfr2S252W and β-galactosidase in either the neural crest or mesoderm of the skull. We show that mutation of the mesoderm alone is necessary and sufficient to cause craniosynostosis, while mutation of the neural crest is neither. The lineage border is not disrupted by aberrant cell migration during fusion. Instead, the suture mesenchyme itself remains intact and is induced to undergo osteogenesis. We eliminate postulated roles for dura mater or skull base changes in craniosynostosis. The viability of conditionally mutant mice also allows post-natal assessment of other aspects of Apert syndrome.
Keywords: Apert syndrome, Fgfr2S252W, Craniosynostosis, Mesoderm, Neural crest
Introduction
The mammalian skull is a complex structure that can be broadly divided into the viscerocranium, or facial skeleton, and the neurocranium, comprised of the chondrocranium (cranial base) and calvarium (vault). In much of the facial skeleton and in the calvarium, osteoblasts differentiate directly from mesenchymal condensations to form bones by intramembranous ossification. These bones are separated by sutures, within which osteoblast proliferation along the bone edges drives their expansion, while an intervening, non-ossifying suture mesenchyme maintains their separation (Morriss-Kay and Wilkie, 2005). Lineage mapping shows that the skull is derived from neural crest and mesoderm. The facial skeleton, most of the cranial base, and the frontal bones within the calvarium are derived from neural crest, while the basioccipital and part of the basisphenoid of the posterior cranial base, and the parietal bones of the calvarium, are derived from mesoderm. The interparietal bone of the calvarium is a composite of both neural crest and mesoderm (Jiang et al., 2002; McBratney-Owen et al., 2008; Yoshida et al., 2008). Calvarial sutures form within the apparently homogenous mesenchyme between bones of either the same or different lineages. In the coronal suture, an asymmetrical lineage border exists between the neural crest-derived frontal bone and the mesoderm-derived parietal bone and intervening suture mesenchyme (Jiang et al., 2002; Yoshida et al., 2008). The molecular mechanisms controlling lineage separation and suture formation are poorly understood. The loss of sutures by pathologic bone growth between adjacent bones (synostosis) underlies a variety of common non-syndromic and syndromic craniosynostotic human diseases, collectively occurring in 1 in 2500 newborns (Muenke and Wilkie, 2000). While these syndromic conditions affect tissues throughout the body, the altered growth of the skull and its effect on brain growth are especially significant to the development and health of the afflicted individual (Morriss-Kay and Wilkie, 2005).
Activating mutations in FGFRs-1,-2 and -3, and inactivating mutations in TWIST1 and EFNB1, have been identified in a significant proportion of syndromic craniosynostosis (CS), all of which involve the coronal suture (Wilkie et al., 2010). Fgfr2 expression coincides with osteoprogenitor proliferation along the growing edges (osteogenic fronts) of the frontal and parietal bones within the developing coronal suture, but is replaced by Fgfr1 expression as osteoblasts mature and begin to secrete the extracellular matrix of bone (Iseki et al., 1999). A model of Fgf signaling in the suture proposes that excess Fgfr2 activity ultimately promotes maturation and matrix production at the expense of osteoprogenitors, leading to suture fusion, but does not account for the presence or fate of the intervening undifferentiated suture mesenchyme, or the timing of osteoprogenitor loss (Morriss-Kay and Wilkie, 2005). TWIST1 is expressed within the suture mesenchyme and osteogenic fronts (Johnson et al., 2000). In TWIST1+/−andEphA4−/−mice, neural crest and mesoderm cells mix across the lineage border, and this is proposed to cause CS (Merrill et al., 2006; Ting et al., 2009; Yen et al., 2010). This mechanism is distinct from the model of Fgf signaling above, but Fgfr2 expression is also upregulated in the suture mesenchyme of TWIST1+/−mutants (Rice et al., 2000), and so the role of cell mixing, and its general importance to the etiology of CS syndromes, remains undetermined. However, these two models of suture fusion make it clear that CS must be understood not only in terms of the molecular consequences of genetic mutations on individual cells, but also in terms of the interactions between cell populations of different lineages.
Apert syndrome is principally caused by activating mutations of FGFR2 (S252W and P253R) that increase the affinity and decrease the specificity of the receptor for FGF ligands (Park et al., 1995; Wilkie et al., 1995; Yu et al., 2000). This condition is characterized by fusion of the frontal and parietal bones across the coronal suture, and digital fusions in the hands and feet (Cunningham et al., 2007). In a mouse model of Apert syndrome bearing a Cre-inducible Fgfr2S252W (Chen et al., 2003), we have previously shown that ectopic osteogenesis replaces the coronal suture mesenchyme over the course of fetal suture development. This occurred rapidly in the early basal mesenchyme, preempting the formation of a recognizable suture (suture agenesis). In more apical locations the typical sutural architecture of overlapping osteogenic fronts was initially established, but bony fusion inevitably occurred (Holmes et al., 2009). We therefore sought to understand in more detail the fate of the suture mesenchyme in CS.
While either osteoprogenitor depletion or cell mixing could be involved in Apert CS, the asymmetric division of neural crest from mesoderm in the coronal suture suggests a novel hypothesis—that an activating mutation of Fgfr2 in the neural crest will have little or no effect on suture development, as neural crest cells form only the frontal bone, while the same mutation in mesoderm (therefore including the suture mesenchyme) will cause CS. We have used mesoderm- and neural crest-specific Cre drivers (Mesp1Cre and Wnt1Cre, respectively) (Danielian et al., 1998; Saga et al., 1999), in combination with the Cre-inducible Apert mouse model (Fgfr2NeoS252W) (Chen et al., 2003; Holmes et al., 2009) and the R26R reporter mouse (Soriano, 1999), to generate lacZ-traceable mesoderm or neural crest bearing the Apert mutation. This allows us to test our specific hypothesis and simultaneously assess any contribution of cell mixing to the CS phenotype. We have found that mesodermal expression of Fgfr2S252W is sufficient and necessary for CS in the Apert mouse model, which occurs in the absence of cell mixing. Mutant suture mesenchyme is not lost, but is induced to form the bone that joins the frontal and parietal bones.
Materials and methods
Mice
Wnt1Cre (Danielian et al., 1998) and Mesp1Cre (Saga et al., 1999) mice were kind gifts from Drs Chai and Maxson (University of Southern California, Los Angeles, CA, USA), respectively. R26R lacZ reporter mice (Soriano, 1999) were from The Jackson Laboratory (USA). The Apert mouse model Fgfr2NeoS252W (Chen et al., 2003) and the EIIaCre mouse (Lakso et al., 1996) have been described. Mice were bred on a mixed background, and genotyped by PCR of genomic DNA prepared from tail-tips for Fgfr2 and Cre as described (Holmes et al., 2009). The PCR primers for Fgfr2 flank the location of the LoxP/Neo/LoxP cassette, but do not amplify a product from this cassette under the conditions used; the sizes of the WT and Cre-excised mutant products are 393 bp and 457 bp, respectively.Neo specific to the Fgfr2NeoS252W allele was detected with primers within Fgfr2 (5′-TAGGTAGTCCATAACTCGG-3′) and Neo (5′-AGGATCTCCTGTCATCTCACCTTGCTCCTG-3′).
To simplify the genotype nomenclature in the text, we have used the following abbreviations: Mesp1Cre/Fgfr2NeoS252W/+ mice are referred to as MesoS252W/+ in the text and MesoAp in the figures; likewise, Wnt1Cre/Fgfr2NeoS252W/+ mice are referred to as NCS252W/+ or NCAp. The presence of R26R is indicated where relevant, and both the mutant and wild-type (WT) mice contain the appropriate Cre. WT control mice include both Fgfr2+/+ and Fgfr2NeoS252W/+ (Cre-negative), but only Fgfr2+/+ mice are shown in the figures, except for control mice in the PCR determination of Fgfr2NeoS252W recombination in frontal and parietal bones. Fully heterozygous mutant mice generated with EIIaCre are referred to as Fgfr2S252W/+. Animal protocols were approved by the NYU School of Medicine Institutional Animal Care and Use Committee (IACUC).
Skeletal preparations
Skulls were prepared by incubating skinned and eviscerated mice in daily changes of 1–2% KOH, at room temperature with occasional agitation, until soft tissue was separated from the skeleton. Skeletons were washed thoroughly in water and the skulls dried for photography. Prolonged incubation in KOH was used to dissociate the individual unfused bones of the skull.
Histology and histochemistry
LacZ activity was detected as described (Sanes et al., 1986). For lacZ staining of sections, embryonic or newborn (P0) heads were skinned and fixed in glutarate/formaldehyde buffer for 1.5–2 h at 4° C, washed in PBS, equilibrated in 30% sucrose/PBS, then embedded in Tissue-Tek OTC Compound (Sakura, Japan). For BrdU detection, heads were fixed in 4% PFA overnight before washing in PBS and embedding as above; glutarate/formaldehyde-fixed sections were also used for BrdU assays. Frozen sections of 10 μm thickness were cut on a Microm Cryostat and transferred to Superfrost Plus microscope slides (Fisher Scientific, USA). Alkaline phosphatase (ALP) activity was detected using Fast Red TR (F-2768, Sigma, USA) or Fast Blue RR (F-0500, Sigma, USA) as described previously (Holmes et al., 2009). BrdU administration, and detection within 70 μm of each osteogenic front at E16.5, was as described previously (Holmes et al., 2009). Alcian Blue (0.1% in 3% acetic acid) and von Kossa staining were performed by standard procedures. Counterstaining was with Eosin Y (E4382, Sigma, USA).
Results
Mesodermal expression of Fgfr2S252W is sufficient and necessary for coronal suture fusion
Fgfr2 is expressed at very low levels in coronal suture mesenchyme, but is upregulated in the proliferating cells of the osteogenic fronts of the frontal and parietal bones (Iseki et al., 1999; Johnson et al., 2000). Given that neural crest and mesoderm form an asymmetric lineage border within the coronal suture, while Fgfr2 is expressed in both osteogenic fronts symmetrically, we first sought to determine whether coronal fusion in the Apert mouse model requires expression of Fgfr2S252W in both lineages, or occurs even if expression is restricted to a single lineage. To test this, we created conditional mutants in which expression of the Apert allele is restricted to one lineage by crossing mice containing either the Mesp1Cre mesoderm-specific driver (Saga et al., 1999) or the Wnt1Cre neural crest-specific driver (Danielian et al., 1998) with mice heterozygous for the Cre-inducible Fgfr2NeoS252W Apert allele (Chen et al., 2003). Expression of this allele is repressed by the neomycin(neo) sequence flanked by loxP sites, and is restored upon Cre recombination (Chen et al., 2003). In conditionally mutant offspring of these crosses, only the Cre-expressing mesoderm or neural crest will have the Fgfr2S252W/+ genotype, while the adjacent lineage in the suture (neural crest or mesoderm, respectively) will be functionally unaffected (Fgfr2NeoS252W/+) (Chen et al., 2003; Holmes et al., 2009). To facilitate the tracking of cells from the Cre-expressing lineage in later experiments, Mesp1Cre and Wnt1Cre mice also contained the R26R lacZ reporter gene, in which β-galactosidase expression is enabled by Cre activity (Soriano, 1999). In conditionally mutant offspring containing R26R, the Cre+Fgfr2S252W/+ lineage will therefore also be β-galactosidase-positive, while the adjacent unaffected Cre−Fgfr2NeoS252W/+ lineage will be β-galactosidase-negative. In true wild-type (Fgfr2+/+; WT) offspring, the presence of the R26R reporter will allow tracking of the Cre+ lineage. The incidence of craniosynostosis in mice containing both the Fgfr2NeoS252W allele and either of the Cre drivers was initially scored by inspection in newborns (P0), and later in embryonic (by sectioning) and viable post-natal animals at various ages. Separately, the frequency of R26R recombination was scored for each Cre driver in newborns. Our findings are summarized in Table 1.
Table 1.
Frequencies of craniosynostosis and lacZ expression resulting from tissue-specific Cre recombination.
| Frequency (%) | |||
|---|---|---|---|
|
| |||
| Mesp1Cre/R26R × Fgfr2NeoS252W/+ | E16.5 | P0 | Post-natala |
| Craniosynostosis (Mesp1Cre/Fgfr2NeoS252W/+) | 6/6 (100) | 25/26(96)b | 15/15 (100) |
| lacZ+ mesoderm (Mesp1Cre/R26R; WT and conditional mutant) | - | 18/19 (95)b | - |
|
| |||
| Wnt1Cre/R26R × Fgfr2NeoS252W/+ | |||
|
| |||
| Craniosynostosis (Wnt1Cre/Fgfr2NeoS252W/+) | 0/4 (0) | 0/16 (0) | 2/12 (17)c |
| lacZ+ neural crest (Wnt1Cre/R262R; WT and conditional mutant) | - | 17/17 (100) | - |
Age ranges are 2 weeks -1 year for Mesp1Cre recombinants, and 2-3 weeks for Wnt1Cre recombinants.
One Mesp1Cre/R26R/Fgfr2NeoS252W/+ pup showed no recombination.
Atypical fusion at the midline between frontal and parietal bones.
Mesodermal expression of Fgfr2S252W (referred to as MesoS252W/+ in the text, MesoAp in figures) was sufficient to cause varying degrees of unilateral or bilateral coronal suture fusion, evident by embryonic day (E) 16.5 (Table 1). Unlike fully heterozygous Fgfr2S252W/+ mice, MesoS252W/+ mice of both sexes were viable and fertile. This viability is likely due to the lack of Fgfr2S252W expression in the ectodermal and endodermal epithelia of the palate, lung, and trachea, which are otherwise malformed at birth (Wang et al., 2005). Post-natal skull development varied amongst conditionally mutant individuals, which ranged in appearance from near WT to having clear facial shortening (Fig. 1A). Despite coronal suture fusion, parietal bone growth was not greatly reduced compared to WT, while frontal and facial bone lengths were variably shortened (Fig. 1B). Coronal sutures were typically fused along their length (Fig. 1C, middle panel), but some showed patency towards the midline and interrupted fusion (Fig. 1C, lower panel). The palatal extensions of the maxillary and premaxillary bones were often malformed and misaligned in more severely affected mice (Fig. 1D). Dissociation of skulls in KOH showed no fusions between facial bones, but within the cranial base the presphenoid bone was frequently broadened and malformed, and varying degrees of fusion of the intersphenoidal suture were found in 12/18 MesoS252W/+ animals, compared to 1/19 WTs (Fig. 1E). The frontoethmoidal suture often showed partial fusion (Fig. 1F). Mesodermal expression of Fgfr2S252W therefore not only causes coronal suture fusion, but also has secondary effects on bone growth within the neural crest-derived cranial base and facial skeleton.
Fig. 1.
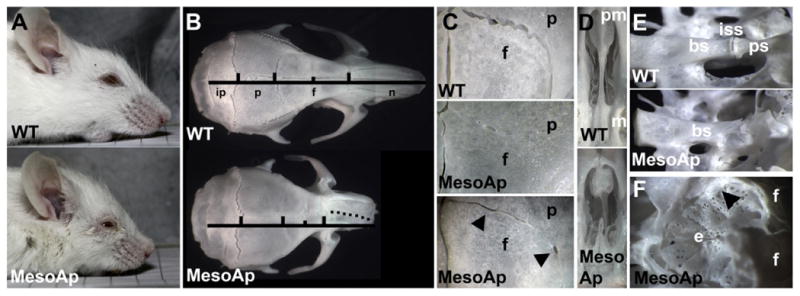
Mesodermal expression of Fgfr2S252W causes CS. (A) Heads of 1-year old wild-type (WT; upper panel) and MesoS252W/+ (MesoAp; lower panel) mice. (B) Dorsal view of skulls from mice in (A). Vertical bars mark the suture position between interparietal (ip), parietal (p), frontal (f) and nasal (n) bones. The short vertical bar marks the jugum limitans of the frontal bone. The dotted line along the MesoAp internasal suture indicates the curvature of the snout. (C) Dorsal view of coronal sutures showing full (middle panel) or partial fusion (lower panel) in MesoAp skulls. The interfrontal suture is at left. In the lower panel, lateral fusion is interrupted by patent regions (arrowheads). The upper and lower panels are from the same skulls as in (B). (D) Ventral view of palatal extensions of the premaxilla (pm) and maxilla (m) of skulls in (B). (E) Dorso-lateral view of basisphenoid (bs), intersphenoidal synchondrosis (iss), and presphenoid (ps) in a second pair of WT and MesoAp mice. (F) Dorsal view of the ethmoid (e) and anterior frontal (f) bones of the MesoAp mouse in (E). Black arrowhead indicates partial fusion of the frontoethmoidal suture.
In contrast, neural crest expression of Fgfr2S252W (NCS252W/+ in the text, NCAp in figures) did not result in CS in embryonic or newborn mice (Table 1). NCS252W/+ mice survived beyond birth, and in only two of twelve juvenile mice short regions of fusion were found near the midline of the skull, and were not representative of the embryonic fusion typical of Apert syndrome (Holmes et al., 2009; Mathijssen et al., 1996). The absence of CS was not due to a lack of Cre activity or recombination of the Fgfr2NeoS252W allele in neural crest. The R26R reporter was always activated by Wnt1Cre (Table 1) and NCS252W/+ mice clearly showed deformities of varying severity in neural crest-derived frontal and facial bones (Fig. 2A–C), and were often identifiable by a “short face” phenotype at birth (not shown). Examination of skulls at P0 invariably showed synostosis of the frontal-premaxillary, premaxillary–maxillary, and the zygomatico-maxillary and zygomatico-squamosal sutures, so that the frontal bones and major facial bones formed a connected unit (not shown). This pattern of suture fusion within the face conforms with previous reports for fully heterozygous mice in this Apert model (Martinez-Abadias et al., 2010; Purushothaman et al., 2011; Wang et al., 2005). Common features in juvenile NCS252W/+ mice were deviation of the snout (Fig. 2C and E), extreme shortening of the palate (Fig. 2D), and fusion of the maxillary–palatine and frontoethmoidal sutures (Fig. 2D and F). The interpalatine suture was occasionally fused (Fig. 2D). PCR of genomic DNA prepared from separated P0 frontal and parietal bones confirmed directly that Cre recombination of Fgfr2NeoS252W occurred in neural crest-derived frontal bones (Fig. 2G). After two weeks mutant animals failed to thrive and invariably died between 2 and 3 weeks of age, presumably because facial shortening allowed overgrowth of the lower incisors and prevented sufficient feeding (Fig. 2B). Trimming of the lower incisors allowed survival. MesoS252W/+ and Wnt1Cre mice could be crossed to give compound Meso/NCS252W/+ animals that combined the facial and coronal suture fusion of each genotype (not shown).
Fig. 2.
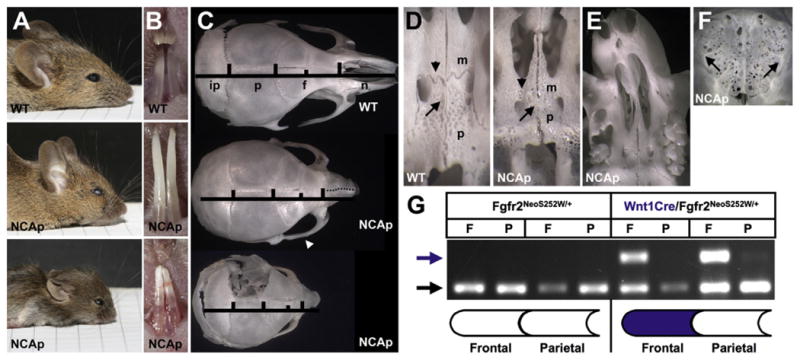
Neural crest expression of Fgfr2S252W causes facial shortening but not CS. (A) Heads of 3-week old WT (upper panel) and mediumly affected (middle panel) or strongly affected (lower panel) NCS252W/+(NCAp) mice. (B) Lower incisors of mice in (A). (C) Dorsal view of skulls from mice in (A). The left parietal is removed in the lower panel. The NCAp skull in the middle panel shows zygomatic arch fusion (arrowhead) and a deviated snout (dotted line). (D) Ventral view of the palate in WT (left panel) and NCAp (right panel) skulls. Shortening of the maxillary (m) and palatine (p) bones and fusion of the maxillary–palatine (arrowhead) and interpalatine (arrow) sutures occurs in NCAp mice. (Same mice as in A, upper and lower panels). (E) Ventral view showing curvature of the snout and palate in an NCAp mouse at 3 weeks. (F) Dorsal view of the ethmoid plate in an NCAp mouse shows extensive fusion of the dorsal frontoethmoidal suture between the two arrows. (G) PCR to detect the wild-type Fgfr2 (black arrow) and Cre-recombined Fgfr2S252W (blue arrow) alleles in genomic DNA prepared from individual frontal (F) and parietal (P) bones of two WT (Fgfr2NeoS252W/+) and two NCS252W/+ (Wnt1Cre/Fgfr2NeoS252W//+) newborn mice. The larger PCR band contains the additional LoxP sequence resulting from Neo excision. The schematic of frontal and parietal bones below indicates the region of expected Wnt1Cre activity in blue.
The results of these crosses therefore show that expression of the Fgfr2S252W allele within mesoderm, but not neural crest, is necessary and sufficient for CS in the Apert mouse model.
Osteogenesis is induced within Fgfr2S252W/+ sutural mesenchyme
The formation of the frontal and parietal bones requires two basic steps. First, their adjacent neural crest and mesoderm territories are established. Second, overt ossification begins within each territory, extending radially outwards and upwards. At the presumptive coronal suture, then, the neural crest and mesoderm must first form a distinct border, and then their respective bone anlagen must form overlapping but separate osteogenic fronts within the separate lineage territories. Coronal suture fusion could result from defects in either of these two processes. In all CS mouse models so far, the initial normal formation of frontal and parietal bones suggest that the overall process of cell migration and proliferation that establishes the neural crest and mesoderm territories is not greatly disturbed. However, significant cell mixing across the coronal suture lineage border in TWIST1+/− mice and other non-Fgfr CS mouse models has been shown (Merrill et al., 2006; Ting et al., 2009; Yen et al., 2010).
To assess any contribution of cell mixing to coronal suture fusion in the Apert mouse model we compared developing sutures from MesoS252W/+/R26R, NCS252W/+/R26R, and WT/R26R (Cre+) embryos. In the sutures of MesoS252W/+ embryos only the mesodermal lineage is mutant, while the neural crest contribution (i.e., the frontal bone) is functionally WT. In MesoS252W/+/R26R embryos at E14.5, before overt fusion, diffuse alkaline phosphatase (ALP) activity, indicating osteogenesis (Candeliere et al., 2001), is seen in the suture mesoderm, while the neural crest/mesoderm border is intact (Fig. 3A–D). Occasional mesoderm-derived endothelial cells are present in WT and mutant neural crest (Fig. 3A and C) (Yoshida et al., 2008). In MesoS252W/+/R26R embryos at E17.5, secreted osteoid uniting the frontal and parietal bones lies between mesodermal cells with no evidence of cell mixing (Fig. 3E–H). No changes were seen in the coronal sutures of NCS252W/+/R26R mice (not shown).
Fig. 3.
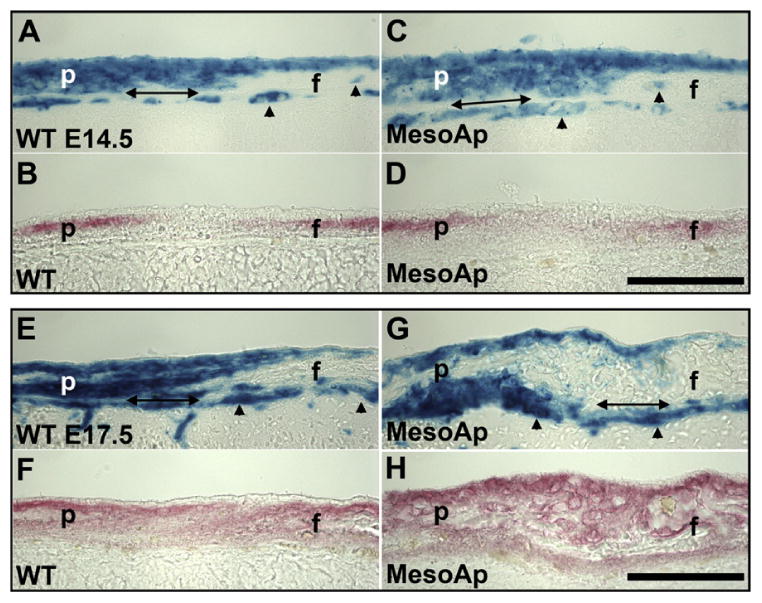
The neural crest/mesoderm border remains intact during coronal suture fusion. (A–D) Coronal sutures of (A and B) WT(R26R/Mesp1Cre/Fgfr2+/+) and (C and D) MesoS252W/+ (MesoAp; R26R/Mesp1Cre/Fgfr2NeoS252W/+) E14.5 littermates. In adjacent sections, mesoderm is stained for β-galactosidase activity (A and C; blue), and osteogenic mesenchyme of the parietal (p) and frontal (f) bones are stained for ALP activity (B and D; red). As diffuse ALP activity rises in the MesoAp suture mesenchyme, the neural crest/mesoderm border is maintained. Double-headed arrows indicate the dura mater. Arrowheads indicate mesoderm-derived endothelial cells. (E–H) Coronal sutures of (E and F) WT and (G and H) MesoAp E17.5 littermates, stained as in (A–D). After fusion, the relative positions of mesoderm and neural crest are maintained. The acellular osteoid lacks β-galactosidase activity. Scale bars=50 μm.
We next examined the progress of ossification in fusing MesoS252W/+ sutures with respect to the neural crest/mesoderm border within the coronal suture. At E14.5, proliferation in the WT suture, determined by BrdU incorporation, is concentrated around the widely separated osteogenic fronts, while in the unfused MesoS252W/+ suture ectopic proliferation extends with mildly increased ALP activity through the mutant mesoderm up to the neural crest/mesoderm border (Fig. 4A–D). However, the rate of proliferation at this age did not differ from the WT suture (not shown), as found in fully heterozygous mutants (Holmes et al., 2009). In more mature MesoS252W/+ sutures undergoing fusion (Fig. 4E–L), ectopic ALP expression within mutant mesenchyme immediately above fused regions displayed two surprising qualities—continuity with the WT neural crest, and separation from the parietal ALP domain. In an E15.5 suture, for example (Fig. 4E–H), a spur of ectopic osteoid contiguous with the WT frontal bone is secreted by mutant suture mesenchyme, extending towards but separate from the mutant parietal bone. In an E17.5 suture (Fig. 4I–L), while ectopic ALP activity is seen adjacent to the mutant parietal osteoid, a second domain of ectopic ALP activity is seen within the mutant mesenchyme, separate from the parietal domain and contiguous with the WT frontal bone. This suggests that, in the more mature MesoS252W/+ suture, osteogenic factors from the edge of the WT frontal bone drive osteogenesis within the suture mesenchyme leading to fusion. This appeared to be a common feature of fusion between overlapping fontal and parietal bones in the more apical suture, because the extension of the WT frontal bone to a point posterior to the leading edge of the mutant parietal bone was seen in all MesoS252W/+ animals examined (Fig. 4M and N), and was commonly seen in fused coronal sutures of Fgfr2S252W/+ animals (Holmes et al., 2009). The dura mater, a membrane underlying the calvarial bones, can influence suture patency (Opperman et al., 1993; Roth et al., 1996). It should be noted that the dura mater underlying the fusing sutures is derived from neural crest (Jiang et al., 2002), and is therefore functionally WT in MesoS252W/+ animals (Figs. 3 and 4).
Fig. 4.
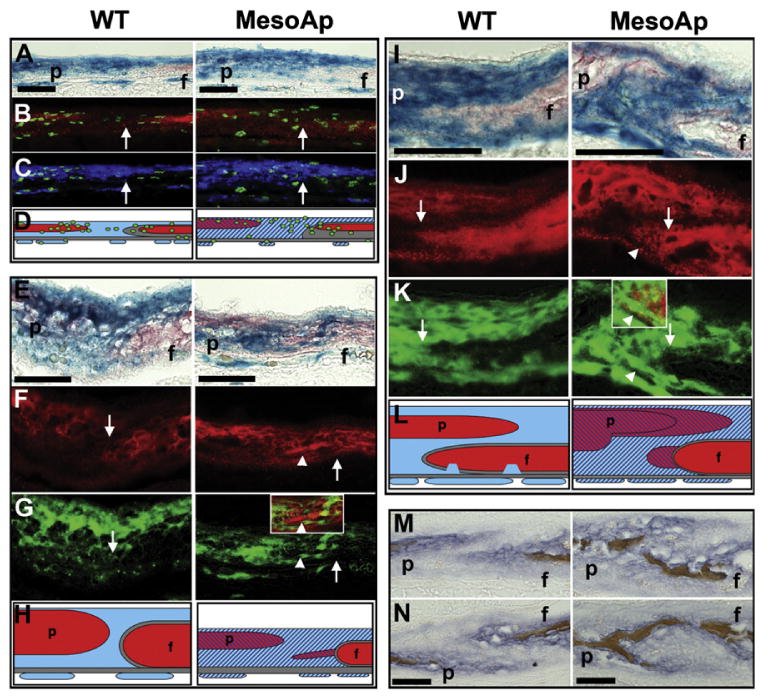
Osteogenic induction within the MesoS252W/+ coronal suture mesenchyme causes fusion. (A–D) Coronal sutures of WT (R26R/Mesp1Cre/Fgfr2+/+; left panels) and MesoS252W/+ (MesoAp; R26R/Mesp1Cre/Fgfr2NeoS252W/+; right panels) BrdU-treated E14.5 littermates double-stained for (A) β-galactosidase activity (blue) and ALP (red). (B) Merged images of the sutures in (A) photographed under red and green fluorescence to show low ALP activity (red) and BrdU incorporation (green) extending across the suture mesenchyme in the MesoAp suture. White arrows (B,C,F,G,J,K) indicate the neural crest/mesoderm border. (C) Merged images of the sutures in (A) with mesodermal β-galactosidase rendered in false color (blue) and BrdU incorporation (green) show ectopic proliferation is within the mesoderm. (D) Schematic summary of A–C. WT (red) and mutant (striped red) osteogenic domains are shown within WT (blue) or mutant (striped blue) mesoderm and WT neural crest (gray), above the dura mater (gray). BrdU-incorporating cells are green. Endothelial cells (blue or striped blue) are shown below the dura mater. (E–H) WT and MesoAp coronal sutures at E15.5 double-stained for (I) β-galactosidase activity (blue) and ALP (red). (F) Sutures in (E) photographed under red fluorescence to show the ectopic osteoid spur extending from the WT frontal bone (arrowhead). (G) Sutures in (E) rendered in false color to show mesodermal β-galactosidase activity in green. The inset in MesoAp shows the region of the ectopic osteoid spur in (F) merged with the corresponding region in (G). (H) Schematic summary of E–G. Colors as in (D). (I–L) WT and MesoAp coronal sutures at E17.5 double-stained for (I) β-galactosidase activity (blue) and ALP (red). (J) Sutures in (I) photographed under red fluorescence to show ectopic ALP activity extending from the WT frontal bone (arrowhead). (K) Sutures in (E) rendered in false color to show mesodermal β-galactosidase activity in green. The inset in MesoAp shows the region of ectopic ALP in (L) merged with the corresponding region in (K). (L) Schematic summary of I–K. Colors as in (D). (M) Left and (N) right coronal sutures from the same WT (left panels) or MesoAp (right panels) embryos at E16.5, double-stained for ALP activity (purple) and mineralized bone (brown). Scale bars=50 μm (A–L), and 100 mm (M and N). f, frontal bone; p, parietal bone.
Differences in coronal suture fusion between MesoS252W/+ and fully heterozygous EIIaCre-induced Fgfr2S252W/+ embryos were noted. As in Fgfr2S252W/+ embryos, bony fusion could be observed by E16.5 in MesoS252W/+ embryos (Fig. 4M and N), but incipient fusion between E13.5 and E14.5 was less obvious than in Fgfr2S252W/+ embryos (not shown) (Holmes et al., 2009). In embryonic Fgfr2S252W/+ sutures, progressive suture fusion originating from a single point was invariably observed (Holmes et al., 2009), while in embryonic MesoS252W/+ coronal sutures we often saw interruptions in fused domains when analyzed by serial sectioning (not shown). In adult MesoS252W/+ skulls, this resulted in areas of bony fusion separated by patent regions along the coronal suture (Fig. 1C, lower panel). Coronal suture fusion in MesoS252W/+ mice was therefore fully penetrant (Table 1), but less robust than in Fgfr2S252W/+ animals.
In summary, loss of the coronal suture mesenchyme in the Apert mouse model is not a result of lineage mixing prior to or during fusion of the adjacent frontal and parietal bones. Rather, the mesoderm-derived suture mesenchyme actively participates in fusion through its osteogenic induction under the influence of both osteogenic fronts. The participation of mutant neural crest tissues is not required in this process.
MesoS252W/+ suture mesenchyme exerts a paracrine influence on the neural crest
We had previously found that proliferation in WT osteogenic fronts decreased gradually between E13.5 and E16.5; in Fgfr2S252W/+ sutures, ectopic proliferation was seen in the early basal suture as the osteogenic fronts approached each other, but the rate of proliferation was not above the WT, and as overt fusion progressed, the decrease was strongly enhanced by E16.5 (Holmes et al., 2009). We re-examined this aspect of CS in the MesoS252W+ animals. Despite ectopic proliferation within the mutant mesoderm at E14.5 (Fig. 4A–D), the rate of proliferation within osteogenic fronts of WT and MesoS252W/+ coronal sutures did not differ (not shown). Later, in mutant parietal osteogenic fronts lying just above or between areas of suture fusion at E16.5, BrdU incorporation was strongly decreased compared to WT (Fig. 5A). Surprisingly, BrdU incorporation was also similarly decreased in the osteogenic front of the adjacent WT frontal bone, showing that an anti-proliferative influence must come from or through the intervening mutant mesoderm of the suture to act on the WT neural crest. This effect was seen in 5/5 sutures examined. In contrast, in the non-fusing coronal sutures of NCS252W/+ animals at E16.5, proliferation decreased mildly in only half (3/6) of the osteogenic fronts of mutant neural crest-derived frontal bones, while proliferation in the adjacent WT parietal bone was always unaffected (Fig. 5B). Further evidence of a paracrine osteogenic effect of mutant mesoderm-derived suture mesenchyme lies in the overgrowth of the WT frontal bone often seen just above actively fusing areas of the coronal suture (Fig. 5C), an effect not seen in NCS252W/+ sutures (Fig. 5D). At birth, mesenchyme was typically depleted in the more apical patent areas of MesoS252W/+ sutures, possibly as a result of decreased proliferation and premature incorporation into the bone edge (Fig. 5C).
Fig. 5.

Paracrine influences from MesoS252W/+ suture mesenchyme affect proliferation and bone deposition. (A) (Upper panel) BrdU incorporation within WT or MesoS252W/+(MesoAp) frontal (f) and parietal (p) osteogenic fronts at E16.5. Black columns=WT (Fgfr2+/+ or Fgfr2NeoS252W/+) tissue; blue column=Fgfr2S252W/+ tissue. P values are indicated. (Lower panel) Representative images of coronal sections from the corresponding sutures co-stained for ALP activity (red fluorescence) and BrdU incorporation (green fluorescence). Scale bar=70 μm. (B) (Upper panel) BrdU incorporation within WT or NCS252W/+(NCAp) frontal (f) and parietal (p) osteogenic fronts at E16.5. P value is indicated. (Lower panel) Representative images of coronal sections from the corresponding sutures, as in (A). (C) Sagittal sections of WT (R26R/Mesp1Cre/Fgfr2-/+) and MesoS252W/+ (MesoAp; R26R/Mesp1Cre/Fgfr2NeoS252W/+) coronal sutures of P0 calvaria double-stained for ALP (red) and β-galactosidase (blue) activity. (D) Sagittal sections of WT (R26R/Wnt1Cre/Fgfr2-/+) and NCS252W/+ (NCAp; R26R/Wnt1Cre/Fgfr2NeoS252W/+) coronal sutures of P0 calvaria double-stained for ALP (red) andβ-galactosidase (blue) activity. Scale bar for C,D=100 μm.
Ectopic midline cartilage results from mesodermal Fgfr2S252W expression
In our previous study of Fgfr2S252W/+ mice, we found ectopic midline cartilage in the interparietal foramen and thickening of the cartilage underlying the basal frontal bone at E16.5 (Holmes et al., 2009). We therefore took advantage of the lineage-specific expression of Fgfr2S252W and R26R to investigate these phenomena further. The sagittal suture separating the parietal bones contains both neural crest anteriorly (extending from the frontal bone domain), and mesoderm posteriorly (Fig. 6A) (Jiang et al., 2002; Yoshida et al., 2008). Careful examination of WT, NCS252W/+, and MesoS252W/+ mice at E16.5–E17.5 showed that small cartilage islands occurred frequently in the sagittal suture in all genotypes, and that their lineage reflected the surrounding mesenchyme, being neural crest-derived anteriorly, mesoderm-derived posteriorly, and of mixed origin in between (Fig. 6B–D). Ectopic cartilage was not seen in NCS252W/+ mutants, but was clearly evident in the interparietal foramen region of MesoS252W/+ animals, and appeared uniformly mesoderm in origin (Fig. 6E–G). All endogenous and ectopic cartilages lay below the plane of osteogenic mesenchyme (Fig. 6B and F). Thickening of the cartilage at the base of the frontal bone was not seen in either lineage-specific mutant, and may depend on the combined effects of Fgfr2S252W activity in more than one lineage.
Fig. 6.
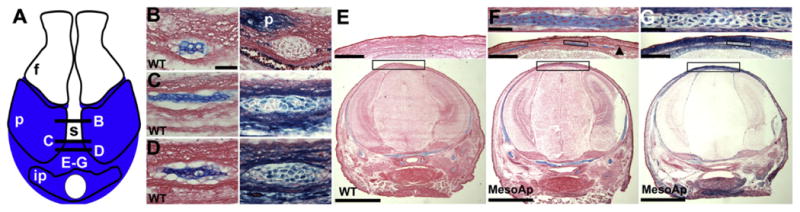
Ectopic midline cartilage is derived from mutant mesoderm. (A) Schematic of calvarium showing neural crest (white) and mesoderm (blue) distribution in the frontal (f), parietal (p), and interparietal (ip) bones and the sagittal suture (s). Locations of coronal plane sections in B–G are indicated. (B) Adjacent sections within the anterior sagittal suture of an E17.5 WT (Mesp1Cre/R26R) calvarium are stained for cartilage (left panel; Alcian blue, counterstained with eosin) and mesodermal β-galactosidase activity (blue, right panel; counterstained with eosin). The mesodermal parietal bone (p) expresses β-galactosidase. The unstained cartilage is derived from the neural crest. (C) As in (B), but a mid-sagittal location of a separate E17.5 WT calvarium. Cartilage is derived from both neural crest and mesoderm. (D) As in (B), from the same calvarium, but within the posterior sagittal suture. Cartilage is derived from mesoderm. Scale bar for B–D=50 μm. (E) (lower panel) Coronal section at the level of the interparietal foramen of a WT skull at E16.5, stained for cartilage. The boxed region shown enlarged in the upper panel is devoid of cartilage. (F) (lower panel) Coronal section at the level of the interparietal foramen of a MesoS252W/+/R26R(MesoAp) skull at E16.5, stained for cartilage. The boxed region shown enlarged in the middle panel contains multiple areas of ectopic cartilage. The black arrowhead indicates the parietal bone edge. An individual cartilage island (boxed) is shown in the upper panel. (G) The section adjacent to (F), stained for β-galactosidase activity. The middle and upper panels show that the ectopic cartilages in (F) are derived from mesoderm. Scale bars for E–G=2 mm (lower panels), 400 μm (middle panels), 50 μm (upper panels).
Cranial base curvature involves both neural crest- and mesoderm-derived mesenchyme
Mid-sagittal sections of heads from P0 NCS252W/+ (n=5) and MesoS252W/+ (n=2) pups revealed retroflexion of the cranial base, compared to WT (Fig. 7A–H). This curvature was even greater in Fgfr2S252W/+ animals (n=3; Fig. 7B and F), suggesting that Fgfr2S252W activity in both the neural crest and mesodermal components of the skull base is additive for this phenotype.
Fig. 7.
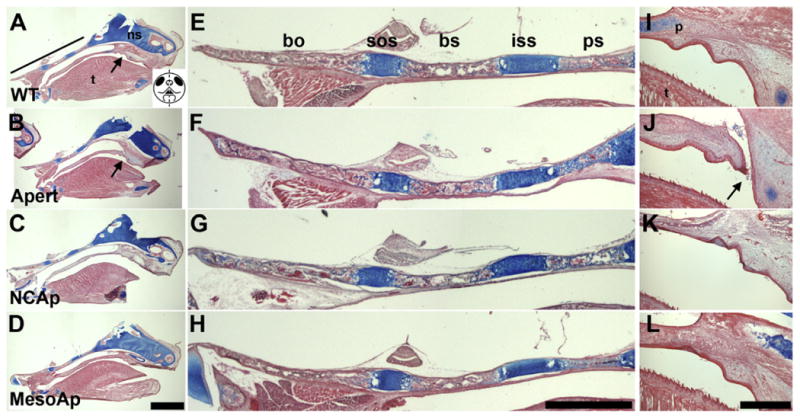
Influence of Fgfr2S252W expression on skull base and palate formation. (A–D) Mid-sagittal sections of (A) WT, (B) Fgfr2S252W/+ (Apert), (C) NCAp, and (D) MesoAp skulls (calvaria removed) at P0, stained for cartilage (Alcian blue, counterstained with eosin). The black line and arrow in (A) indicate the areas of enlargement of the cranial base (E–H) and palate (I–L), respectively, for all genotypes. The schematic in (A) shows a frontal view of a P0 face with the central vertical dashed line indicating the mid-sagittal plane of section for all panels. The arrow in (B) indicates the perforated palate, enlarged in (J). Scale bar=2 mm. (E–H) Enlargement of the cranial bases from (A–D). Scale bar=1 mm. (I–L) Enlargement of the anterior secondary palates from (A–D). Perforation is only present in the Apert palate (J, arrow). Scale bar=400 μm. ns, cartilage primordium of nasal septum; t, tongue; p, palatal extension of maxillary bone; bo, basioccipital; sos, spheno-occipital synchondrosis; bs, basisphenoid; iss, intersphenoidal synchondrosis; ps, presphenoid.
Palatal perforation is absent in conditional Apert animals
Clefting of the soft palate is a common defect in Apert syndrome (Kreiborg and Cohen, 1992). Fgfr2S252W/+ mice had incomplete closure of the anterior end of the secondary palate at P0 (n=3/3; Fig. 7J) (Wang et al., 2005), while NCS252W/+ pups had complete palatal closure at this age (n=5; Fig. 7K). Fgfr2S252W signaling in the neural crest-derived palatal mesenchyme alone is therefore not sufficient to prevent closure, suggesting the importance of aberrant Fgfr2S252W activity in the oral epithelium in this defect. MesoS252W/+ pups also had complete palate closure, as expected (Fig. 7L).
Discussion
The developing mammalian coronal suture contains both a lineage border, between neural crest and mesoderm, and a cell fate border within the mesoderm, between osteoblasts and suture mesenchyme (Jiang et al., 2002; Yoshida et al., 2008). The significance of these borders for the etiology of CS, and the impact of aberrant Fgfr2 signaling on their integrity during fusion in Apert syndrome, remains obscure. We have used Cre/lox technology, introducing the gain-of-function Fgfr2S252W allele and a lacZ reporter gene into the mesoderm or neural crest, to both determine the integrity of these borders during CS, and to test the hypothesis that CS of the coronal suture depends specifically upon mesodermal mutation of Fgfr2 to disrupt the mesenchyme/osteoblast balance within the suture. In agreement with our hypothesis, we find that CS only occurs when Fgfr2S252W is expressed in mesoderm, via the ectopic induction of osteogenesis within the intact sutural mesenchyme. The inductive signals are presumably Fgfs and Bmps derived from the osteogenic fronts (Hajihosseini and Heath, 2002; Iseki et al., 1997; Rice et al., 1999). While Fgfs from neural crest cells of the frontal bone can therefore participate in this induction, the mutant neural crest cells themselves are limited, like WT cells, to producing frontal bone. No increase in cell mixing at the border within the coronal suture is seen when Fgfr2S252W is expressed in either neural crest or mesoderm, including during the process of CS resulting from its mesodermal expression. Expression of Fgfr2S252W in the mesoderm is therefore necessary and sufficient for CS of the coronal suture in the Apert mouse model. This does not preclude an additive role for Fgfr2S252W activity in adjacent tissues derived from neural crest (frontal bone and dura mater) and neuroectoderm (brain), and such a role is in fact likely because the onset of coronal suture fusion in MesoS252W/+ mice is mildly delayed compared to fully heterozygous Fgfr2S252W/+ mice. Fgfr2S252W activity in the frontal bone is probably most important in this regard, as this would more quickly close the gap between the advancing osteogenic fronts, leading to CS beginning earlier in sutural development, when fusion is most efficient (Holmes et al., 2009). Furthermore, as a source of Fgfs, the osteogenic front of the frontal bone appears to play a decisive role in the fusion of the overlapping fronts of the more developed suture from E16.5. At this time it is hemmed in by the dura mater on its inner surface and by mesoderm at its leading edge and outer surface. In WT or NCS252W/+ sutures, neural crest-derived Fgfs act autocrinally, with little or no influence on the adjacent WT suture mesenchyme, derived from mesoderm. However, in MesoS252W/+ sutures, Fgfr2S252W in the suture mesenchyme is sufficiently sensitive to allow juxtacrinal osteogenic induction from the neural crest, resulting in the bony bridging from the frontal bone edge to a point behind the parietal bone edge typically seen. In contrast, growth at the osteogenic front of the mutant parietal bone can still advance within the layer of mesoderm extending beyond it and overlaying the neural crest and frontal bone.
Lineage mixing and maintenance of the suture
Disruption of the neural crest/mesoderm border may be an important mechanism of CS depending on the underlying genetic defect, as evidenced by cell mixing in mouse models of human CS caused by loss-of-function mutations in TWIST1 (Saethre-Chotzen syndrome) and JAGGED1 (Alagille syndrome), and in EphA4 mutants (Merrill et al., 2006; Ting et al., 2009; Yen et al., 2010). TWIST1 plays a critical role in suture organization, as it is upstream not only of Jagged1, and Efna4 and its ligands, EphA2 and EphA4 (which control cell mixing independently of Jagged1), but also of Fgfr2. TWIST1 homodimers upregulate Fgfr2 expression in the osteogenic fronts, while TWIST1/E protein heterodimers downregulate Fgfr2 expression in the suture mesenchyme (Connerney et al., 2008; Connerney et al., 2006). In TWIST1+/− mice, cell mixing across the lineage border can be correlated with the initiation of suture fusion (Merrill et al., 2006), and it is suggested that mixing may cause a dilution of the suture mesenchyme with osteogenic cells (Ting et al., 2009). However, TWIST1 haploinsufficiency also results in the upregulation of Fgfr2 expression within the suture mesenchyme (Rice et al., 2000), which could predispose it to osteogenic induction and, by analogy to the Apert mouse model, lead to CS independent of cell mixing. Indeed, expression of the receptor tyrosine kinase inhibitor Sprouty1 in TWIST1+/−. mice prevents CS (Connerney et al., 2008). The relative importance of each effect to CS in this model remains to be determined. In contrast, no cell mixing was seen during suture fusion caused by mesodermal Fgfr2S252W expression, where the suture mesenchyme cells themselves are induced to synthesize bone matrix.
Differentiation of the suture mesenchyme affects proliferation and bone growth
We have previously shown that, in Fgfr2S252W/+ mice, ectopic proliferation at WT rates coincides with osteogenic front approximation in the early suture mesenchyme, while in the osteogenic fronts of older sutures proliferation decreases significantly compared to the WT just prior to fusion (Holmes et al., 2009). Both of these observations could be reproduced in the MesoS252W/+ coronal suture. In the earlier suture, ectopic proliferation is seen principally in the mutant mesoderm of the suture mesenchyme. In the older sutures, surprisingly, decreased proliferation in both osteogenic fronts is observed when only the mesoderm is mutant, suggesting that an extracellular signal from either the suture mesenchyme or the parietal osteogenic front acts upon the neural crest. In NCS252W/+ sutures, a similar signal affecting the WT parietal is not generated by the mutant frontal, suggesting that the signal is derived from the mutant suture mesenchyme itself. The osteogenic fronts of the coronal suture appear to have the highest concentrations of Fgfs and Bmps, but the suture mesenchyme also expresses Fgf2 and Bmp4 (Hajihosseini and Heath, 2002; Iseki et al., 1997; Rice et al., 1999). The ectopic induction of osteogenesis within the suture mesenchyme in MesoS252W/+ animals would therefore be accompanied by increased expression of these osteogenic signals, which could act back on both adjacent osteogenic fronts, driving down proliferation as differentiation proceeds. This mechanism could also account for the thickening of the osteoid in both osteogenic fronts often seen in older sutures just prior to fusion. The mild decrease in proliferation seen in some frontal osteogenic fronts in NCS252W/+ animals may be an enhancement of the decreased proliferation seen in maturing WT coronal sutures (Holmes et al., 2009), perhaps tied to the age-appropriate expansion rate of the calvaria.
Primary CS and facial dysmorphology
Skull development requires the integrated growth of the calvaria, facial skeleton, cranial base, and brain, all of which express Fgfr2 (Blak et al., 2005; Britto et al., 2001; Orr-Urtreger et al., 1993; Rice et al., 2003). In Apert and other craniosynostotic syndromes all these tissues suffer varying degrees of malformation (Cohen and Kreiborg, 1990, 1996; Cunningham et al., 2007; Raybaud and Di Rocco, 2007). Much research has focused on how coronal fusion may depend on changes elsewhere in the affected skull, and on how changes in the major subdivisions of the skull contribute to the final dysmorphic phenotype. Lineage-specific expression of Fgfr2S252W allows us to assess the influence of these separate regions on coronal suture fusion during fetal development, and, by circumventing the early post-natal lethality of Fgfr2S252W/+ mice, their cross-influences during post-natal growth.
In the WT rodent calvaria, all sutures remain patent except the posterior frontal, which fuses postnatally, and has therefore been a focus of study for its possible relevance to CS. The dura mater, a neural crest-derived membrane underlying the calvaria (Jiang et al., 2002), exerts a positive influence via secreted factors on both suture patency and, in the posterior frontal suture, fusion (Levine et al., 1998; Opperman et al., 1993; Roth et al., 1996). Dural cells transfected with the Apert Fgfr2P253R allele have increased osteogenic influence on co-cultured osteoblasts (Ang et al., 2010). However, our results clearly demonstrate that mutant dura mater in NCS252W/+ mice is unable to induce osteogenic fusion of the overlying coronal suture mesenchyme in the Apert mouse model.
Expansion of the brain and skull are tightly linked (Baer, 1954; Moss, 1954), and it has been suggested that a primary Apert brain defect may result in coronal suture fusion (Faro et al., 2006). However, a recent comparison of mouse models of the two major Apert mutations found no correlation between brain phenotypes and the extent or pattern of coronal suture fusion (Aldridge et al., 2010). Our results demonstrate that coronal suture fusion occurs in the absence of a primary brain defect.
Prior to the identification of genes underlying CS syndromes, there was speculation that coronal suture fusion was a secondary response to shortening of the skull base (Cohen, 1993). In Apert syndrome, for example, it was suggested that cartilage defects leading to hypoplastic development of the anterior cranial base could alter tensile forces transmitted to the brain via the dura mater attached to the base, resulting in coronal suture fusion (Kreiborg et al., 1993). Recent data from mouse models suggests that no such link exists. Mutant calvaria from another Apert mouse model (Fgfr2P253R) dissected from the cranial base and grown in culture still exhibit coronal suture fusion (Yin et al., 2008), while forced expression of the same mutation in cartilage shortens the cranial base without inducing CS (Nagata et al., 2011). In the Fgfr2S252W/+ coronal suture changes begin by E13.5 (Holmes et al., 2009), when the cartilage of the anterior cranial base is not yet fully formed (McBratney-Owen et al., 2008). In our current study, coronal suture fusion occurs in MesoS252W/+ animals by E16.5 in the presence of a WT anterior cranial base, and without any obvious defect of the partly mutant posterior cranial base.
The viability of MesoS252W/+ and NCS252W/+ animals allowed some interesting observations on the contribution of Fgfr2S252W activity to post-natal facial morphology. Not surprisingly, neural crest expression of Fgfr2S252W caused extensive and early fusion of facial sutures, resulting in loss of their growth potential and subsequent mid-face hypoplasia expected of the Apert phenotype.
In contrast, mesodermal expression resulted not only in CS, but also in a widely variable facial phenotype in adult animals, which at its most extreme included significant facial shortening despite the presence of patent sutures between WT neural crest-derived facial bones. Deformities of the presphenoid bone and fusion of the frontoethmoidal suture (both WT) in MesoS252W/+ mice suggests that CS alone, or combined with changes in the posterior cranial base, can exert strong secondary effects on presphenoid and anterior cranial base morphology. This issue has been addressed in mouse models of the two major Apert Fgfr2 mutations, but no correlation between the extent of coronal fusion and facial morphology was seen (Martinez-Abadias et al., 2010). Our data complement this finding, in demonstrating that the facial dysmorphology is a strong primary effect of Fgfr2S252W activity in neural crest alone, while any additive effect of coronal suture fusion or a posterior cranial base defect may be minor in comparison, and masked by the primary defect of neural crest. On the other hand, we also show that full expression of Apert syndrome phenotypes, such as cranial base curvature, result from changes in both mesoderm and neural crest.
Ectopic cartilage
Cartilage overgrowth of the tracheal sleeve is an infrequent but significant factor in the mortality of Apert syndrome (Cohen and Kreiborg, 1992), and this and other cartilage abnormalities are reproduced in the Apert mouse model (Holmes et al., 2009; Wang et al., 2005). Secondary cartilage in the calvarial midline is not uncommon (Cohen, 2006), and their transitory appearance in late fetal, newborn, and juvenile ages has been noted in rats (Pritchard et al., 1956) and mice (Liu et al., 1999; Rice et al., 2000; Saito, 2000), but their significance is unknown. By lineage tracing we demonstrate that this cartilage generally shares the lineage of the surrounding mesenchyme. However, the ectopic cartilage specific to the Apert mouse model and located between the parietal and interparietal bone was dependent on mesodermal expression of Fgfr2S252W and was mesodermal in origin. The ectopic cartilage could arise from aberrant migration from more basal cranial cartilage, or be induced in situ by endogenous Fgfs. The position of endogenous and ectopic midline cartilages below the plane of osteogenic mesenchyme suggests their origin is independent from this mesenchyme.
In conclusion, we have shown that in a mouse model of Apert syndrome, the expression of the gain-of-function Fgfr2S252W allele in the mesoderm is necessary and sufficient for coronal suture fusion. The suture mesenchyme, derived from mesoderm, actively participates in this fusion as it is induced to undergo osteogenesis. Disruption of the lineage border between mesoderm and neural crest by ectopic cell mixing, a characteristic of some mouse models of syndromic CS, does not occur in the Apert syndrome mouse model. The viability of mice with lineage-restricted expression of Fgfr2S252W will allow further exploration of both the craniofacial and other phenotypes of Apert syndrome, and alternative clinical treatments of these phenotypes.
Acknowledgments
The authors thank Dr Ethylin Wang Jabs for critical reading of the manuscript. This work was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases (Grant Number AR051358). Technical assistance was provided by the NYULMC Histopathology Core (NYU Cancer Institute Center Support Grant 5P30CA0016087).
Contributor Information
Greg Holmes, Email: gregory.holmes@exchange.mssm.edu.
Claudio Basilico, Email: claudio.basilico@med.nyu.edu.
References
- Aldridge K, Hill CA, Austin JR, Percival C, Martinez-Abadias N, Neuberger T, Wang Y, Jabs EW, Richtsmeier JT. Brain phenotypes in two FGFR2 mouse models for Apert syndrome. Dev Dyn. 2010;239:987–997. doi: 10.1002/dvdy.22218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ang BU, Spivak RM, Nah HD, Kirschner RE. Dura in the pathogenesis of syndromic craniosynostosis: fibroblast growth factor receptor 2 mutations in dural cells promote osteogenic proliferation and differentiation of osteoblasts. J Craniofac Surg. 2010;21:462–467. doi: 10.1097/SCS.0b013e3181cfe9a0. [DOI] [PubMed] [Google Scholar]
- Baer MJ. Patterns of growth of the skull as revealed by vital staining. Hum Biol. 1954;26:80–126. [PubMed] [Google Scholar]
- Blak AA, Naserke T, Weisenhorn DM, Prakash N, Partanen J, Wurst W. Expression of Fgf receptors 1, 2, and 3 in the developing mid- and hindbrain of the mouse. Dev Dyn. 2005;233:1023–1030. doi: 10.1002/dvdy.20386. [DOI] [PubMed] [Google Scholar]
- Britto JA, Evans RD, Hayward RD, Jones BM. From genotype to phenotype: the differential expression of FGF, FGFR, and TGFbeta genes characterizes human cranioskeletal development and reflects clinical presentation in FGFR syndromes. Plast Reconstr Surg. 2001;108:2026–2039. doi: 10.1097/00006534-200112000-00030. [DOI] [PubMed] [Google Scholar]
- Candeliere GA, Liu F, Aubin JE. Individual osteoblasts in the developing calvaria express different gene repertoires. Bone. 2001;28:351–361. doi: 10.1016/s8756-3282(01)00410-0. [DOI] [PubMed] [Google Scholar]
- Chen L, Li D, Li C, Engel A, Deng CX. A Ser252Trp [corrected] substitution in mouse fibroblast growth factor receptor 2 (Fgfr2) results in craniosynostosis. Bone. 2003;33:169–178. doi: 10.1016/s8756-3282(03)00222-9. [DOI] [PubMed] [Google Scholar]
- Cohen MM. The new bone biology: pathologic, molecular, and clinical correlates. Am J Med Genet. 2006;140:2646–2706. doi: 10.1002/ajmg.a.31368. [DOI] [PubMed] [Google Scholar]
- Cohen MM., Jr Sutural biology and the correlates of craniosynostosis. Am J Med Genet. 1993;47:581–616. doi: 10.1002/ajmg.1320470507. [DOI] [PubMed] [Google Scholar]
- Cohen MM, Jr, Kreiborg S. The central nervous system in the Apert syndrome. Am J Med Genet. 1990;35:36–45. doi: 10.1002/ajmg.1320350108. [DOI] [PubMed] [Google Scholar]
- Cohen MM, Jr, Kreiborg S. Upper and lower airway compromise in the Apert syndrome. Am J Med Genet. 1992;44:90–93. doi: 10.1002/ajmg.1320440121. [DOI] [PubMed] [Google Scholar]
- Cohen MM, Jr, Kreiborg S. A clinical study of the craniofacial features in Apert syndrome. Int J Oral Maxillofac Surg. 1996;25:45–53. doi: 10.1016/s0901-5027(96)80011-7. [DOI] [PubMed] [Google Scholar]
- Connerney J, Andreeva V, Leshem Y, Mercado MA, Dowell K, Yang X, Lindner V, Friesel RE, Spicer DB. TWIST1 homodimers enhance FGF responsiveness of the cranial sutures and promote suture closure. Dev Biol. 2008;318:323–334. doi: 10.1016/j.ydbio.2008.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connerney J, Andreeva V, Leshem Y, Muentener C, Mercado MA, Spicer DB. TWIST1 dimer selection regulates cranial suture patterning and fusion. Dev Dyn. 2006;235:1345–1357. doi: 10.1002/dvdy.20717. [DOI] [PubMed] [Google Scholar]
- Cunningham ML, Seto ML, Ratisoontorn C, Heike CL, Hing AV. Syndromic craniosynostosis: from history to hydrogen bonds. Orthod Craniofac Res. 2007;10:67–81. doi: 10.1111/j.1601-6343.2007.00389.x. [DOI] [PubMed] [Google Scholar]
- Danielian PS, Muccino D, Rowitch DH, Michael SK, McMahon AP. Modification of gene activity in mouse embryos in utero by a tamoxifen-inducible form of Cre recombinase. Curr Biol. 1998;8:1323–1326. doi: 10.1016/s0960-9822(07)00562-3. [DOI] [PubMed] [Google Scholar]
- Faro C, Chaoui R, Wegrzyn P, Levaillant JM, Benoit B, Nicolaides KH. Metopic suture in fetuses with Apert syndrome at 22-27 weeks of gestation. Ultrasound Obstet Gynecol. 2006;27:28–33. doi: 10.1002/uog.2648. [DOI] [PubMed] [Google Scholar]
- Hajihosseini MK, Heath JK. Expression patterns of fibroblast growth factors —18 and —20 in mouse embryos is suggestive of novel roles in calvarial and limb development. Mech Dev. 2002;113:79–83. doi: 10.1016/s0925-4773(01)00656-6. [DOI] [PubMed] [Google Scholar]
- Holmes G, Rothschild G, Roy UB, Deng CX, Mansukhani A, Basilico C. Early onset of craniosynostosis in an Apert mouse model reveals critical features of this pathology. Dev Biol. 2009;328:273–284. doi: 10.1016/j.ydbio.2009.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iseki S, Wilkie AO, Heath JK, Ishimaru T, Eto K, Morriss-Kay GM. Fgfr2 and osteopontin domains in the developing skull vault are mutually exclusive and can be altered by locally applied FGF2. Development. 1997;124:3375–3384. doi: 10.1242/dev.124.17.3375. [DOI] [PubMed] [Google Scholar]
- Iseki S, Wilkie AO, Morriss-Kay GM. Fgfr1 and Fgfr2 have distinct differentiation−and proliferation-related roles in the developing mouse skull vault. Development. 1999;126:5611–5620. doi: 10.1242/dev.126.24.5611. [DOI] [PubMed] [Google Scholar]
- Jiang X, Iseki S, Maxson RE, Sucov HM, Morriss-Kay GM. Tissue origins and interactions in the mammalian skull vault. Dev Biol. 2002;241:106–116. doi: 10.1006/dbio.2001.0487. [DOI] [PubMed] [Google Scholar]
- Johnson D, Iseki S, Wilkie AO, Morriss-Kay GM. Expression patterns of Twist and Fgfr1, -2 and -3 in the developing mouse coronal suture suggest a key role for twist in suture initiation and biogenesis. Mech Dev. 2000;91:341–345. doi: 10.1016/s0925-4773(99)00278-6. [DOI] [PubMed] [Google Scholar]
- Kreiborg S, Cohen MM., Jr The oral manifestations of Apert syndrome. J Craniofac Genet Dev Biol. 1992;12:41–48. [PubMed] [Google Scholar]
- Kreiborg S, Marsh JL, Cohen MM, Jr, Liversage M, Pedersen H, Skovby F, Borgesen SE, Vannier MW. Comparative three-dimensional analysis of CT-scans of the calvaria and cranial base in Apert and Crouzon syndromes. J Craniomaxillofac Surg. 1993;21:181–188. doi: 10.1016/s1010-5182(05)80478-0. [DOI] [PubMed] [Google Scholar]
- Lakso M, Pichel JG, Gorman JR, Sauer B, Okamoto Y, Lee E, Alt FW, Westphal H. Efficient in vivo manipulation of mouse genomic sequences at the zygote stage. Proc Nat Acad Sci USA. 1996;93:5860–5865. doi: 10.1073/pnas.93.12.5860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine JP, Bradley JP, Roth DA, McCarthy JG, Longaker MT. Studies in cranial suture biology: regional dura mater determines overlying suture biology. Plast Reconstr Surg. 1998;101:1441–1447. doi: 10.1097/00006534-199805000-00002. [DOI] [PubMed] [Google Scholar]
- Liu YH, Tang Z, Kundu RK, Wu L, Luo W, Zhu D, Sangiorgi F, Snead ML, Maxson RE. Msx2 gene dosage influences the number of proliferative osteogenic cells in growth centers of the developing murine skull: a possible mechanism for MSX2-mediated craniosynostosis in humans. Dev Biol. 1999;205:260–274. doi: 10.1006/dbio.1998.9114. [DOI] [PubMed] [Google Scholar]
- Martinez-Abadias N, Percival C, Aldridge K, Hill CA, Ryan T, Sirivunnabood S, Wang Y, Jabs EW, Richtsmeier JT. Beyond the closed suture in apert syndrome mouse models: evidence of primary effects of FGFR2 signaling on facial shape at birth. Dev Dyn. 2010;239:3058–3071. doi: 10.1002/dvdy.22414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathijssen IM, Vaandrager JM, van der Meulen JC, Pieterman H, Zonneveld FW, Kreiborg S, Vermeij-Keers C. The role of bone centers in the pathogenesis of craniosynostosis: an embryologic approach using CT measurements in isolated craniosynostosis and Apert and Crouzon syndromes. Plast Reconstr Surg. 1996;98:17–26. doi: 10.1097/00006534-199607000-00004. [DOI] [PubMed] [Google Scholar]
- McBratney-Owen B, Iseki S, Bamforth SD, Olsen BR, Morriss-Kay GM. Development and tissue origins of the mammalian cranial base. Dev Biol. 2008;322:121–132. doi: 10.1016/j.ydbio.2008.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrill AE, Bochukova EG, Brugger SM, Ishii M, Pilz DT, Wall SA, Lyons KM, Wilkie AO, Maxson RE. Cell mixing at a neural crest-mesoderm boundary and deficient ephrin-Eph signaling in the pathogenesis of craniosy-nostosis. Hum Mol Genet. 2006;15:1319–1328. doi: 10.1093/hmg/ddl052. [DOI] [PubMed] [Google Scholar]
- Morriss-Kay GM, Wilkie AO. Growth of the normal skull vaultnihms382465 and its alteration in craniosynostosis: insights from human genetics and experimental studies. J Anat. 2005;207:637–653. doi: 10.1111/j.1469-7580.2005.00475.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss ML. Growth of the calvaria in the rat; the determination of osseous morphology. Am J Anat. 1954;94:333–361. doi: 10.1002/aja.1000940302. [DOI] [PubMed] [Google Scholar]
- Muenke M, Wilkie AO. Craniosynostosis syndromes. In: Scriver CR, Beaudet AL, Valle D, Sly WS, Childs B, Kinzler K, Vogelstein B, editors. The Metabolic and Molecular Bases of Inherited Disease. McGraw-Hill; New York, NY: 2000. pp. 6117–6146. [Google Scholar]
- Nagata M, Nuckolls GH, Wang X, Shum L, Seki Y, Kawase T, Takahashi K, Nonaka K, Takahashi I, Noman AA, Suzuki K, Slavkin HC. The primary site of the acrocephalic feature in Apert syndrome is a dwarf cranial base with accelerated chondrocytic differentiation due to aberrant activation of the FGFR2 signaling. Bone. 2011;48:847–856. doi: 10.1016/j.bone.2010.11.014. [DOI] [PubMed] [Google Scholar]
- Opperman LA, Sweeney TM, Redmon J, Persing JA, Ogle RC. Tissue interactions with underlying dura mater inhibit osseous obliteration of developing cranial sutures. Dev Dyn. 1993;198:312–322. doi: 10.1002/aja.1001980408. [DOI] [PubMed] [Google Scholar]
- Orr-Urtreger A, Bedford MT, Burakova T, Arman E, Zimmer Y, Yayon A, Givol D, Lonai P. Developmental localization of the splicing alternatives of fibroblast growth factor receptor-2 (FGFR2) Dev Biol. 1993;158:475–486. doi: 10.1006/dbio.1993.1205. [DOI] [PubMed] [Google Scholar]
- Park WJ, Theda C, Maestri NE, Meyers GA, Fryburg JS, Dufresne C, Cohen MM, Jr, Jabs EW. Analysis of phenotypic features and FGFR2 mutations in Apert syndrome. Am J Hum Genet. 1995;57:321–328. [PMC free article] [PubMed] [Google Scholar]
- Pritchard JJ, Scott JH, Girgis FG. The structure and development of cranial and facial sutures. J Anat. 1956;90:73–86. [PMC free article] [PubMed] [Google Scholar]
- Purushothaman R, Cox TC, Muga AM, Cunningham ML. Facial suture synostosis of newborn Fgfr1(P250R+) and Fgfr2(S252W+) mouse models of Pfeiffer and Apert syndromes. Birth Defects Res A Clin Mol Teratol. 2011;91:603–609. doi: 10.1002/bdra.20811. [DOI] [PubMed] [Google Scholar]
- Raybaud C, Di Rocco C. Brain malformation in syndromic craniosynos-toses, a primary disorder of white matter: a review. Childs Nerv Syst. 2007;23:1379–1388. doi: 10.1007/s00381-007-0474-7. [DOI] [PubMed] [Google Scholar]
- Rice DP, Aberg T, Chan Y, Tang Z, Kettunen PJ, Pakarinen L, Maxson RE, Thesleff I. Integration of FGF and TWIST in calvarial bone and suture development. Development. 2000;127:1845–1855. doi: 10.1242/dev.127.9.1845. [DOI] [PubMed] [Google Scholar]
- Rice DP, Kim HJ, Thesleff I. Apoptosis in murine calvarial bone and suture development. Eur J Oral Sci. 1999;107:265–275. doi: 10.1046/j.0909-8836.1999.eos107406.x. [DOI] [PubMed] [Google Scholar]
- Rice DP, Rice R, Thesleff I. Fgfr mRNA isoforms in craniofacial bone development. Bone. 2003;33:14–27. doi: 10.1016/s8756-3282(03)00163-7. [DOI] [PubMed] [Google Scholar]
- Roth DA, Bradley JP, Levine JP, McMullen HF, McCarthy JG, Longaker MT. Studies in cranial suture biology: part II. Role of the dura in cranial suture fusion. Plast Reconstr Surg. 1996;97:693–699. doi: 10.1097/00006534-199604000-00001. [DOI] [PubMed] [Google Scholar]
- Saga Y, Miyagawa-Tomita S, Takagi A, Kitajima S, Miyazaki J, Inoue T. MesP1 is expressed in the heart precursor cells and required for the formation of a single heart tube. Development. 1999;126:3437–3447. doi: 10.1242/dev.126.15.3437. [DOI] [PubMed] [Google Scholar]
- Saito H. Histological studies of cartilage appearing under the sagittal suture. Dent J Iwate Med Univ. 2000;25:28–36. [Google Scholar]
- Sanes JR, Rubenstein JL, Nicolas JF. Use of a recombinant retrovirus to study post-implantation cell lineage in mouse embryos. EMBO J. 1986;5:3133–3142. doi: 10.1002/j.1460-2075.1986.tb04620.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21:70–71. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- Ting MC, Wu NL, Roybal PG, Sun J, Liu L, Yen Y, Maxson RE., Jr EphA4 as an effector of TWIST1 in the guidance of osteogenic precursor cells during calvarial bone growth and in craniosynostosis. Development. 2009;136:855–864. doi: 10.1242/dev.028605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Xiao R, Yang F, Karim BO, Iacovelli AJ, Cai J, Lerner CP, Richtsmeier JT, Leszl JM, Hill CA, Yu K, Ornitz DM, Elisseeff J, Huso DL, Jabs EW. Abnormalities in cartilage and bone development in the Apert syndrome FGFR2(+/S252W) mouse. Development. 2005;132:3537–3548. doi: 10.1242/dev.01914. [DOI] [PubMed] [Google Scholar]
- Wilkie AO, Byren JC, Hurst JA, Jayamohan J, Johnson D, Knight SJ, Lester T, Richards PG, Twigg SR, Wall SA. Prevalence and complications of single-gene and chromosomal disorders in craniosynostosis. Pediatrics. 2010;126:391–400. doi: 10.1542/peds.2009-3491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkie AO, Slaney SF, Oldridge M, Poole MD, Ashworth GJ, Hockley AD, Hayward RD, David DJ, Pulleyn LJ, Rutland P, et al. Apert syndrome results from localized mutations of FGFR2 and is allelic with Crouzon syndrome. Nat Genet. 1995;9:165–172. doi: 10.1038/ng0295-165. [DOI] [PubMed] [Google Scholar]
- Yen HY, Ting MC, Maxson RE. Jagged1 functions downstream of TWIST1 in the specification of the coronal suture and the formation of a boundary between osteogenic and non-osteogenic cells. Dev Biol. 2010;347:258–270. doi: 10.1016/j.ydbio.2010.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin L, Du X, Li C, Xu X, Chen Z, Su N, Zhao L, Qi H, Li F, Xue J, Yang J, Jin M, Deng C, Chen L. A Pro253Arg mutation in fibroblast growth factor receptor 2 (Fgfr2) causes skeleton malformation mimicking human Apert syndrome by affecting both chondrogenesis and osteogenesis. Bone. 2008;42:631–643. doi: 10.1016/j.bone.2007.11.019. [DOI] [PubMed] [Google Scholar]
- Yoshida T, Vivatbutsiri P, Morriss-Kay G, Saga Y, Iseki S. Cell lineage in mammalian craniofacial mesenchyme. Mech Dev. 2008;125:797–808. doi: 10.1016/j.mod.2008.06.007. [DOI] [PubMed] [Google Scholar]
- Yu K, Herr AB, Waksman G, Ornitz DM. Loss of fibroblast growth factor receptor 2 ligand-binding specificity in Apert syndrome. Proc Nat Acad Sci USA. 2000;97:14536–14541. doi: 10.1073/pnas.97.26.14536. [DOI] [PMC free article] [PubMed] [Google Scholar]