

Abstract
Neighboring genes are often coordinately expressed within cis-regulatory modules, but evidence that nonparalogous genes share functions in mammals is lacking. Here, we report that mutation of either TMEM138 or TMEM216 causes a phenotypically indistinguishable human ciliopathy, Joubert syndrome. Despite a lack of sequence homology, the genes are aligned in a head-to-tail configuration and joined by chromosomal rearrangement at the amphibian-to-reptile evolutionary transition. Expression of the two genes is mediated by a conserved regulatory element in the noncoding intergenic region. Coordinated expression is important for their interdependent cellular role in vesicular transport to primary cilia. Hence, during vertebrate evolution of genes involved in ciliogenesis, nonparalogous genes were arranged to a functional gene cluster with shared regulatory elements.
Cis-regulatory modules (CRMs) provide binding sites for transcription factors that regulate the expression of neighboring genes (). Relatively little is known about the evolution of these regulatory elements, such as how CRMs arise or how the regulated genes cofunction, other than the rare instance such as Hox gene clusters evolved by gene duplication and the addition of regulatory elements to regulate body patterning (2).
Joubert syndrome (JBTS) is the most common neurodevelopmental disorder among the ciliopathy spectrum, which is thought to encompass disorders of structure or function of cellular primary (nonmotile) cilia (3). Affected JBTS patients show hypotonia, ataxia, abnormal eye movement, and a distinct mid-hindbrain malformation presenting the “molar tooth” sign on brain magnetic resonance images (MTI) (fig. S1A) (4). Mounting evidence suggests that primary cilia as cellular antennae sense a wide variety of signals, including Shh signaling, and play a crucial role in vertebrate development (5).
Recently, we reported deleterious mutations of the transmembrane protein (TMEM) 216 gene, linking to the JBTS2 locus on chromosome 11, in about half of the 10 JBTS2-linked families (Fig. 1A) (6–8). However, the remaining half of the JBTS2 families (verified by the pathognomonic MTI) were phenotypically indistinguishable (displaying optic coloboma, retinal dysplasia, nephronophthisis, and occasional occipital encephalocele) but were negative for mutations in TMEM216 (fig. S1, A and B, and table S1). Furthermore, fibroblasts from these latter patients contained intact TMEM216 mRNA and protein expression (fig. S2, A and B), thereby suggesting another JBTS causative gene at the JBTS2 locus.
Fig. 1.

Genetic heterogeneity at JBTS2 locus and the evolutionary location of the TMEM138 and TMEM216 genes mutated in JBTS2-linked families. (A) JBTS2 locus (red box) on chromosome 11. TMEM138 encoding a trispan membrane protein is aligned in a head-to-tail configuration with TMEM216 encoding a tetraspan membrane protein. All missense and splicing mutations of TMEM138 and TMEM216 found in JBTS2-linked families are indicated on the predicted transcript and protein. (B) Schematic synteny representation of TMEM138, TMEM216, and the intergenic region showing that the head-to-tail configuration of TMEM138 and TMEM216 on the same chromosome is conserved from reptiles to humans, but not in lower vertebrates, in which the two TMEMs are located on different chromosomes (table S2). Conserved regions have >50% sequence similarity to humans. UTR, untranslated region.
We thus performed resequencing of all known and predicted exonic and promoter genetic elements within the minimal 17-Mb candidate interval defined by TMEM216 mutation-negative families (9). From these data, we identified four missense mutations and one splicing homozygous deleterious mutation in evolutionarily conserved residues of the nearby TMEM138 gene of unknown function, thus accounting for all JBTS2-linked families (Fig. 1A, fig. S2C, and table S1). All mutations segregated according to a single recessive disease mode and were not present in 400 ethnically matched chromosomes. Among missense mutations in transmembrane domains, TMEM138 p.H96R led to unstable protein when transfected into heterologous cells (fig. S2D), suggesting loss of function as the disease mechanism.
Although both TMEM genes encode trans-membrane proteins (Fig. 1A), neither the genes nor the proteins demonstrated sequence homology or shared any functional domains. Phylogenetic analysis showed that they represented two distinct protein families, which have evolved separately from invertebrates (figs. S4 and S5), excluding a gene-duplication event. In all higher vertebrates, the two genes aligned in a head-to-tail configuration, with a conserved ~23–kilo–base pair noncoding intergenic interval (Fig. 1B). A synteny map of the genomic locus in a species with deposited reference sequence indicated that they were joined by an ancient chromosomal rearrangement at the amphibian-to-reptile evolutionary transition ~340 million years ago (Fig. 1B and fig. S3). Adjacent genes often exhibit correlated expression (10) and, thus, confer a selective advantage as a genetic module for a certain biological function (11). Therefore, the arrangement of two TMEM genes causing indistinguishable phenotypes prompted us to test whether these genes represent co-regulated and cofunctioning genes within a CRM.
To test for coordinated expression, we examined tissue-expression patterns of human TMEM138 and TMEM216 using the microarray database and in situ hybridization of human embryos. We found tight coexpression values of human TMEM138 and TMEM216 across the major tissues, including the brain and kidneys (fig. S6B), and similar expression patterns in various tissues, including the kidneys, cerebellar buds, and telencephalon, at 4 to 8 gestational weeks (gw) of human embryos (Fig. 2A and fig. S6C). To test whether this coordinated expression was due to the adjacent localization, we compared mRNA levels in zebrafish versus mice, representing species before and after the gene rearrangement event. Using quantitative polymerase chain reaction (qPCR), we detected tightly coordinated expression levels in mice compared with those in zebrafish (correlation coefficient r = 0.984 versus 0.386) (Fig. 2B and fig. S6D), which suggests that TMEM138 and TMEM216 might share regulatory elements (REs) within the ~23-kb intergenic region. We further examined several experimental features and found that regulatory factor X 4, a transcription factor regulating ciliary genes, binds a RE conserved in the noncoding intergenic region to mediate coordinated expressions of TMEM138 and TMEM216 (see SOM text and fig. S7)
Fig. 2.

Coordinated expression of adjacent TMEM138 and TMEM216 mediated by the noncoding intergenic region. (A) Similar expression patterns of TMEM138 and TMEM216 based on in situ hybridization at 8 gw in human embryonic tissues. TMEM138 antisense (a and b), TMEM216 antisense (c and d), and sense control probes (TMEM138) (e and f) are shown. TMEM138 and TMEM216 are strongly expressed in kidney, gonad (go), and adrenal gland (ad) as well as in the central nervous system, in particular in the cerebellar bud (cb), telencephalon (tel), rhombencephalon (rh), and cranial nerve ganglia such as trigeminal (V). (B) Real-time qPCR of TMEM138 and TMEM216 in selected tissues indicates tightly coordinated mRNA expressions in mouse tissues (having head-to-tail configuration), but not zebrafish (having two genes on different chromosomes). Housekeeping genes 36B4 (mouse) and Rpl13a (zebrafish) are used for normalization.
The coordinated expression and indistinguishable ciliopathy phenotype caused by either mutation of TMEM138 and TMEM216 suggests possible functional relations at the protein level, possibly co-regulating ciliogenesis. We noticed short cilia and failure of ciliogenesis in TMEM138-(p.A126T) and TMEM216- (p.R73L) mutated fibroblasts, respectively (Fig. 3A), as well as defects of ciliogenesis after knockdown of TMEM138 and TMEM216 in IMCD3 cells (fig. S8), suggesting that both TMEM138 and TMEM216 are required for ciliogenesis. Immunostaining of endogenous TMEM138 with a marker (Arl13b) of the ciliary axoneme and transition zone demonstrated closely adjacent localization with TMEM216 at the base of cilia (γ-tubulin), as reported previously (Fig. 3B and fig. S9C) (6). We also noted prominent TMEM138 and TMEM216 adjacent but nonoverlapping vesicular staining around the base of cilia (Fig. 3B and fig. S9, C and D).
Fig. 3.
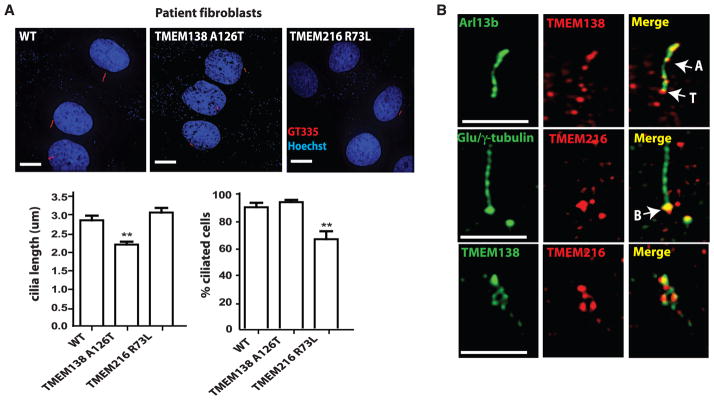
Tethered vesicular trafficking of TMEM138 and TMEM216 to primary cilia is required for ciliogenesis. (A) In patient fibroblasts under 48-hour serum starvation, TMEM138 p.A126T (MTI-656) mutations caused short cilia, and TMEM216 p.R73L mutations disrupt ciliogenesis (defined as having cilia <1 μm long).*P < 0.05, **P < 0.01 [versus wild type (WT) by one-way analysis of variance (ANOVA) with Bonferroni posttest, n = 40 to 50 cells]. Error bars indicate SEM. (B) In IMCD3 cells, high-resolution images of endogenous TMEM138/216 staining show that TMEM138 localized to ciliary axonemes and the base of cilia, whereas TMEM216 localized primarily to basal bodies. Both TMEMs also show closely adjacent vesicular patterns around the base of cilia (also see fig. S9C). Anti-Arl13b (cilia), anti-Glu/γ tubulin (cilia plus centrosome), polyclonal mouse anti-TMEM138 (fig. S9, A and B), and rabbit anti-TMEM216 antibodies were used. A, ciliary axoneme; T, transition zone; B, basal body. Scale bars, 5 μm.
Earlier electron-microscopic findings suggested that vesicles transporting to the cilium play a crucial role for targeted delivery of membrane proteins from the Golgi apparatus (12, 13). It is noteworthy that TMEM216 localized to post-Golgi vesicles along microtubules, as well as the Golgi apparatus surrounding the base of cilium, whereas TMEM138 localized to adjacent but nonoverlapping distinct vesicles, shown by immunoelectron microscopic analysis (figs. S10, S11, and S12A). This observation suggests that both TMEM138 and TMEM216 might mark vesicles en route to the base of cilium, which is known to be crucial for ciliary assembly (14). Using time-lapse analysis with or without fluorescence recovery after photobleaching, we found that the net flux of TMEM216-tagged vesicular movements is toward the centrosome in dsRed-Centrin2 (centrosomal marker) expressing cells (fig. S12B and movies S1 and S2). TMEM138-tagged vesicles displayed tethered vesicular movement with TMEM216-tagged vesicles (fig. S12C and movie S3). This observation prompted us to ask whether the two distinct vesicles differentially carry known ciliary proteins essential for ciliary assembly. We further examined several experimental features showing that TMEM138 and TMEM216 mark two distinct but linked vesicle pools, each associated with unique cilia-targeted proteins, including CEP290 (see SOM text and fig S13).
To understand the adjacency of the two vesicular pools, we tested whether either was required to move vesicles containing the other. Although TMEM216 vesicular movement was not notably affected by disruption of TMEM138 by small interfering RNA (siRNA), the knockdown of TMEM216 disrupted the trafficking of TMEM138, as well as CEP290 (figs. S13B and S14, A and B, and movies S5 and S6), suggesting functional dependence on TMEM216 for trafficking TMEM138 and associated proteins.
To determine the mechanism of this functional dependence, we considered the potential role of tethering proteins in linking these two distinct vesicular pools. Several vesicle tethering factors, including the transport protein particle (TRAPP) II complex, p115, and the conserved oligomeric Golgi complex are known to be involved in tethering Golgi vesicles (15). Among these potential tethering proteins, we found that TRAPPII mediated the tethering of TMEM138 and TMEM216 vesicles crucial for ciliary assembly (Fig. 4A, SOM text, figs. S14C to S16, and movies S7 and S8).
Fig. 4.
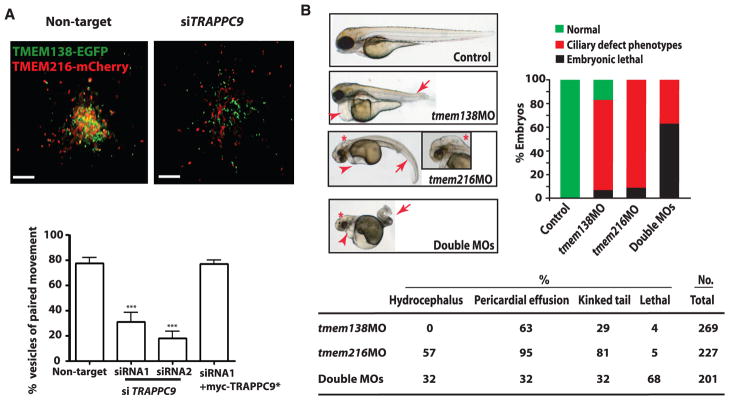
Functional relatedness of TMEM138 and TMEM216. (A) Live-cell imaging shows that knockdown of TRAPPC9, a major subunit of the TRAPPII complex, detached the tethered TMEM138 and TMEM216 vesicles (movies S7 and S8). Defects in tethering were largely rescued by myc-TRAPPC9* (9). ***P < 0.001 (by one-way ANOVA with Bonferroni posttest, n = 90 to 110). Scale bars, 5 μm. Error bars indicate SEM. (B) Injection of translation-blocking antisense morpholinos (MOs) to tmem138 (6 ng), tmem216 (4 ng), or double MOs in WT (AB) zebrafish embryos leads to ciliary phenotypes of curved or kinked tail (arrows) and heart edema (arrowheads) at 3 days post-fertilization in a synergistic and dose-dependent manner (fig. S18A). Only tmem216 morphants present hydrocephalus (*) (>50 embryos for each condition) (fig. S18B).
We questioned whether the protein localization or mutant phenotype of the two TMEMs in species that diverged before the coordinated gene regulation should be distinct. We found the retention of the coordinated vesicular movement in a zebrafish cell line (ZF4) expressing the tagged version of the two TMEMs zebrafish orthologs, suggesting that the coordinated localization of proteins evolutionarily preceded the coordinated gene expression (movie S11). We next tested for conserved genetic function in zebrafish by comparing the morpholino knockdown phenotypes of each. Both knockdowns shared some typical ciliary phenotypes such as pericardial effusion, a curved or kinked tail (16) in a synergistic and dose-dependent manner, as well as gastrulation defects (Fig. 4B and fig. S18, A to D). However, only tmem216 morphants presented hydrocephalic brains [ciliary phenotype (17)] and more severe defects of the left/right heart axis (Fig. 4B and figs. S17 and S18A). Together, the data indicate that before their adjacent genomic localization, the proteins’ organismal functions were not completely congruent and were associated with distinguishable phenotypes, unlike JBTS2-linked Joubert patients.
Our findings suggest that nonparalogous genes not only can be chromosomally rearranged into a functional gene cluster during vertebrate evolution, but also can be assembled into a new CRM by evolving regulatory elements, which correlate with their coordinated expression. We are aware of few other examples in which mutations in adjacent genes lead to indistinguishable or similar human phenotypes: mutations in EVC or EVC2 cause another ciliopathy, Ellis–van Creveld Syndrome; mutations in ABCG5 and ABCG8 cause sitosterolemia; and disruptions of PKD1 and TSC2 cause renal cysts (18–20). Our results provide insight into the evolved coordinated expression and functional relatedness of adjacent nonparalogous genes as a pathogenesis of phenotypically indistinguishable genetic disorders caused by mutations at a single locus.
Supplementary Material
Acknowledgments
We thank the patients and families for invaluable contributions; the Broad Sequencing Plaform, the UCSD Neuroscience Core (grant P30NS047101), and T. Meerloo at the UCSD ImmunoEM Core; M. G. Rosenfeld, B. Ren, P. Novick, K. Frazer, and S. Rifkin for discussion; and C. Janke (Centre de Recherche de Biochimie Macromoléculaire, Montpellier, France, GT335 antibody), T. Caspary (Emory Arl13b antibody), P. Aza-Blanc (Sanford-Burnham Institute, siRNAs), and D. Traver (UCSD) for ZF4 cells. This work was supported by the Daland Fellowship from the American Philosophical Society (J.H.L.), the Italian Ministry of Health (RC2010, Ricerca Finalizzata 2006), Telethon Foundation Italy (GGP08145, E.M.V.), the Pierfranco and Luisa Mariani Foundation (E.M.V.), l’Agence National pour la Recherche (grant ANR-2010-BLAN-1122 01- Foetocilpath, T.A.-B.), the American Heart Association (grant 09POST2250641, J.E.L.), Newlife Foundation for Disabled Children, the Medical Research Council (grant G0700073), the Sir Jules Thorn Charitable Trust (09/JTA, C.A.J.), European Community’s Seventh Framework Programme FP7/2009 (241955), SYSCILIA (E.E.D., N.K., C.A.J.), Distinguished George W. Brumley Professorship (N.K.), the NIH (grants NS052455 and NS04843, J.G.G.; DK068306 and DK090917, F.H.; U54 HG003067, E.S. Lander; EY021872, E.E.D.; HD042601, DK075972, DK072301, N.K.), Simons Foundation Autism Research Initiative (J.G.G.), and HHMI (F.H., S.F.-N., J.G.G.).
Footnotes
References and Notes
- 1.Jeziorska DM, Jordan KW, Vance KW. Semin Cell Dev Biol. 2009;20:856. doi: 10.1016/j.semcdb.2009.07.007. [DOI] [PubMed] [Google Scholar]
- 2.Manzanares M, et al. Nature. 2000;408:854. doi: 10.1038/35048570. [DOI] [PubMed] [Google Scholar]
- 3.Tobin JL, Beales PL. Genet Med. 2009;11:386. doi: 10.1097/GIM.0b013e3181a02882. [DOI] [PubMed] [Google Scholar]
- 4.Doherty D. Semin Pediatr Neurol. 2009;16:143. doi: 10.1016/j.spen.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Singla V, Reiter JF. Science. 2006;313:629. doi: 10.1126/science.1124534. [DOI] [PubMed] [Google Scholar]
- 6.Valente EM, et al. Nat Genet. 2010;42:619. doi: 10.1038/ng.594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Keeler LC, et al. Am J Hum Genet. 2003;73:656. doi: 10.1086/378206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Valente EM, et al. Am J Hum Genet. 2003;73:663. doi: 10.1086/378241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Materials and methods are available as supporting material on Science Online.
- 10.Hurst LD, Pál C, Lercher MJ. Nat Rev Genet. 2004;5:299. doi: 10.1038/nrg1319. [DOI] [PubMed] [Google Scholar]
- 11.Stuart JM, Segal E, Koller D, Kim SK. Science. 2003;302:249. doi: 10.1126/science.1087447. [DOI] [PubMed] [Google Scholar]
- 12.Sorokin S. J Cell Biol. 1962;15:363. doi: 10.1083/jcb.15.2.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Papermaster DS, Schneider BG, Besharse JC. Invest Ophthalmol Vis Sci. 1985;26:1386. [PubMed] [Google Scholar]
- 14.Nachury MV, Seeley ES, Jin H. Annu Rev Cell Dev Biol. 2010;26:59. doi: 10.1146/annurev.cellbio.042308.113337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cai H, Reinisch K, Ferro-Novick S. Dev Cell. 2007;12:671. doi: 10.1016/j.devcel.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 16.Khanna H, et al. Nat Genet. 2009;41:739. doi: 10.1038/ng.366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pathak N, Obara T, Mangos S, Liu Y, Drummond IA. Mol Biol Cell. 2007;18:4353. doi: 10.1091/mbc.E07-06-0537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ruiz-Perez VL, et al. Am J Hum Genet. 2003;72:728. doi: 10.1086/368063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Berge KE, et al. Science. 2000;290:1771. doi: 10.1126/science.290.5497.1771. [DOI] [PubMed] [Google Scholar]
- 20.Kleymenova E, et al. Mol Cell. 2001;7:823. doi: 10.1016/s1097-2765(01)00226-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.