

Abstract
The neoclerodane diterpene salvinorin A is the major active component of the hallucinogenic mint plant Salvia divinorum Epling & Játiva (Lamiaceae). Since the finding that salvinorin A exerts its potent psychotropic actions through the activation of opioid receptors, the site of action of morphine and related analogues, there has been much interest in elucidating the underlying mechanisms behind its effects. These effects are particularly remarkable, because (1) salvinorin A is the first reported non-nitrogenous opioid receptor agonist, and (2) its effects are not mediated through the previously investigated targets of psychotomimetics. This perspective outlines our research program, illustrating a new direction to the development of tools to further elucidate the biological mechanisms of drug tolerance and dependence. The information gained from these efforts is expected to facilitate the design of novel agents to treat pain, drug abuse, and other CNS disorders.
Keywords: Opioids, salvinorin A, Salvia divinorum, herkinorin, neoclerodanes
Introduction
The inability to adequately control pain is a major problem in medicine and society.1–5 According to the Institute of Medicine, chronic pain affects about 100 million American adults. This is more people than those affected by diabetes, heart disease and cancer combined. It has been estimated that the associated costs of pain in the United States exceed $600 billion dollars per year primarily due to medical treatment and lost productivity. While these expenidtures are significant, the costs in terms of suffering and quality of life cannot be adequately quantitated.
Opioid analgesics, such as morphine and its analogs, have been the mainstay for treatment of pain for thousands of years and are currently the “gold-standard” for pain management. However, clinicians are conservative in prescribing, and patients are conservative in taking, opioids due to valid concerns about adverse effects (constipation, respiratory depression, nausea, tolerance and dependence) as well as social and legal issues. As a result, pain is often undertreated (more than 65% of patients in nursing homes report inadequate treatment of pain)6 and patients continue to suffer. Thus, the development of improved opioid analgesics represents a critically important research objective.
Opioid analgesics produce their effects by binding to and activitating opioid receptors. Initially, it was believed that there was one opiate receptor.7 As a result of intensive work over the last 40 years, we now know that there are three major types of opioid receptors, mu (μ), delta (δ), and kappa (κ), and all are members of the G-protein coupled receptor superfamily.8 Each opioid receptor type plays a role in antinociception, in addition to other biological responses.9 In addition, there is pharmacological evidence for the existence of additional opioid receptor subtypes, as well as the formation of opioid receptor heterodimers.10–15 However, current thinking is that clinically used opioids exert their analgesic effectiveness mostly through their interactions with μ receptors.16
In addition to μ agonists, selective κ and δ receptor agonists have been explored as potential analgesics that would not have the side effects of morphine and other μ agonists. A number of clinically used opioids have κ agonist activity in addition to their effects at μ receptors. However, the use of selective κ agonists is limited due to dose dependent neuropsychiatric effects including sedation and dysphoria.17–21 The development of chemical and biological tools has helped to clarify the role of δ receptors in various pain states, as well as mood disorders.22, 23 However, no δ selective agonist has reached the clinic but progress is being made towards its development.24
Need for a New Direction
The chemistry and pharmacology of the opium alkaloids morphine (1), codeine (2), and thebaine (3) (Figure 1) have been extensively investigated for many years.25, 26 While these studies have produced many clinically useful agents and essential chemical probes for opioid receptors, new agents are needed to provide greater insight into the mechanisms of opioid antinociception and opioid addiction. At present, nearly all nonpeptide opioids are derived from morphine. While the degree and severity may vary among individual members, agents derived from morphine generally suffer from the same side effects including tolerance, constipation, and respiratory depression.
Figure 1.
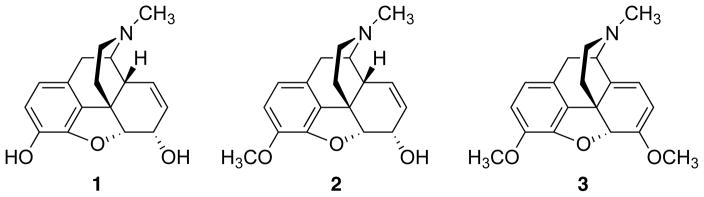
Structures of opium alkaloids morphine (1), codeine (2), and thebaine (3).
As described elsewhere, the investigation of natural products has proven to be an excellent source of clinical agents for a number of therapeutic areas including pain.27 In addition, much of what we know about pain processing can be directly attributed to the extensive investigation of morphine and related compounds. Given that past accomplishments are often a great predictor of future success, it was surmised that exploring nature might be a fruitful approach for identifying new opioid receptor probes with the greatest potential for reduced side effects. Assuming that a new natural product scaffold could be identified with opioid activity, this would provide an opportunity for new chemical investigation.
Salvinorin A as an Atypical Opioid
Salvia divinorum Epling & Játiva (Lamiaceae) is a mint plant native to Oaxaca, Mexico that has been used by the Mazatec Indians living there as a divinatory or psychotomimetic agent.28, 29 The term hallucinogenic or psychotomimetic has been given to several classes of agents including cannabinoids, lysergic acid derivatives, phenethylamines, indolalkylamines, other indolic compounds, piperidyl benzilate esters, as well as phenylcyclohexyl compounds.30 The common theme of all these classes of pharmacologically active substances is that they alter consciousness, often in dramatic and unpredictable ways, and in high doses may produce delirium, true hallucinations, loss of contact with reality, and in some cases death.31 Although the effects of S. divinorum have been known for centuries, it was not until 2002 that opioid receptors were implicated in its actions.32
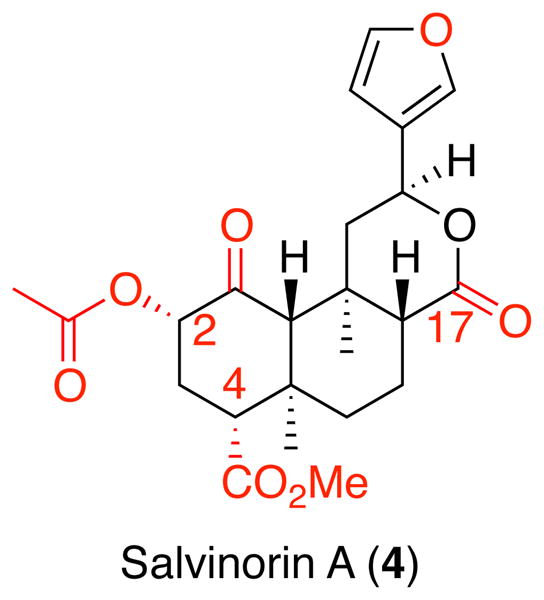
The principal psychoactive constituent found in S. divinorum is the neoclerodane diterpene salvinorin A (4) (Figure 2).33, 34 Through large scale screening of human cloned GPCRs 4 was identified as a potent and selective κ opioid receptor agonist.32 This activity was striking, as it was the first example of a naturally occurring small molecule selective for κ opioid receptors. In addition, it did not structurally resemble other major classes of non-peptide opioid receptor ligands, such as the epoxymorphinans, morphinans, orvinols, benzomorphans, phenylpiperidines, anilidopiperidines, phenylmorphans, arylacetamides, and nitrobenzimidazoles.35 The most striking structural difference between 4 and these chemotypes was the lack of a basic nitrogen atom. Up until the discovery of 4, it was generally accepted that the presence of a positively charged nitrogen atom represented an absolute requirement for high affinity with opioid receptors.36 The conventional thinking was that the cationic amino charge of the opioid ligand would form a salt bridge with the side chain carboxyl group of an aspartate residue located in TM III of the opioid receptor. However, 4 lacked the potential for this interaction. This suggested that there was a high probability 4 interacts with opioid receptors in a manner qualitatively different than all other known opioids. In fact, several recent crystal structures of the opioid receptors with selective antagonists have nicely corroborated this thinking.37–39
Figure 2.

Structure of salvinorin A (4) and major classes of non-peptide opioid receptor ligands.
The structure and potential mechanism of 4 as a psychotomimetic was also fascinating. Previously known targets of psychotomimetics include cannabinoid receptors, NMDA receptors, cholinergic receptors, and most notably serotonin receptors. As mentioned above, 4 had no appreciable affinity for these known targets. Furthermore, 4 had no structural similarity to other psychotomimetics such as Δ9-THC, hyoscyamine, phencyclidine (PCP), lysergic acid diethylamide (LSD), mescaline, dimethyltryptamine (DMT), and ibogaine (Figure 3).
Figure 3.

Structures of salvinorin A (4), Δ9-THC, hyoscyamine, phencyclidine (PCP), lysergic acid diethylamide (LSD), mescaline, dimethyltryptamine (DMT), and ibogaine.
On the basis of its unique structure as an opioid and as a psychotomimetic, a program was established to better understand the biological actions of 4 and related neoclerodanes as opioid ligands and psychotomimetics. It was envisioned that by better understanding the chemistry and pharmacology of neoclerodanes related to 4, one might be able to develop novel treatments for pain, drug abuse, and other CNS disorders that lack the detrimental effects associated with morphine like scaffolds.
Rationale
It is well known that ionic bonds often provide a key anchoring interaction between ligands and their target. The lack of a readily ionizable group in 4 suggested that hydrophobic interactions and/or hydrogen bonding were likely to play an important role its interaction with the κ opioid receptor. However, it was not readily clear what, if any, groups were necessary for the nature of the high affinity and selectivity of 4. Furthermore, given the lack of a definitive binding site, as well as its relationship to known ligands, it was apparent that previous structure-activity relationships of opioids were not likely to be useful. Thus, a series of chemical probes based on 4 would need to be prepared and evaluated at opioid receptors to investigate for their usefulness as potential opioid ligands.
In order to conduct a thorough medicinal chemistry campaign, the first initial question that needed to be answered was how to obtain multigram quantities of the natural product needed for chemical diversification. Two approaches were considered: (1) total synthesis and (2) isolation from S. divinorum. Total synthesis is often a fruitful technique and has been used previously to probe the structure-function of many different natural products. The major advantage of this approach would be analogues not accessible by an isolation route could be prepared for testing. However, the major disadvantage of the total synthesis approach was that it would likely require many steps with a low overall yield. This has subsequently been shown in two published routes.40–42 Furthermore, it was anticipated that this approach would be time-consuming and the ability to more fully investigate the pharmacological effects of 4 in vivo compared to traditional opioids or other hallucinogens would also be inhibited. The alternative approach was isolation of the natural product from S. divinorum. This approach has been used successfully as a starting point with the opium alkaloids and many natural products with antibiotic or anticancer activity. The advantage of isolation relies on using readily available S. divinorum leaves due to its largely unscheduled nature at the time. This is no longer the case as several states including Kansas now treat S. divinorum as a controlled substance. Further, extracting the source has the benefit of identifying other naturally occurring congeners to add to structure-activity relationship studies. Finally, this method was expected to provide information on the basic pharmacology of 4 in a faster timeframe than total synthesis. Thus, it was decided to explore methods to isolate 4 and other naturally occurring secondary metabolites from S. divinorum.
The second initial question that needed to be answered was what analogues could or should be prepared based on 4. As mentioned above, the lack of a readily ionizable group in 4 suggested that hydrophobic interactions and/or hydrogen bonding were likely to play an important role. Initial analogue design ideas focused on preparing analogues to selectively probe the importance of existing hydrogen bonding groups, such as the 1-ketone, 2-position acetyl group, 4-carbomethoxy group, 17-lactone carbonyl, and furanyl oxygen (Chart 1). Unfortunately, the lack of a readily ionizable group and the presence of several ester moieties in 4 also indicated that water solubility and metabolic stability were going to be problematic and analogues with enhanced properties should be prioritized. Given there were published methods for solubilizing CNS active molecules with poor water solubility identified from the study of phytocannabinoids, we focused initially on finding analogues with enhanced stability.
Chart 1.
Development of Neoclerodanes
Initial phytochemical investigation of S. divinorum identified the neoclerodane diterpenes salvinorin A (4) and salvinorin B (5) (Figure 4).33, 34 Later work by several different groups of investigators isolated salvinorins C – I,43–47 divinatorins A – F,45, 46, 48 and and salvidivins A – D.46 Our own efforts working with commercially available S. divinorum leaves identified salvinicins A (6) and B (7).49 The structures of these congeners were elucidated by spectroscopic techniques and the absolute stereochemistry was assigned on the basis of single-crystal X-ray crystallographic analysis of 6 and a 3,4-dichlorobenzoate derivative of 5. Interestingly, 7 exhibited antagonist activity at μ receptors with a Ki of > 1.9 μM. This was the first report of a neoclerodane diterpene with opioid antagonist activity.
Figure 4.

Structures of neoclerodanes 5 – 12 related to 4.
Having identified a practical method for extracting 4,50 we set out to explore the chemistry associated with the natural product. Previous reports had indicated that it would be possible to selectively hydrolyze the C-2 ester of 4 to 5.34, 51 We were able to identify conditions whereby the C-2 acetate is selectively removed while retaining the configuration of the stereogenic centers by treating 4 with Na2CO3 in MeOH.50 This particular transformation is challenging/low yielding as epimerization of the C-8 position can occur due to breaking of the C-8/C-9 bond via base-promoted cleavage.51, 52 A number of diverse organic and inorganic bases have been tried but to date have not resulted in an overall improvement in the synthesis of 5.52, 53
With a reliable method for the conversion of 4 to 5 in hand, we set out to explore the structure-activity relationships of the C-2 position. We initially focused on modifications to explore the steric tolerance of the C-2 position.54, 55 Lacking a receptor-ligand crystal structure to aid analogue design, we took the approach of systematically changing the structure of 4 and observing its effects on opioid affinity and efficacy. Our over-arching hypothesis was that each neoclerodane was binding in an identical manner at the κ opioid receptor. While this may or may not be the case, we felt it was a good starting point for our structure-activity relationship explorations. It was quickly learned that modification of the C-2 position has clear effects on opioid receptor affinity and activity.56 In particular, we found that the incorporation of an aromatic group to the C-2 position decreases affinity at κ receptors but increases affinity for μ receptors.54, 57 This work identified herkinorin (8) as a neoclerodane with μ agonist activity. In addition, we and others found that the C-2 ester could be effectively replaced by sulfonates and alkyloxymethyl ethers.52, 54, 55, 58 Interestingly, the sulfonates appear to be binding a manner different than the corresponding esters based on the observation that parallel changes in structure did not result in parallel changes in affinity.55 Also, the replacement of the C-2 ester moiety with an ethoxymethyl ether (9) results in the most potent neoclerodane at κ receptors to date.58
In an attempt to generate more stable salvinorin A analogues, we explored the incorporation of amides into the C-2 position.57 It was expected that the replacement of the ester linkage with a corresponding amide would result in enhanced stability to plasma esterases as had been indicated as a site of ex vivo metabolism.59 In addition, we felt this substitution was likely to enhance aqueous solubility. Using a several step procedure, we were able to convert 5 to the 2-amino analogue.57 This then allowed the preparation of various amides and sulfonamides. Generally, we found that this biosiosteric replacement descreased affinity for κ opioid receptors. However, this modification did increase affinity for μ opioid receptors. Combining previous C-2 SAR, we identified benzamide 10 as the highest affinity and most μ selective neoclerodane described to date.
More recently, we have investigated the structural basis that underlies the increases in affinity and potency seen with ethoxymethyl ether 9.60 Noting that the ether moiety at C-2 is relatively flexible and can adopt different conformations when interacting with κ receptors, we applied the concept of conformational constraint in order to probe this phenomenon.61 We found that constraining the ethoxymethyl ether into a tetrahydropyran ring decreased affinity for κ opioid receptors. However when constraining the ether into a tetrahydropyran ring, a new stereocenter is formed. Using a mixture of spectroscopic methods and X-ray crystallography, we were able to assign the absolute stereochemistry for each epimer. Biological evaluation revealed the eutomer (11) and distomer (12) and indicated a preference for the hydrogen of the new stereocenter to be in the β position or R configuration. Further, we found tetrahydropyran 11 attenuated cocaine-induced drug seeking behavior comparably to 4 representing the first modified neoclerodane that has demonstrated anti-addictive capabilities.
It is widely known in medicinal chemistry that furan rings should be avoided in drug development campaigns due to their potential for hepatotoxicity.62 Due to the presence of a furan ring, 4 possesses the potential for toxicity. Furthermore, previous studies showed that teucrin A (Figure 5), a neoclerodane present in germander (Teucrium chamaedrys L.; Lamiaceae), produced hepatotoxicity in humans likely resulting from the formation of an enedial formed during metabolism of the furan ring by cytochrome P450 enzymes.63, 64 In order to reduce the potential of forming reactive metabolites and increase the value of neoclerodanes as in vivo biological probes for opioid receptors, we sought to find replacements for the furan ring. Our studies and those of others indicate that the furan ring is not required for biological activity.56 It should be noted, however, that complete removal of the furan moiety results in a large reduction in κ opioid receptor affinity compared to 4.65 We have identified conditions that enhance the reactivity of the furan ring in 4 to participate in a Diels-Alder reaction.66 Further, several of the cycloadduct analogues were themselves useful as synthetic intermediates as they were able to undergo reductive elimination to afford their phenyl ring counterparts. More recently, we found a palladium catalyzed Liebeskind-Srogl cross-coupling reaction of a thioester derived from 4 and a boronic acid that occurs at neutral pH and ambient temperature to produce ketone analogs at C-12.67 To the best of our knowledge, this was the first reported usage of the Liebeskind-Srogl reaction68 to diversify a natural product scaffold. Using this chemistry, we were able to prepare the furan-2-yl analog of salvinorin A (13). Interestingly, 13 has similar affinity to 4 suggesting that a hydrogen bond exists from the furanyl oxygen of 13 to the same residue on the κ receptor as the furanyl oxygen of 4.
Figure 5.
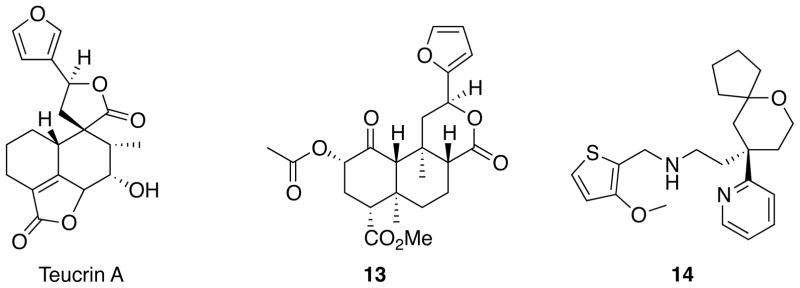
Structures of teucrin A, neoclerodane 13, and TRV130 (14).
Having established a facile isolation of 4, we sought to further explore its in vitro and in vivo pharmacology. Monkeys trained to discriminate 4 from saline generalized to a number of structurally diverse κ agonists.69 However, these animals did not generalize to μ or δ opioid agonists, the classical hallucinogen psilocybin or the dissociative NMDA antagonist, ketamine. The discriminative effects of 4 were blocked by the opioid antagonist quadazocine, but not by the serotonergic antagonist ketanserin. This indicates that the discriminative stimulus produced by 4 is mediated by agonism at κ opioid receptors, and is different from that elicited by classical hallucinogens. Recently, we found that the p-glycoprotein inhibitor tariquidar enhances the concentration of 4 in the cerebrospinal fluid as determined by LC/MS/MS.70 These are the first studies in vivo showing sensitivity of 4 to modulation by the p-glycoprotein transporter, a major functional component of the blood-brain barrier.
In collaboration with researchers at Johns Hopkins University, we have begun to evaluate the dose-related effects of inhaled 4 in individuals with histories of hallucinogen use.71, 72 In a double-blind, placebo-controlled study, inhaled doses of 4 from 0.375 μg/kg to 21 μg/kg resulted in orderly dose- and time-related participant ratings of drug strength.71 More recently, 4 was found to produce a unique profile of subjective and cognitive effects, including strong dissociative effects and memory impairment, which only partially overlap with classical hallucinogens.72 As seen previously, dose-related effects peaked at 2 min and then rapidly dissipated. Collectively, the effects in humans complement those seen in non-human primates and are relevant to understanding the neurobiology of the kappa opioid system. In addition, these findings suggest that future studies of 4 can be conducted without appreciable risk.
Herkinorin
A growing body of pharmacological evidence has shown that structurally similar ligands acting at the same receptor can elicit different signaling pathways.73 This has been termed “biased agonism” or “functional selectivity” and is thought to be due to differences in ligand-induced receptor conformations.74, 75 Such differences at the mu opioid receptor regulation are physiologically relevant as mice lacking β-arrestin2 display enhanced antinociception, decreased tolerance, and greatly diminished side effects (constipation and respiratory depression) following morphine treatment.76 Therefore, the development of ligands that activate mu receptors in the absence of β-arrestin – μ receptor interactions may provide valuable tools for studying this pharmacology further and could possibly lead to the discovery of novel compounds for the treatment chronic pain.
As described above, herkinorin (8) was identified as the first μ selective ligand from the neoclerodane scaffold.54 Surprisingly, 8 was also found to activate G protein coupling and ERK1/2 in a naloxone reversible manner yet does not induce receptor-β-arrestin interactions.77 Additional studies in non-human primates showed that 8 has opioid receptor mediated effects using prolactin release as a neuroendocrine biomarker of opioid activity in vivo.78 More recently, we reported that 8 has antinociceptive properties in the rat formalin paw withdrawal test, a model for peripheral antinociception in inflammatory pain.79 Further, we found that 8 has a reduced tolerance profile and remains efficacious in rats made tolerant to chronic morphine. These initial findings suggest that therapeutic efficacies may be attainable and that herkinorin-like compounds may be useful in morphine-tolerant peripheral pain treatment.
Future Perspective
The goal of identifying opioid analgesics with greatly reduced side effects relative to morphine has remained elusive likely due to vast efforts exploring the morphine scaffold. Even with the diversity of structures exhibiting affinity and activity at opioid receptors, there is still ample opportunity for chemical investigation. With the recent publishing of the crystal structures of the κ, μ, and δ opioid receptors,37–39 as well as the nociception receptor,80 a new era in opioid receptor research has begun. The ability to conduct structure-based drug design on this important class of GPCRs is now possible. At present, all of the structures have been established with antagonists crystalizing the inactive state of the receptor. This will be especially useful for the identification of new selective antagonists. Unfortunately, it may not be as useful for the identification of new agonists at opioid receptors. However, the concominant use of molecular dynamics and the corresponding crystal structure is likely to be a more fruitful approach. Regardless, it is easy to envision new scaffolds being developed, as molecular probes to better understand the mechanisms of opioid addiction.
The biological basis of how 4 exerts its potent psychotomimetic effects is not completely understood. It is clear the discriminative stimulus effects of 4 are different than those elicited by classical hallucinogens and dissociatives and similar to other κ agonists. At present, there are no animal models that selectively model the psychotomimetic effects of κ agonists. Whether, all neoclerodanes produce the same type of psychotomimetic effects is an unresolved question.
As stated above, centrally active κ agonists are currently limited by sedation and dysphoria. One approach to circumventing these side effects is to identify peripherally restricted compounds.81–85 The rationale for this approach is that peripherally restricted κ agonists would be devoid of the dysphoria and sedation seen with centrally acting agents but analgesic efficacy would be maintained given that activation of peripheral κ receptors also produces antinociception.86, 87 Given that 4 appears to be a substrate for the p-glycoprotein, neoclerodanes related to 4 might be a new scaffold for the development of peripherally restricted κ agonists.
Although the concept of functional selectivity was proposed almost 20 years ago,88 there are few studies in physiologically relevant cell systems and in vivo. Just as selectivity for different receptor subtypes became a valuable pharmacological property exploited by medicinal chemists to specifically target a therapeutic effect and reduce off-target adverse effects, ligand functional selectivity may become the next major advance in drug development. In addition to receptor selectivity, ligands that have selectivity for certain signaling pathways over others would be expected to have enhanced therapeutic efficacy and fewer adverse effects.
As seen with 8, the development of opioid ligands that direct signaling of the receptors toward G protein coupling without recruiting β-arrestins may be therapeutically advantages for producing pain relief with reduced side effects. More recently, TRV130 (14) was found to have robust G protein signaling, with potency and efficacy similar to morphine, but less β-arrestin recruitment and receptor internalization.89 Interestingly, 14 was found to have less gastrointestinal dysfunction and respiratory suppression than morphine at equianalgesic doses. The further development of functionally selective opioid ligands offers a new approach to pain management, as well as to other disorders where opioid receptors have been implicated.90, 91
In 1929, the eminent pharmacologist Reid Hunt stated that, “A thorough study of the morphine molecule might show a possibility of separating the analgesic from the habit forming property … work along these lines would involve cooperation between the highest type of organic chemists and pharmacologists.”92–94 While much excellent work has been done under this directive, there are still many unanswered questions and research opportunities. It is my sincere hope that continued research in this field may yet provide the holy grail of opioids, a powerful analgesic drug devoid of the side effects associated with morphine.95
Acknowledgments
I am deeply honored to have been the recipient of the 2012 David W. Robertson Award for Excellence in Medicinal Chemistry this past August in Philadelphia. I never had the pleasure of meeting Dr. Robertson but I became very familiar with his excellent work with the selective serotonin reuptake inhibitor fluoxetine during my time as a graduate student in the laboratory of Dr. Richard Glennon. A special thanks goes to Dr. Kenner Rice for his support, insights, and for first introducing me to opioid research. As a medicinal and natural product chemist, I have benefited greatly from working with a number of outstanding pharmacologists on several different research projects. However, I will always be indebted to my two longtime collaborators, Drs. Richard B. Rothman and Eduardo R. Butelman for taking a chance and collaborating with a young assistant professor. I would also like to thank my past and present group members for all their hard work and the National Institute on Drug Abuse for their financial support of our research efforts. Lastly and most importantly, I would like to thank my wife Deanna for her love, confidence in my abilities, and unwavering support.
Biography
Thomas E. Prisinzano, Ph.D. received his B.S. degree in Chemistry from the University of Delaware, Newark, DE in 1995 and was awarded a Ph.D. in Pharmaceutical Sciences from the School of Pharmacy, Virginia Commonwealth University, Richmond, VA in 2000. He was an Intramural Research Training Award Fellow in the National Institute of Diabetes and Digestive and Kidney Diseases in Bethesda, MD from 2000 – 2003. Currently, he is Professor and Chair of the Department of Medicinal Chemistry at the University of Kansas. His research focuses on the development of novel agents to treat pain, substance abuse and other CNS disorders through the identification, structure elucidation, and synthesis of natural products.
Footnotes
Adapted from a David W. Robertson Award for Excellence in Medicinal Chemistry address, 244th National Meeting of the American Chemical Society, Philadelphia, Pennsylvania, August 19 – 23, 2012.
References
- 1.Fairchild A. Under-treatment of cancer pain. Curr Opin Support Palliat Care. 2010;4:11–15. doi: 10.1097/SPC.0b013e328336289c. [DOI] [PubMed] [Google Scholar]
- 2.Mitera G, Fairchild A, DeAngelis C, Emmenegger U, Zurawel-Balaura L, Zhang L, Bezjak A, Levin W, McLean M, Zeiadin N, Pang J, Nguyen J, Sinclair E, Chow E, Wong R. A multicenter assessment of the adequacy of cancer pain treatment using the pain management index. J Palliat Med. 2010;13:589–593. doi: 10.1089/jpm.2009.0342. [DOI] [PubMed] [Google Scholar]
- 3.Phillips DM. JCAHO pain management standards are unveiled. Joint Commission on Accreditation of Healthcare Organizations. JAMA. 2000;284:428–429. doi: 10.1001/jama.284.4.423b. [DOI] [PubMed] [Google Scholar]
- 4.Richardson P, Mustard L. The management of pain in the burns unit. Burns. 2009;35:921–936. doi: 10.1016/j.burns.2009.03.003. [DOI] [PubMed] [Google Scholar]
- 5.Stewart WF, Ricci JA, Chee E, Morganstein D, Lipton R. Lost productive time and cost due to common pain conditions in the US workforce. JAMA. 2003;290:2443–2454. doi: 10.1001/jama.290.18.2443. [DOI] [PubMed] [Google Scholar]
- 6.Pizzo PA, Clark NM. Alleviating suffering 101--pain relief in the United States. N Engl J Med. 2012;366:197–199. doi: 10.1056/NEJMp1109084. [DOI] [PubMed] [Google Scholar]
- 7.Pert CB, Snyder SH. Opiate receptor: demonstration in nervous tissue. Science. 1973;179:1011–1014. doi: 10.1126/science.179.4077.1011. [DOI] [PubMed] [Google Scholar]
- 8.Waldhoer M, Bartlett SE, Whistler JL. Opioid receptors. Annu Rev Biochem. 2004;73:953–990. doi: 10.1146/annurev.biochem.73.011303.073940. [DOI] [PubMed] [Google Scholar]
- 9.Kieffer BL, Gaveriaux-Ruff C. Exploring the opioid system by gene knockout. Prog Neurobiol. 2002;66:285–306. doi: 10.1016/s0301-0082(02)00008-4. [DOI] [PubMed] [Google Scholar]
- 10.Pasternak GW. Multiple opiate receptors: deja vu all over again. Neuropharmacology. 2004;47(Suppl 1):312–323. doi: 10.1016/j.neuropharm.2004.07.004. [DOI] [PubMed] [Google Scholar]
- 11.Zaki PA, Bilsky EJ, Vanderah TW, Lai J, Evans CJ, Porreca F. Opioid receptor types and subtypes: the delta receptor as a model. Annu Rev Pharmacol Toxicol. 1996;36:379–401. doi: 10.1146/annurev.pa.36.040196.002115. [DOI] [PubMed] [Google Scholar]
- 12.Rothman RB. Evidence for heterogeneity of kappa-opioid binding sites: a review of data obtained using brain membranes depleted of mu and delta binding sites with irreversible agents. Analgesia. 1994;1:27–49. [Google Scholar]
- 13.Rios CD, Jordan BA, Gomes I, Devi LA. G-protein-coupled receptor dimerization: modulation of receptor function. Pharmacol Ther. 2001;92:71–87. doi: 10.1016/s0163-7258(01)00160-7. [DOI] [PubMed] [Google Scholar]
- 14.Costantino CM, Gomes I, Stockton SD, Lim MP, Devi LA. Opioid receptor heteromers in analgesia. Expert Rev Mol Med. 2012;14:e9. doi: 10.1017/erm.2012.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pasternak GW. Preclinical pharmacology and opioid combinations. Pain Med. 2012;13(Suppl 1):S4–11. doi: 10.1111/j.1526-4637.2012.01335.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pasternak GW. The pharmacology of mu analgesics: from patients to genes. Neuroscientist. 2001;7:220–231. doi: 10.1177/107385840100700307. [DOI] [PubMed] [Google Scholar]
- 17.Pfeiffer A, Brantl V, Herz A, Emrich HM. Psychotomimesis Mediated by Kappa Opiate Receptors. Science. 1986;233:774–776. doi: 10.1126/science.3016896. [DOI] [PubMed] [Google Scholar]
- 18.Martin WR, Fraser HF, Gorodetzky CW, Rosenberg DE. Studies of the Dependence-Producing Potential of the Narcotic Antagonist 2-Cyclopropylmethyl-2′-hydroxy-5,9-dimethyl-6,7-benzomorphan (Cyclazocine, WIN-20,740: ARC II-C-3) J Pharmacol Exp Ther. 1965;150:426–436. [PubMed] [Google Scholar]
- 19.Kumor KM, Haertzen CA, Johnson RE, Kocher T, Jasinski D. Human psychopharmacology of ketocyclazocine as compared with cyclazocine, morphine and placebo. J Pharmacol Exp Ther. 1986;238:960–968. [PubMed] [Google Scholar]
- 20.Rimoy GH, Wright DM, Bhaskar NK, Rubin PC. The cardiovascular and central nervous system effects in the human of U-62066E. A selective opioid receptor agonist. Eur J Clin Pharmacol. 1994;46:203–207. doi: 10.1007/BF00192549. [DOI] [PubMed] [Google Scholar]
- 21.Walsh SL, Strain EC, Abreu ME, Bigelow GE. Enadoline, a Selective Kappa Opioid Agonist: Comparison with Butorphanol and Hydromorphone in Humans. Psychopharmacology. 2001;157:151–162. doi: 10.1007/s002130100788. [DOI] [PubMed] [Google Scholar]
- 22.Pradhan AA, Befort K, Nozaki C, Gaveriaux-Ruff C, Kieffer BL. The delta opioid receptor: an evolving target for the treatment of brain disorders. Trends Pharmacol Sci. 2011;32:581–590. doi: 10.1016/j.tips.2011.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gaveriaux-Ruff C, Kieffer BL. Delta opioid receptor analgesia: recent contributions from pharmacology and molecular approaches. Behav Pharmacol. 2011;22:405–414. doi: 10.1097/FBP.0b013e32834a1f2c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nozaki C, Le Bourdonnec B, Reiss D, Windh RT, Little PJ, Dolle RE, Kieffer BL, Gaveriaux-Ruff C. delta-Opioid mechanisms for ADL5747 and ADL5859 effects in mice: analgesia, locomotion, and receptor internalization. J Pharmacol Exp Ther. 2012;342:799–807. doi: 10.1124/jpet.111.188987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Casy AF, Parfitt RT. Opioid analgesics: chemistry and receptors. Plenum Press; New York: 1986. p. xv.p. 518. [Google Scholar]
- 26.Nagase H. Chemistry of Opioids Preface. Chemistry of Opioids. 2011;299:Ix–Xi. [PubMed] [Google Scholar]
- 27.Newman DJ, Cragg GM. Natural products as sources of new drugs over the 30 years from 1981 to 2010. J Nat Prod. 2012;75:311–335. doi: 10.1021/np200906s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Epling C, Jativa-M CD. A New Species of Salvia from Mexico. Bot Museum Leaflets, Harvard Univ. 1962;20:75–76. [Google Scholar]
- 29.Tyler VE. The Physiological Properties and Chemical Constituents of Some Habit-Forming Plants. Lloydia. 1966;29:275–292. [Google Scholar]
- 30.Glennon RA. Hallucinogens, Stimulants, and Related Drugs of Abuse and their Therapeutic Potential. In: Lemke TL, Williams DA, Roche VF, Zito SW, editors. Foye’s Principles of Medicinal Chemistry. 7. Wolters Kluwer, Lippincott Williams & Wilkins; Philadelphia: 2013. pp. 632–657. [Google Scholar]
- 31.Nichols DE. Hallucinogens. Pharmacol Ther. 2004;101:131–181. doi: 10.1016/j.pharmthera.2003.11.002. [DOI] [PubMed] [Google Scholar]
- 32.Roth BL, Baner K, Westkaemper R, Siebert D, Rice KC, Steinberg S, Ernsberger P, Rothman RB. Salvinorin A: A Potent Naturally Occurring Nonnitrogenous Kappa Opioid Selective Agonist. Proc Natl Acad Sci USA. 2002;99:11934–11939. doi: 10.1073/pnas.182234399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ortega A, Blount JF, Manchand PS. Salvinorin, a New Trans-Neoclerodane Diterpene from Salvia-Divinorum (Labiatae) J Chem Soc, Perkin Trans. 1982;1:2505–2508. [Google Scholar]
- 34.Valdes LJ, III, Butler WM, Hatfield GM, Paul AG, Koreeda M. Divinorin A, a Psychotropic Terpenoid, and Divinorin B from the Hallucinogenic Mexican MintSalvia divinorum. J Org Chem. 1984;49:4716–4720. [Google Scholar]
- 35.McCurdy CR, Prisinzano TE. Opioid Receptor Ligands. In: Abraham DJ, Rotella DP, editors. Burger’s Medicinal Chemistry, Drug Discovery, and Development. 7. Vol. 8. John Wiley & Sons, Inc; 2010. pp. 569–735. [Google Scholar]
- 36.Rees DC, Hunter JC. In: Comprehensive Medicinal Chemistry. Emmet JC, editor. Pergamon; New York: 1990. pp. 805–846. [Google Scholar]
- 37.Wu H, Wacker D, Mileni M, Katritch V, Han GW, Vardy E, Liu W, Thompson AA, Huang XP, Carroll FI, Mascarella SW, Westkaemper RB, Mosier PD, Roth BL, Cherezov V, Stevens RC. Structure of the human kappa-opioid receptor in complex with JDTic. Nature. 2012;485:327–332. doi: 10.1038/nature10939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Granier S, Manglik A, Kruse AC, Kobilka TS, Thian FS, Weis WI, Kobilka BK. Structure of the delta-opioid receptor bound to naltrindole. Nature. 2012;485:400–404. doi: 10.1038/nature11111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Manglik A, Kruse AC, Kobilka TS, Thian FS, Mathiesen JM, Sunahara RK, Pardo L, Weis WI, Kobilka BK, Granier S. Crystal structure of the micro-opioid receptor bound to a morphinan antagonist. Nature. 2012;485:321–326. doi: 10.1038/nature10954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Scheerer JR, Lawrence JF, Wang GC, Evans DA. Asymmetric synthesis of salvinorin A, a potent kappa opioid receptor agonist. J Am Chem Soc. 2007;129:8968–8969. doi: 10.1021/ja073590a. [DOI] [PubMed] [Google Scholar]
- 41.Hagiwara H, Suka Y, Nojima T, Hoshi T, Suzuki T. Second-generation synthesis of salvinorin A. Tetrahedron. 2009;65:4820–4825. [Google Scholar]
- 42.Nozawa M, Suka Y, Hoshi T, Suzuki T, Hagiwara H. Total synthesis of the hallucinogenic neoclerodane diterpenoid salvinorin A. Org Lett. 2008;10:1365–1368. doi: 10.1021/ol800101v. [DOI] [PubMed] [Google Scholar]
- 43.Valdes LJ, III, Chang HM, Visger DC, Koreeda M. Salvinorin C, a New Neoclerodane Diterpene from a Bioactive Fraction of the Hallucinogenic Mexican Mint Salvia Divinorum. Org Lett. 2001;3:3935–3937. doi: 10.1021/ol016820d. [DOI] [PubMed] [Google Scholar]
- 44.Munro TA, Rizzacasa MA. Salvinorins D-F, New Neoclerodane Diterpenoids from Salvia divinorum, and an Improved Method for the Isolation of Salvinorin A. J Nat Prod. 2003;66:703–705. doi: 10.1021/np0205699. [DOI] [PubMed] [Google Scholar]
- 45.Lee DY, Ma Z, Liu-Chen LY, Wang Y, Chen Y, Carlezon WA, Jr, Cohen B. New neoclerodane diterpenoids isolated from the leaves of Salvia divinorum and their binding affinities for human kappa opioid receptors. Bioorg Med Chem. 2005;13:5635–5639. doi: 10.1016/j.bmc.2005.05.054. [DOI] [PubMed] [Google Scholar]
- 46.Shirota O, Nagamatsu K, Sekita S. Neo-clerodane diterpenes from the hallucinogenic sage Salvia divinorum. J Nat Prod. 2006;69:1782–1786. doi: 10.1021/np060456f. [DOI] [PubMed] [Google Scholar]
- 47.Ma Z, Lee DY. Revised structure of deacetyl-1,10-didehydrosalvinorin G. Tetrahedron Lett. 2007;48:5461–5464. doi: 10.1016/j.tetlet.2007.05.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bigham AK, Munro TA, Rizzacasa MA, Robins-Browne RM. Divinatorins A-C, new neoclerodane diterpenoids from the controlled sage Salvia divinorum. J Nat Prod. 2003;66:1242–1244. doi: 10.1021/np030313i. [DOI] [PubMed] [Google Scholar]
- 49.Harding WW, Tidgewell K, Schmidt M, Shah K, Dersch CM, Snyder J, Parrish D, Deschamps JR, Rothman RB, Prisinzano TE. Salvinicins A and B, New Neoclerodane Diterpenes from Salvia divinorum. Org Lett. 2005;7:3017–3020. doi: 10.1021/ol0510522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tidgewell K, Harding WW, Schmidt M, Holden KG, Murry DJ, Prisinzano TE. A facile method for the preparation of deuterium labeled salvinorin A: synthesis of [2,2,2-2H3]-salvinorin A. Bioorg Med Chem Lett. 2004;14:5099–5102. doi: 10.1016/j.bmcl.2004.07.081. [DOI] [PubMed] [Google Scholar]
- 51.Koreeda M, Brown L, Valdes LJ., III The Absolute Stereochemistry of Salvinorins. Chem Lett. 1990:2015–2018. [Google Scholar]
- 52.Lee DYW, Karnati VVR, He M, Liu-Chen LY, Kondaveti L, Ma Z, Wang Y, Chen Y, Beguin C. Synthesis and in vitro pharmacological studies of new C(2) modified salvinorin A analogues. Bioorg Med Chem Lett. 2005;15:3744–3747. doi: 10.1016/j.bmcl.2005.05.048. [DOI] [PubMed] [Google Scholar]
- 53.Munro TA, Goetchius GW, Roth BL, Vortherms TA, Rizzacasa MA. Autoxidation of Salvinorin A under Basic Conditions. J Org Chem. 2005;70:10057–10061. doi: 10.1021/jo051813e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Harding WW, Tidgewell K, Byrd N, Cobb H, Dersch CM, Butelman ER, Rothman RB, Prisinzano TE. Neoclerodane diterpenes as a novel scaffold for mu opioid receptor ligands. J Med Chem. 2005;48:4765–4771. doi: 10.1021/jm048963m. [DOI] [PubMed] [Google Scholar]
- 55.Tidgewell K, Harding WW, Lozama A, Cobb H, Shah K, Kannan P, Dersch CM, Parrish D, Deschamps JR, Rothman RB, Prisinzano TE. Synthesis of Salvinorin A Analogues as Opioid Receptor Probes. J Nat Prod. 2006;69:914–918. doi: 10.1021/np060094b. [DOI] [PubMed] [Google Scholar]
- 56.Cunningham CW, Rothman RB, Prisinzano TE. Neuropharmacology of the naturally occurring kappa-opioid hallucinogen salvinorin A. Pharmacol Rev. 2011;63:316–347. doi: 10.1124/pr.110.003244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tidgewell K, Groer CE, Harding WW, Lozama A, Schmidt M, Marquam A, Hiemstra J, Partilla JS, Dersch CM, Rothman RB, Bohn LM, Prisinzano TE. Herkinorin analogues with differential beta-arrestin-2 interactions. J Med Chem. 2008;51:2421–2431. doi: 10.1021/jm701162g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Munro TA, Duncan KK, Xu W, Wang Y, Liu-Chen LY, Carlezon WA, Jr, Cohen BM, Beguin C. Standard protecting groups create potent and selective kappa opioids: salvinorin B alkoxymethyl ethers. Bioorg Med Chem. 2008;16:1279–1286. doi: 10.1016/j.bmc.2007.10.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schmidt MS, Prisinzano TE, Tidgewell K, Harding W, Butelman ER, Kreek MJ, Murry DJ. Determination of Salvinorin A in body fluids by high performance liquid chromatography-atmospheric pressure chemical ionization. J Chromatogr B Analyt Technol Biomed Life Sci. 2005;818:221–225. doi: 10.1016/j.jchromb.2004.12.041. [DOI] [PubMed] [Google Scholar]
- 60.Prevatt-Smith KM, Lovell KM, Simpson DS, Day VW, Douglas JT, Bosch P, Dersch CM, Rothman RB, Kivell B, Prisinzano TE. Potential Drug Abuse Therapeutics Derived from the Hallucinogenic Natural Product Salvinorin A. MedChemComm. 2011;2:1217–1222. doi: 10.1039/C1MD00192B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Smissman EE, Nelson WL, LaPidus JB, Day JL. Conformational Aspects of Acetylcholine Receptor Sites. The Isomeric 3-Trimethylammonium-2-acetoxy-trans-decalin Halides1 and the Isomeric α,β-Dimethylacetylcholine Halides2. J Med Chem. 1966;9:458–465. doi: 10.1021/jm00322a002. [DOI] [PubMed] [Google Scholar]
- 62.Dalvie DK, Kalgutkar AS, Khojasteh-Bakht SC, Obach RS, O’Donnell JP. Biotransformation Reactions of Five-Membered Aromatic Heterocyclic Rings. Chem Res Toxicol. 2002;15:269–299. doi: 10.1021/tx015574b. [DOI] [PubMed] [Google Scholar]
- 63.Peterson LA. Reactive Metabolites in the Biotransformation of Molecules Containing a Furan Ring. Chem Res Toxicol. 2013;26:6–25. doi: 10.1021/tx3003824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kouzi SA, McMurtry RJ, Nelson SD. Hepatotoxicity of germander (Teucrium chamaedrys L.) and one of its constituent neoclerodane diterpenes teucrin A in the mouse. Chem Res Toxicol. 1994;7:850–856. doi: 10.1021/tx00042a020. [DOI] [PubMed] [Google Scholar]
- 65.Simpson DS, Katavic PL, Lozama A, Harding WW, Parrish D, Deschamps JR, Dersch CM, Partilla JS, Rothman RB, Navarro H, Prisinzano TE. Synthetic studies of neoclerodane diterpenes from Salvia divinorum: preparation and opioid receptor activity of salvinicin analogues. J Med Chem. 2007;50:3596–3603. doi: 10.1021/jm070393d. [DOI] [PubMed] [Google Scholar]
- 66.Lozama A, Cunningham CW, Caspers MJ, Douglas JT, Dersch CM, Rothman RB, Prisinzano TE. Opioid receptor probes derived from cycloaddition of the hallucinogen natural product salvinorin A. J Nat Prod. 2011;74:718–726. doi: 10.1021/np1007872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lovell KM, Vasiljevik T, Araya JJ, Lozama A, Prevatt-Smith KM, Day VW, Dersch CM, Rothman RB, Butelman ER, Kreek MJ, Prisinzano TE. Semisynthetic neoclerodanes as kappa opioid receptor probes. Bioorg Med Chem. 2012;20:3100–3110. doi: 10.1016/j.bmc.2012.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Liebeskind LS, Srogl J. Heteroaromatic thioether-boronic acid cross-coupling under neutral reaction conditions. Organic Letters. 2002;4:979–981. doi: 10.1021/ol0200091. [DOI] [PubMed] [Google Scholar]
- 69.Butelman ER, Rus S, Prisinzano TE, Kreek MJ. The discriminative effects of the kappa-opioid hallucinogen salvinorin A in nonhuman primates: dissociation from classic hallucinogen effects. Psychopharmacology (Berl) 2010;210:253–262. doi: 10.1007/s00213-009-1771-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Butelman ER, Caspers M, Lovell KM, Kreek MJ, Prisinzano TE. Behavioral effects and central nervous system levels of the broadly available kappa-agonist hallucinogen salvinorin A are affected by P-glycoprotein modulation in vivo. J Pharmacol Exp Ther. 2012;341:802–808. doi: 10.1124/jpet.112.193227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Johnson MW, MacLean KA, Reissig CJ, Prisinzano TE, Griffiths RR. Human psychopharmacology and dose-effects of salvinorin A, a kappa opioid agonist hallucinogen present in the plant Salvia divinorum. Drug Alcohol Depend. 2011;115:150–155. doi: 10.1016/j.drugalcdep.2010.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Maclean KA, Johnson MW, Reissig CJ, Prisinzano TE, Griffiths RR. Dose-related effects of salvinorin A in humans: dissociative, hallucinogenic, and memory effects. Psychopharmacology (Berl) 2013;226:381–392. doi: 10.1007/s00213-012-2912-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Urban JD, Clarke WP, von Zastrow M, Nichols DE, Kobilka B, Weinstein H, Javitch JA, Roth BL, Christopoulos A, Sexton PM, Miller KJ, Spedding M, Mailman RB. Functional Selectivity and Classical Concepts of Quantitative Pharmacology. J Pharmacol Exp Ther. 2007;320:1–13. doi: 10.1124/jpet.106.104463. [DOI] [PubMed] [Google Scholar]
- 74.Stallaert W, Christopoulos A, Bouvier M. Ligand functional selectivity and quantitative pharmacology at G protein-coupled receptors. Expert Opin Drug Discov. 2011;6:811–825. doi: 10.1517/17460441.2011.586691. [DOI] [PubMed] [Google Scholar]
- 75.Goupil E, Laporte SA, Hebert TE. Functional selectivity in GPCR signaling: understanding the full spectrum of receptor conformations. Mini reviews in medicinal chemistry. 2012;12:817–830. doi: 10.2174/138955712800959143. [DOI] [PubMed] [Google Scholar]
- 76.Schmid CL, Bohn LM. Physiological and pharmacological implications of beta-arrestin regulation. Pharmacol Ther. 2009;121:285–293. doi: 10.1016/j.pharmthera.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Groer CE, Tidgewell K, Moyer RA, Harding WW, Rothman RB, Prisinzano TE, Bohn LM. An opioid agonist that does not induce mu-opioid receptor-arrestin interactions or receptor internalization. Mol Pharmacol. 2007;71:549–557. doi: 10.1124/mol.106.028258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Butelman ER, Rus S, Simpson DS, Wolf A, Prisinzano TE, Kreek MJ. The effects of herkinorin, the first mu-selective ligand from a salvinorin A-derived scaffold, in a neuroendocrine biomarker assay in nonhuman primates. J Pharmacol Exp Ther. 2008;327:154–160. doi: 10.1124/jpet.108.140079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lamb K, Tidgewell K, Simpson DS, Bohn LM, Prisinzano TE. Antinociceptive effects of herkinorin, a MOP receptor agonist derived from salvinorin A in the formalin test in rats: new concepts in mu opioid receptor pharmacology: from a symposium on new concepts in mu-opioid pharmacology. Drug Alcohol Depend. 2012;121:181–188. doi: 10.1016/j.drugalcdep.2011.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Thompson AA, Liu W, Chun E, Katritch V, Wu H, Vardy E, Huang XP, Trapella C, Guerrini R, Calo G, Roth BL, Cherezov V, Stevens RC. Structure of the nociceptin/orphanin FQ receptor in complex with a peptide mimetic. Nature. 2012;485:395–399. doi: 10.1038/nature11085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.DeHaven-Hudkins DL, Dolle RE. Peripherally restricted opioid agonists as novel analgesic agents. Curr Pharm Des. 2004;10:743–757. doi: 10.2174/1381612043453036. [DOI] [PubMed] [Google Scholar]
- 82.Vanderah TW, Largent-Milnes T, Lai J, Porreca F, Houghten RA, Menzaghi F, Wisniewski K, Stalewski J, Sueiras-Diaz J, Galyean R, Schteingart C, Junien JL, Trojnar J, Rivière PJM. Novel d-amino acid tetrapeptides produce potent antinociception by selectively acting at peripheral κ-opioid receptors. Eur J Pharmacol. 2008;583:62–72. doi: 10.1016/j.ejphar.2008.01.011. [DOI] [PubMed] [Google Scholar]
- 83.Floyd BN, Camilleri M, Busciglio I, Sweetser S, Burton D, Wong GY, Kell S, Khanna S, Hwang S, Zinsmeister AR. Effect of a κ-opioid agonist, i.v. JNJ-38488502; on sensation of colonic distensions in healthy male volunteers. Neurogastroenterol Motil. 2009;21:281–290. doi: 10.1111/j.1365-2982.2008.01202.x. [DOI] [PubMed] [Google Scholar]
- 84.Camilleri M. Novel pharmacology: asimadoline, a κ-opioid agonist, and visceral sensation. Neurogastroent Motil. 2008;20:971–979. doi: 10.1111/j.1365-2982.2008.01183.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Arendt-Nielsen L, Olesen AE, Staahl C, Menzaghi F, Kell S, Wong GY, Drewes AM. Analgesic Efficacy of Peripheral k-Opioid Receptor Agonist CR665 Compared to Oxycodone in a Multi-modal, Multi-tissue Experimental Human Pain Model: Selective Effect on Visceral Pain. Anesthesiology. 2009;111:616–624. doi: 10.1097/ALN.0b013e3181af6356. [DOI] [PubMed] [Google Scholar]
- 86.Stein C. Peripheral Mechanisms of Opioid Analgesia. Anesth Analg. 1993;76:182–191. doi: 10.1213/00000539-199301000-00031. [DOI] [PubMed] [Google Scholar]
- 87.Stein C, Cabot PJ, Schafer M. Peripheral opioid analgesia: Mechanisms and clinical implications. In: Stein C, editor. Opioids in pain control : basic and clinical aspects. Cambridge University Press; New York: 1999. pp. 96–108. [Google Scholar]
- 88.Kenakin T. Agonist-receptor efficacy. II. Agonist trafficking of receptor signals. Trends Pharmacol Sci. 1995;16:232–238. doi: 10.1016/s0165-6147(00)89032-x. [DOI] [PubMed] [Google Scholar]
- 89.Dewire SM, Yamashita DS, Rominger DH, Liu G, Cowan CL, Graczyk TM, Chen XT, Pitis PM, Gotchev D, Yuan C, Koblish M, Lark MW, Violin JD. A G Protein-Biased Ligand at the mu-Opioid Receptor Is Potently Analgesic with Reduced Gastrointestinal and Respiratory Dysfunction Compared with Morphine. J Pharmacol Exp Ther. 2013;344:708–717. doi: 10.1124/jpet.112.201616. [DOI] [PubMed] [Google Scholar]
- 90.Carroll FI, Carlezon WA., Jr Development of kappa Opioid Receptor Antagonists. J Med Chem. 2013;56:2178–2195. doi: 10.1021/jm301783x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lutz PE, Kieffer BL. Opioid receptors: distinct roles in mood disorders. Trends Neurosci. 2013;36:195–206. doi: 10.1016/j.tins.2012.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.White WC. Report of the Committee on Drug Addiction, 1929–1941. National Research Council; 1941. [DOI] [PubMed] [Google Scholar]
- 93.Eddy NB. The National Research Council Involvement in the Opiate Problem 1928–1971. National Academy of Sciences; Washington, DC: 1973. [Google Scholar]
- 94.Rice KC. Analgesic Research at the National Institutes of Health: State of the Art 1930s to the Present. In: Meldrum ML, editor. Opioids and Pain Relief: A Historical Perspective, Progress in Pain Research and Management. IASP Press; Seattle: 2003. pp. 57–83. [Google Scholar]
- 95.Corbett AD, Henderson G, McKnight AT, Paterson SJ. 75 years of opioid research: the exciting but vain quest for the Holy Grail. Br J Pharmacol. 2006;147(Suppl 1):S153–S162. doi: 10.1038/sj.bjp.0706435. [DOI] [PMC free article] [PubMed] [Google Scholar]