

Abstract
Significance: Mitochondria are fundamental to the life and proper functioning of cells. These organelles play a key role in energy production, in maintaining homeostatic levels of second messengers (e.g., reactive oxygen species and calcium), and in the coordination of apoptotic cell death. The role of mitochondria in aging and in pathophysiological processes is constantly being unraveled, and their involvement in neurodegenerative processes, such as Alzheimer's disease (AD), is very well known. Recent Advances: A considerable amount of evidence points to oxidative damage to mitochondrial DNA (mtDNA) as a determinant event that occurs during aging, which may cause or potentiate mitochondrial dysfunction favoring neurodegenerative events. Concomitantly to reactive oxygen species production, an inefficient mitochondrial base excision repair (BER) machinery has also been pointed to favor the accumulation of oxidized bases in mtDNA during aging and AD progression. Critical Issues: The accumulation of oxidized mtDNA bases during aging increases the risk of sporadic AD, an event that is much less relevant in the familial forms of the disease. This aspect is critical for the interpretation of data arising from tissue of AD patients and animal models of AD, as the major part of animal models rely on mutations in genes associated with familial forms of the disease. Future Directions: Further investigation is important to unveil the role of mtDNA and BER in aging brain and AD in order to design more effective preventive and therapeutic strategies. Antioxid. Redox Signal. 18, 2444–2457.
Introduction
The susceptibility of mitochondrial DNA (mtDNA) to damage is much higher than that of nuclear DNA (nDNA), resulting in higher mutation rates in mtDNA (135). mtDNA has multiple copies, and each mitochondrion contains 2 to 10 molecules of DNA, which are organized as nucleoids (51). The existence of several copies of mtDNA means that mutated and wild-type mtDNA can co-exist, a condition known as heteroplasmy (188). The ratio between wild-type and mutant mtDNA may define a threshold where a biochemical abnormality may determine a pathological phenotype. Indeed, it is estimated that in many patients with clinical manifestations of mitochondrial disorders, the proportion of mutant mtDNA exceeds 50% (138). The mitochondrial genome contains 37 genes, 13 of which encode for subunits of electron transport chain (ETC) complexes, 22 for transfer RNAs, and 2 for ribosomal RNAs (115, 59). Therefore, mtDNA integrity is mandatory for normal function of ETC, as it encodes several subunits of mitochondrial respiratory chain complexes as well as other mitochondrial proteins (19). If a proper ETC function is not ensured, reactive oxygen species (ROS) production is largely increased, as observed in mitochondrial diseases, or in experimental and animal models of oxidative phosphorylation (OXPHOS) deficiencies (125, 196). Increased generation of ROS and oxidative damage occur during aging as well as several age-related degenerative diseases, including Alzheimer's disease (AD) (20, 10, 165). Furthermore, it has been suggested that age-associated deficiencies in the repair of oxidative DNA damage correlate with cognitive decline and neurodegenerative diseases that are prominent in the aged population (199, 177).
This review addresses several aspects of mitochondrial (dys) function and the involvement of mitochondria in aging, and AD is also discussed. Special attention is given to mtDNA and its repair mechanisms.
Mitochondria: Cell Keepers or Executioners?
The survival of eukaryotic cells greatly relies on mitochondrial function. The classical appraisal of mitochondrial function is based on energy-producing capacity. Nevertheless, the importance of mitochondria to the cells is far more complex and includes a number of functions that span from energy production, calcium (Ca2+) homeostasis, and production of second messengers, to the control of apoptotic cell death (Fig. 1). In addition, the canonical view of mitochondria as bean-shaped organelles has been revoked and redefined to a more dynamic perspective, fusing, dividing, and moving within cells (50). Mitochondria are able to change from a network-like appearance, forming long tubules, to a more individualized state, appearing similar to small round vesicles. The stimuli that alter this equilibrium toward highly branched or completely fragmented morphology are linked to the cell compartmentalization, developmental stage, stress stimulus, and the functional state of the mitochondria, among others (14). Disturbing either mitochondrial fission or fusion may affect mitochondrial membrane stability with possible negative consequences for ETC functionality (38, 116, 40).
FIG. 1.

Physiological functions of mitochondria. Mitochondria are centrally positioned in diverse aspects of cellular physiology such as homeostasis of second messengers [e.g., reactive oxygen species (ROS), calcium (Ca2+)], apoptosis and energy production. MPTP, mitochondrial permeability transition pore, mitochondrial permeability transition pore; CI, CII, CIII, and CIV, complexes I, II, III, and IV of the respiratory chain; FoF1, ATP synthase.
Mitochondrial bioenergetic production depends on the formation of a “protonmotive force,” which is generated through the extrusion of protons to the intermembrane space driven by the electron flow throughout ETC, from complexes with lower to complexes with higher oxidation potentials. Protons are driven back to the matrix through the ATP synthase during ATP production (52). Although the electron flow through ETC complexes is a very efficient process, a small amount of superoxide anions (O2•−) is produced, due to electron leak mostly from complexes I and III (163, 56, 33, 10). At low/moderate levels, ROS act as second messengers within cells; however, exacerbated ROS production is deleterious for the cell, contributing to a variety of pathological processes (192, 1). Redox imbalance will be further discussed in a subsequent section of the article.
Mitochondria are also intracellular buffers of cytoplasmic Ca2+, thus playing a key role in normal neurotransmission, short- and long-term plasticity, excitotoxicity, and regulation of gene transcription, processes that are highly dependent on Ca2+ levels (35, 152, 208, 153, 210, 169, 154, 203). Ca2+ is internalized into mitochondria via the Ca2+uniporter, a protein that is still to be fully identified and biochemically characterized. Nevertheless, a candidate protein, which was named MCU (from “mitochondrial Ca2+uniporter”), proved to be essential for high-capacity Ca2+ transport into mitochondria in a number of in vitro and in vivo experimental models (13, 48). On the other hand, Ca2+ release is mediated by Na+/Ca2+ or H+/Ca2+ exchangers (203). It was shown that mitochondria are involved in cells' Ca2+ buffering impairment, a situation which occurs in the aging brain and AD (26, 29). The impairment of Ca2+ homeostasis is intimately associated with mitochondrial permeability transition (MPT). MPT is potentiated by oxidative stress, high phosphate concentrations, and adenine nucleotide depletion and is characterized by the opening of a high conductance pore known as mitochondrial permeability transition pore (MPTP) that enables the release of ions and solutes from the matrix to the cytosol (209, 44). The MPTP eventually culminates in cell death due to the release of proapoptotic factors such as cytochrome c and apoptosis-inducing factor (80, 189, 150).
ROS Imbalance in Aging and Alzheimer's Disease
Endogenous production and scavenging of ROS
The balance between ROS production and scavenging enables cells to achieve a physiological equilibrium where the levels of free radicals might play a role in cell transduction (178). ROS interfere with the macromolecules of cells; however, under physiological conditions, the cells' quality control systems are able to overcome this damage, avoiding the development of a pathological state (159). During aging, the quality control systems become defective, resulting in an accumulation of damaged components, which, accompanied by a redox disequilibrium, may elicit a pathological condition (156).
In cells, there are multiple sources of ROS, including mitochondria, nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX), xanthine oxidase, and nitric oxide synthase (NOS) (147). Mitochondria are recognized as the hub of ROS production during normal aerobic activity. The electrons leak from the ETC directly to molecular oxygen, producing short-lived free radicals such as O2•−(133, 191). While complex I releases O2•− only to the matrix, complex III releases O2•− to both the matrix and intermembrane space (23). O2•− can be converted into nonradical derivates such as hydrogen peroxide (H2O2) either by a spontaneous dismutation reaction or catalyzed by the manganese superoxide dismutase that resides in the mitochondrial matrix (73). H2O2 can be converted into hydroxyl radicals (•OH) through the Fenton reaction. In the Fenton reaction, a molecule of H2O2 reacts with ferrous iron (Fe2+) to generate ferric iron (Fe3+), hydroxide anion (OH−), and •OH. Fe3+ can be reduced by O2•−, generating a redox cycle in which the O2•− facilitates the Fenton reaction by making Fe2+ available (92). Similar to iron, copper also participates in the Fenton reaction, which exacerbates ROS production (60, 85, 157). •OH can also be produced by a direct reaction of O2•− with H2O2, a reaction known as the Haber–Weiss reaction (92). Mitochondria consubstantiate a microenvironment that is highly enriched in iron, as many mitochondrial enzymes possess heme groups and iron-sulfur clusters in their active centers, making them favorable locations of •OH production (128). Hence, mitochondria are prone to oxidative damage and particularly susceptible to •OH-mediated oxidation, which plays a major role in DNA oxidation. Apart from the ETC, several other sites in the mitochondria have also been reported to generate O2•−, including pyruvate dehydrogenase, α-ketoglutarate dehydrogenase (171), glycerol-3-phosphate dehydrogenase, and fatty acid β-oxidation (23). Recently, important advances toward understanding mitochondrial ROS generation have been made. Transient quantal O2•− flashes were observed in excitable cells such as neurons, which are associated with and required for the opening of MPTP, which represents a new facet of mitochondrial ROS (57, 198). To counteract an exaggerated production of ROS, mitochondria possess a very efficient antioxidant system, including glutathione peroxidase, catalase, and peroxiredoxin III, which are responsible for converting H2O2 to water (71).
As mentioned earlier, another source of cellular ROS is the NOX family proteins that are enzymatic complexes which catalyze the electron transfer from NADPH to molecular oxygen and generate O2•− and its downstream reactive species (16, 65). NADPH oxidase is composed of cytochrome b558 (an heterodimer comprising a 22-KDa alpha-subunit-p22phox-and a glycosylated approximately 91-KDa beta-subunit-gp91phox), several cytosolic proteins (p47phox, p67phox, and p40phox), and the Rac G-protein. According to the new terminology, the NOX family refers to the catalytic subunit of NADPH oxidase, and these include NOX2 and its six homologs (NOX1, NOX3, NOX4, NOX5, DUOX1, and DUOX2)(65). It is known that NOX1, NOX2, and NOX4 are expressed in neurons, astrocytes, and microglia. Under normal circumstances, NOX is latent. However, on stimulation, NOX is translocated to the membrane and forms an heterodimeric enzymatic complex with cytochrome b558 that catalyzes the reduction of molecular oxygen to O2•−(16).
In this way, compromised mitochondrial functioning, NOX overactivation, or the failure of free radical-scavenging systems could constitute critical events underlying oxidative damage in brain aging and AD.
Oxidative stress in the aging brain
Aging is an inevitable biological process that is characterized by a progressive decline in physiological function, including cognition, and by the increased susceptibility to disease, representing a major risk factor for the development of AD (36, 76). Oxidative stress and mitochondrial malfunction are two interdependent mechanisms that play a central role in brain aging (36). The brain is particularly vulnerable to oxidative damage as a consequence of its high levels of polyunsaturated fatty acids, high oxygen consumption, high content in transition metals, and poor antioxidant defenses (139). Compelling evidence reports that the aging brain is associated with the accumulation of markers of proteins, lipids, and DNA oxidative damage (37, 61, 66, 78, 166, 28). It was previously shown that the aged brain is characterized by increased levels of protein carbonyls, 3-nitrotyrosine, thiobarbituric acid reactive substances (TBARS), and diminished content of cardiolipin and protein thiols (41, 67, 160).
Along with oxidative stress, mitochondrial dysfunction also contributes to the aging brain. The most important functional deficits documented in aged brain are the loss of the mitochondrial membrane potential and OXPHOS capacity, decreased respiration and ATP synthesis, and increased susceptibility to MPTP opening (9, 22, 41, 58, 144).
Oxidative stress in Alzheimer's disease
AD is the most prevalent age-related neurodegenerative disorder that affects approximately 35 million people worldwide (149). Clinically, AD is characterized by the progressive loss of cognitive function and behavioral disturbances (149). These traits are accompanied by two distinctive pathological features, the massive deposition of aggregated amyloid-β (Aβ) peptide in the extracellular space as senile plaques, and the presence of intracellular neurofibrillary tangles, mainly composed of hyperphosphorylated tau protein (34, 131).
The pathogenic road map leading to AD pathology is still not entirely understood; however, multiple pieces of evidence support the key involvement of oxidative stress and mitochondrial malfunction in the onset and progression of the disease (129, 155, 180, 181). Oxidative stress is manifested by the occurrence of elevated levels of oxidatively modified lipids, proteins, and nucleic acids in vulnerable brain regions of AD subjects when compared with age-matched controls (18, 107, 120, 122, 141). Indeed, increased levels of lipid peroxidation products, such as TBARS, malondialdehyde, 4-hydroxy-2-nonenal (HNE), and F2-isoprostanes, were documented in the AD brain, particularly in regions where senile plaques and neurofibrillary tangles typically accumulate (106, 118, 119, 148, 158, 206). With regard to protein oxidation, AD is characterized by increased levels of protein carbonyls and widespread nitration of tyrosine residues in brain cortex and hippocampus (81, 112, 167). An increase in 8-hydroxyguanine (8OHG) and 8-hydroxy-2-deoxyguanosine (8OHdG), markers of RNA and DNA oxidation, respectively, and protein adducts were observed in brain regions that were most affected by AD pathology (108, 140, 142, 156). The role of oxidative stress in AD is further reinforced by the existence of a defective antioxidant defense system (5, 11). A decrease was documented in the activities of the antioxidant enzymes copper/zinc superoxide dismutase (Cu/ZnSOD) and catalase in the frontal and temporal cortex of AD subjects (118). AD subjects also exhibit reduced total antioxidant capacity (176), and a negative correlation was observed between the total antioxidant capacity and the duration of the disease (72).
During the course of AD, oxidative damage is also coupled to a progressive decline of the mitochondrial function (127). This notion is supported by an extensive literature which reports that AD is characterized by reduced cerebral energy metabolism (8), impaired activities of the tricarboxylic acid cycle enzymes (25, 121, 162), and defects in the mitochondrial ETC (21, 32, 45, 146, 96, 193). The most consistent defect at ETC level is the decline in cytochrome c oxidase (COX) activity, an effect that is positively correlated with Aβ concentration, as determined by in vitro studies (30). During AD progression, Aβ is translocated toward mitochondria (88, 186), enabling its interaction with critical redox centers of the subunit I of COX (6, 7) and Aβ-binding alcohol dehydrogenase (ABAD) (111, 187). The interaction of Aβ with the subunit I of COX and ABAD potentiates mitochondrial dysfunction and further increases ROS production in a vicious cycle. There is also evidence which supports a role for mtDNA mutations in the development and progression of AD (83).
Another important aspect is the role of redox-active metals in AD-related oxidative damage. Indeed, disruption of iron homeostasis has been suggested to be a trigger of oxidative stress and an early neuropathological event in AD (64). It was demonstrated that iron-mediated enhancement of oxidative stress occurs in preclinical AD (168), and increased redox-active iron is found in the cerebrospinal fluid from AD subjects (103). Besides its effects on oxidative status, redox-active metals also potentiate Aβ aggregation, aggravating AD pathology (27). Indeed, iron, zinc, and copper participate in the initiation of Aβ-mediated seeding process and Aβ oligomerization (86).
NOX overactivation is another pathogenic step underlying exacerbated oxidative damage in AD pathology (65). Mounting evidence suggests that the NOX system may be altered in AD, as indicated by the increased levels of p47phox and p67phox in the membrane fraction of AD brains, which foster the idea that NOX is overactivated in AD (55). Microglial expression of NOX subunit p22phox is also enhanced in the AD brain (3). A deficiency of NOX2 in transgenic AD mice reduces oxidative stress and improves cerebrovascular function and memory deficits without affecting Aβ levels or senile plaques (145), which reinforces the role of NOX in AD-associated oxidative damage. Importantly, aggregated Aβ stimulates O2•− and H2O2 production in microglial cells and induces the translocation of Rac from the cytosol to the membrane, supporting the idea that Aβ can affect NOX2-mediated pathways (126, 200).
Overall, these findings indicate that mitochondria, NOX, and oxidative stress are important contributors in AD-related neurodegeneration.
mtDNA Oxidation and Repair Deficiency
mtDNA oxidation and repair mechanisms
Mitochondrial dysfunction and apoptosis can also be triggered by point mutations, nucleic acid modifications, and large-scale deletions in mtDNA (104, 100). It has been reported that mtDNA damage is 10- to 20-fold higher, more extensive and persists longer when compared with nDNA (175, 204). mtDNA is particularly susceptible to oxidative damage, because it is not compacted around histones and is localized near the ETC, which is a major source of ROS. In addition, mtDNA has none or few noncoding regions, increasing the chances of mutagenicity in coding regions (4, 156). Mitochondria are highly enriched in iron microenvironments, thus favoring the formation of •OH that, due to its short half-life, preferentially reacts with mitochondrial components, including mtDNA (192, 130). In addition, the oxidation of HNE can originate epoxide forms that interact with DNA bases (110, 91). During aging and in neurodegenerative disorders, nitric oxide (NO) interacts with O2•−, resulting in the formation of peroxynitrite (ONOO−), which contributes to mtDNA damage, including single-strand breaks (182, 194, 190).
All four bases (purines- adenine, guanine; pyrimidines- cytosine, thymine) and the respective deoxynucleosides are highly susceptible to oxidative damage. The main products of DNA oxidation include 8-hydroxyadenine (8OHA), 8-hydroxyguanine (8OHG, and its deoxynucleoside equivalent, 8OHdG), 5,6-dihydroxy-5,6-dihydrothymine, and ring-opened lesions (4,6-diamino-5-formamidopyrimidine, FapyA, and 2,6-diamino-4-hydroxy-5-formamidopyrimidine, FapyG) (Fig. 2) (117). Overall, more than 20 oxidized base adducts can be formed from ROS attack on the DNA (42, 172). Nevertheless, guanine has the lowest oxidation potential, being the most readily oxidized base (130). 8OHG and 8OHdG, along with FapyG, are the most studied and common forms of oxidized DNA bases (53, 95). While mutagenesis is stimulated by the accumulation of 8OHdG by pairing with adenine as well as cytosine (113), the FapyG lesions inhibit DNA synthesis (143, 114).
FIG. 2.

Molecular structures of some oxidation products of DNA. The products of DNA oxidation result from the attack of reactive oxygen species, mainly •OH, to DNA bases.
Base excision repair (BER) is the primary nuclear and mitochondrial repair pathway for oxidative DNA damage. BER is evolutionarily conserved and is responsible for recognizing, excising, and replacing a wide number of DNA modifications that are characterized by small base modifications (99, 82). Generally, the BER machinery consists of several proteins that act in an ordered multistep cascade:(i) the recognition and excision of the damaged base;(ii) the incision of the DNA backbone in the abasic (AP) site; (iii) the generation of a 3′-OH and a 5′-P moieties in the DNA termini; (iv) the synthesis of the missing nucleotide; and (v) the sealing of the remaining DNA nick (Fig. 3) (205). This mechanism is essentially the same for nDNA and mtDNA repair; however, the isoforms of some enzymes involved in the process may differ from the nucleus to the mitochondria, even though all of them are nuclear encoded (199). The initial removal of the damaged base is accomplished by substrate-specific DNA glycosylases that hydrolyze the N-glycosidic bond between the modified base and the DNA backbone (54, 87). DNA glycosylases can be divided into two distinct functional groups: (i) a monofunctional group of enzymes with glycosylase activity only, which includes hydroxymethyl-uracyl DNA glycosylase (UDG) whose mitochondrial isoform UDG1 is generated by alternative splicing (31, 136); (ii) a byfunctional group of enzymes with intrinsic 3′AP lyase activity, in addition to glycosylase activity, which include 8OHG DNA glycosylase (OGG1), the human endonuclease III homolog (NTH1), and Nei-like homologs (NEILs)(68). Oxidized bases are generally removed by bifunctional DNA glycosylases. OGG1 has two isoforms, α-OGG1 that localizes to both the nucleus and mitochondria and β-OGG1that localizes in mitochondria (137). NTH1 has a putative mitochondrial targeting sequence, which allows its localization to mitochondria (185, 173, 94). NEILs are localized in the nucleus and mitochondria (77, 84, 132). In human cells, oxidative pyrimidine lesions are generally excised by NTH1 or NEILs; whereas oxidative purine lesions are excised by OGG1 (132, 77, 89). 8OHG lesions are primarily repaired by OGG1 (97, 47). The step after the removal of the damaged base by glycosylases is the incision of the DNA backbone in an adjacent site to the AP site. This stage is characterized by different types of lyase activity, either occurring immediately 5′ to the AP site or 3′ to the AP site depending on whether the excision step was accomplished by monofunctional or byfunctional glycosylases, respectively (49, 201). Indeed, AP endonuclease (APE1) is responsible for the incision of the DNA backbone after UDG1 removal of the modified base (49, 201). APE1 localizes to both the nucleus and mitochondria (62, 151, 202). Moreover, the byfunctional glycosylases are capable of incising the DNA backbone, leaving a DNA single-strand break. The final steps of the repairing process may undergo two distinct subpathways, the short- or long-patch BER (SP-BER or LP-BER, respectively). The SP-BER involves the incorporation of a single nucleotide into the gap by DNA polymerase. The LP-BER involves the incorporation of several nucleotides, typically 2 to 7, followed by the cleavage of the resulting 5′(91). Finally, the nick left behind by DNA polymerases needs to be sealed, a process performed by ligases, ligase I (nucleus) in the case of LP-BER, and ligase III (nucleus and mitochondria) in the case of SP-BER (70). The polymerase responsible for the mtDNA repair synthesis is polymerase γ (74, 93).
FIG. 3.

Base excision repair (BER) machinery. An oxidative lesion (herein represented by the lightning symbol) is removed by DNA glycosylases, which excise the oxidized base from the DNA backbone, leaving an abasic site (AP). Afterward, the DNA backbone is incised in order to create a single-nucleotide gap that is ready for subsequent filling by DNA polymerases. In this step, de novo synthesis can follow one of two sub-pathways; in short-patch BER (SP-BER), 1 nucleotide is inserted and in long-patch BER (LP-BER), 2–7 nucleotides are inserted. The last step involves the ligation of the nick by DNA ligases. APE1, AP endonuclease. See text for further details.
Despite the current knowledge on the mechanisms that maintain the genomic integrity, particularly mitochondrial genome, it is of great interest to gain more insight into the real importance of each enzyme and each subpathway involved in the repair process. Indeed, the repair of mtDNA oxidative damage was thought to be mediated solely by SP-BER (17, 174); however, in recent years, LP-BER was also demonstrated to counteract the accumulation of oxidative damage to mtDNA (Fig. 3)(2, 105, 184, 207).
Aging
Aging has been established as being the main risk factor for the development of late-onset neurodegenerative disorders such as AD. The accumulation of oxidative damage plays a key role in the aging process, as postulated by the free radical theory of aging (75). Age-associated oxidation of mtDNA results from an increased oxidative attack to the nucleic acids and a reduced efficacy in mtDNA repair machinery, namely BER (Fig. 4). Indeed, the aging brain is characterized by an increased oxidative damage to mtDNA noticed by the formation of 8OHdG, which is the most common marker of oxidative DNA damage (123). Notably, in human subjects (42–97 years), a progressive augment in 8OHdG was reported in both nDNA and mtDNA with aging; however, the extent of increase of 8OHdG is ten-fold more in mtDNA compared with nDNA (123). The increased susceptibility of mtDNA, compared with nDNA, was also observed in aged brains of other mammalian species (12). An age-associated impairment of mitochondrial BER machinery, particularly OGG1, UDG, APE1, and polymerase γ, has been reported (90, 39). Moreover, five specific brain areas were shown to have deficits in mitochondrial BER, namely caudate nucleus, frontal cortex, hippocampus, cerebellum, and brain stem (90). A recent study demonstrated that brain cortical and hippocampal mtDNA glycosylases behave differently in cortical and hippocampal mitochondria of rodents (68). Hippocampal mtDNA glycosylases present lower activity when compared with cortical glycosylases. Importantly, brain cortical mtDNA glycosylases show an age-dependent decrease in their activity; while hippocampal glycosylases present only minor alterations (68). These findings highlight how mitochondrial heterogeneity influences the susceptibility of these organelles to damage. In fact, it was also shown that synaptic mitochondria are more susceptible to Ca2+ overload and the induction of MPTP than nonsynaptic mitochondria (24, 134), which reinforces the idea that synaptic mitochondria, including their DNA, are more vulnerable to injury.
FIG. 4.
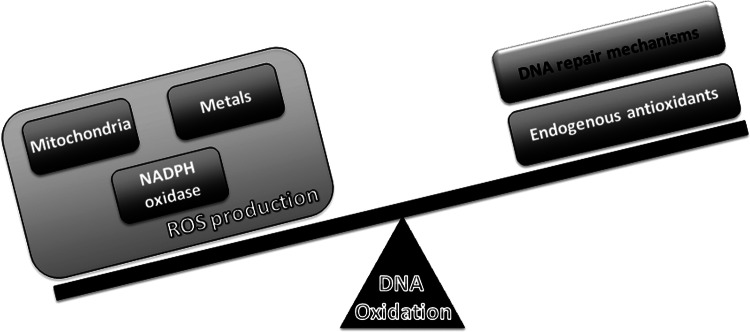
Redox imbalance, DNA repair, and oxidation. Increased DNA oxidation in aging and AD results from an imbalance between ROS production and ROS scavenging as well as from the failure of DNA repair mechanisms.
BER enzymes are negatively modulated by covalent modifications in an age-dependent manner putatively due to decreased acetylation (183) or iron/copper dyshomeostasis (79). Notably, a general trend toward increasing heteroplasmy with the aging process has been observed, due to a gradual accumulation of alterations in mtDNA throughout life (170). These results are in accordance with previous observations of increased mtDNA deletions (15, 98) and somatic mutations (164) with age in the substantia nigra. A causal relation between the malfunction of BER machinery and neurodegeneration has been established, which is further associated with behavioral alterations (102).
Alzheimer's disease
Mitochondrial dysfunction and exacerbated generation of ROS are well known features of AD. Moreira (130) and de la Monte (46) reported that AD brains present increased fragmentation of mtDNA, reduced mtDNA content and mass, reduced level of COX, and evidence of apoptotic cell loss. Despite no causative mtDNA mutations being linked to AD, some polymorphic variations can occur, having implications in enzymatic activities, such as COX (109). Some mtDNA mutations have been associated with increased incidence of AD (195, 43). Likewise, a reduction in the level of ND6 complex I transcript in AD has been reported (43). AD brains present increased mtDNA mutations that are enhanced in an age-dependent manner, when compared with control cases (43). Nevertheless, and despite no causative mutations in mtDNA being currently known, mitochondrial dysfunction has been proposed to precipitate Aβ deposition, neurofibrillary tangle formation, and, ultimately, neurodegeneration (Fig. 5) (179, 180).
FIG. 5.
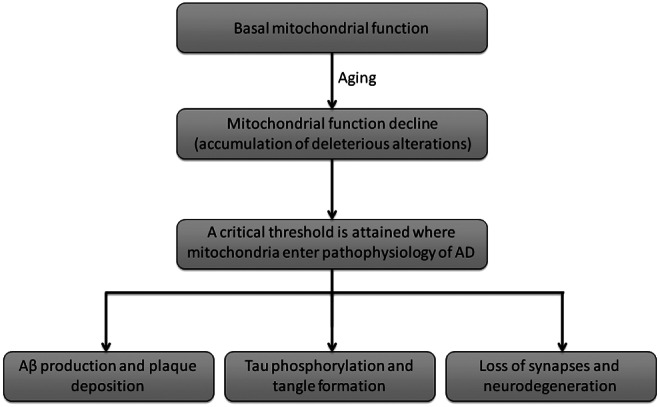
Mitochondrial cascade hypothesis for AD. The accumulation of damage and consequent decline of mitochondrial function with aging are hypothesized to be the triggers of sporadic (late onset) AD. This hypothesis postulates that amyloidosis, tangle formation, synapse, and neuronal loss are consequences of mitochondrial defects (Swerdlow and Khan 2009).
Although several studies demonstrate that oxidation of both nDNA and mtDNA is increased in AD brains (63, 124, 197), mtDNA oxidation has been found to be 10-fold higher than nDNA in frontal, parietal, and temporal lobes of AD patients (197). The simultaneous increased oxidation of mtDNA and deficiency of DNA repair could enhance the lesion to mitochondrial genome, potentially leading to neuronal loss. Indeed, Shao et al. (161) demonstrated that mitochondrial OGG1 activity is decreased in the frontal and temporal lobe of late-stage AD, and in the temporal lobe of MCI patients, compromising the removal of oxidatively damaged bases from mtDNA. Opposing results were recently reported in the brains of the triple transgenic model of AD (3×Tg-AD), in which no changes between the synaptosomal BER activities of presymptomatic and symptomatic AD mice were found (69). The contradictory observations reported in human and mice AD brains can be easily explained by the fact that the disease process in 3×Tg-AD mice is the result of a genetic manipulation, as those animals harbor the human amyloid precursor Swedish mutation, presenilin-1 M146V (PS1(M146V)) knock-in mutation, and tau (P301L) mutation; whereas in sporadic AD patients, mitochondria malfunctioning and oxidative stress are considered causative agents (155). Notably, rodents that were engineered to express an inducible mutant form of UDG1 show a decline in cognitive performance, as evaluated by the Morris water maze test (102). Furthermore, and similarly to that described in AD (198), rodents expressing mutant UDG1 also display abnormal mitochondrial dynamics (101), which supports the idea that impaired BER machinery may also play a role in AD.
More studies are needed to clarify the involvement of defects in mtDNA and its repair mechanisms in AD development. Furthermore, caution should be taken in the analysis and interpretation of results obtained with AD transgenic mice, as these animals mimic the familial cases of the disease, which represent less than 5% of all AD cases. In this line, it would be interesting to perform studies in rodents subjected to the intracerebroventricular administration of streptozotocin (icvSTZ), which are considered animal models of sporadic AD.
Conclusion
Mitochondria are major producers of ROS that under low/moderate levels act as second messengers. However, during aging and age-related diseases, an increased production of mitochondrial ROS associated with a defective scavenging system culminate in a redox imbalance and high levels of oxidatively damaged biomolecules. Mitochondrial dysfunction is currently accepted as a pathological hallmark of AD, which is considered an early event in disease pathogenesis. The accumulation of oxidative lesions to mtDNA occurs during aging and is also a prominent feature in AD, along with the failure in BER machinery. The observation that mtDNA oxidation occurs during aging and in the prodromal stage of AD strongly supports the idea that mitochondrial abnormalities are causative agents in AD. Whether mtDNA oxidation is a determinant for the onset of disease is yet to be clarified, namely if there is any threshold that triggers the disease process. Nonetheless, it is tempting to propose that the impairment in OXPHOS results in an exacerbation of ROS generation that increases the probability of mtDNA mutations in a positive feedback loop, a situation which is potentiated by a defective BER machinery (Fig. 6). The clarification of BER in AD also opens new windows for therapeutic intervention that are aimed at effectively repairing damaged mtDNA.
FIG. 6.

Putative vicious cycle of mitochondrial ROS production in aging and AD. Since 13 subunits of the electron transport chain (ETC) are encoded by mtDNA, it is likely that mtDNA oxidation resulting from the increased ROS production leads to ETC dysfunction, which exacerbates ROS production. This vicious cycle is potentiated by the decline in BER efficiency that occurs in the aged and AD brains.
Abbreviations Used
- 8OHA
8-hydroxyadenine
- 8OHdG
8-hydroxydeoxyguanosine
- 8OHG
8-hydroxyguanine
- Aβ
amyloid beta
- ABAD
Aβ-binding alcohol dehydrogenase
- AD
Alzheimer's disease
- APE1
AP endonuclease
- BER
base excision repair
- COX
cytochrome c oxidase
- ETC
electron transport chain
- HNE
4-hydroxy-2-nonenal
- H2O2
hydrogen peroxide
- LP-BER
long-patch BER
- MCU
mitochondrial Ca2+ uniporter
- MPT
mitochondrial permeability transition
- MPTP
mitochondrial permeability transition pore
- mtDNA
mitochondrial DNA
- NADPH
nicotinamide adenine dinucleotide phosphate
- nDNA
nuclear DNA
- NEILS
Nei-like homologs
- NOS
nitric oxide synthase
- NOX
nicotinamide adenine dinucleotide phosphate oxidase
- NTH1
human endonuclease III homologue
- OGG1
80HG DNA glycosylase
- OXPHOS
oxidative phosphorylation
- ROS
reactive oxygen species
- SP-BER
short-patch BER
- TBARS
thiobarbituric acid reactive substances
- UDG
hyderoxymethyl-uracyl DNA glycosylase
Acknowledgments
Renato X. Santos is the recipient of a PhD fellowship from the Fundação para a Ciência e a Tecnologia (SFRH/BD/43972/2008). Work in the authors' laboratories is supported by Fundação para a Ciência e a Tecnologia and Fundo Europeu de Desenvolvimento Regional (PTDC/SAU-NEU/103325/2008 and PTDC/SAU-NMC/110990/2009). This project was also supported by a grant from the National Institute on Minority Health and Health Disparities (G12MD007591) from the National Institutes of Health.
References
- 1.Addabbo F. Montagnani M. Goligorsky MS. Mitochondria and reactive oxygen species. Hypertens. 2009;53:885–892. doi: 10.1161/HYPERTENSIONAHA.109.130054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Akbari M. Visnes T. Krokan HE. Otterlei M. Mitochondrial base excision repair of uracil and AP sites takes place by single-nucleotide insertion and long-patch DNA synthesis. DNA Repair (Amst) 2008;7:605–616. doi: 10.1016/j.dnarep.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 3.Akiyama H. Barger S. Barnum S, et al. Inflammation and Alzheimer's disease. Neurobiol Aging. 2000;21:383–421. doi: 10.1016/s0197-4580(00)00124-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Anderson S. Bankier AT. Barrell BG, et al. Sequence and organization of the human mitochondrial genome. Nature. 1981;290:457–465. doi: 10.1038/290457a0. [DOI] [PubMed] [Google Scholar]
- 5.Ansari MA. Scheff SW. Oxidative stress in the progression of alzheimer disease in the frontal cortex. J Neuropathol Exp Neurol. 2010;69:155–167. doi: 10.1097/NEN.0b013e3181cb5af4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Atamna H. Frey WH., 2nd A role for heme in Alzheimer's disease: heme binds amyloid beta and has altered metabolism. Proc Natl Acad Sci USA. 2004;101:11153–11158. doi: 10.1073/pnas.0404349101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Atamna H. Heme binding to Amyloid-beta peptide: mechanistic role in Alzheimer's disease. J Alzheimers Dis. 2006;10:255–266. doi: 10.3233/jad-2006-102-310. [DOI] [PubMed] [Google Scholar]
- 8.Azari NP. Pettigrew KD. Schapiro MB, et al. Early detection of Alzheimer's disease: a statistical approach using positron emission tomographic data. J Cereb Blood Flow Metab. 1993;13:438–447. doi: 10.1038/jcbfm.1993.58. [DOI] [PubMed] [Google Scholar]
- 9.Bagh MB. Thakurta IG. Biswas M. Behera P. Chakrabarti S. Age-related oxidative decline of mitochondrial functions in rat brain is prevented by long term oral antioxidant supplementation. Biogerontology. 2010;12:119–131. doi: 10.1007/s10522-010-9301-8. [DOI] [PubMed] [Google Scholar]
- 10.Balaban RS. Nemoto S. Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120:483–495. doi: 10.1016/j.cell.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 11.Baldeiras I. Santana I. Proença MT, et al. Oxidative damage and progression to Alzheimer's disease in patients with mild cognitive impairment. J Alzheimers Dis. 2010;21:1165–1177. doi: 10.3233/jad-2010-091723. [DOI] [PubMed] [Google Scholar]
- 12.Barja G. Herrero I. Oxidative damage to mitochondrial DNA is inversely related to maximum life span in the heart and brain of mammals. FASEB J. 2000;14:312–318. doi: 10.1096/fasebj.14.2.312. [DOI] [PubMed] [Google Scholar]
- 13.Baughman JM. Perocchi F. Girgis HS. Plovanich M, et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature. 2011;476:341–345. doi: 10.1038/nature10234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Benard G. Karbowski M. Mitochondrial fusion and division: regulation and role in cell viability. Semin Cell Dev Biol. 2009;20:365–374. doi: 10.1016/j.semcdb.2008.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bender A. Krishnan KJ. Morris CM, et al. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat Genet. 2006;38:515–517. doi: 10.1038/ng1769. [DOI] [PubMed] [Google Scholar]
- 16.Block ML. NADPH oxidase as a therapeutic target in Alzheimer's disease. BMC Neurosci Suppl. 2008;2:S8. doi: 10.1186/1471-2202-9-S2-S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bogenhagen DF. Repair of mtDNA in vertebrates. Am J Hum Genet. 1999;64:1276–1281. doi: 10.1086/302392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bohr VA. Defective DNA base excision repair in brain from individuals with Alzheimer's disease and amnestic mild cognitive impairment. Nucleic Acids Res. 2007;35:5545–5555. doi: 10.1093/nar/gkm605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bohr VA. Repair of oxidative DNA damage in nuclear and mitochondrial DNA, and some changes with aging in mammalian cells. Free Radic Biol Med. 2002;32:804–812. doi: 10.1016/s0891-5849(02)00787-6. [DOI] [PubMed] [Google Scholar]
- 20.Bokov A. Chaudhuri A. Richardson A. The role of oxidative damage and stress in aging. Mech Ageing Dev. 2004;125:811–826. doi: 10.1016/j.mad.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 21.Bosetti F. Brizzi F. Barogi S, et al. Cytochrome c oxidase and mitochondrial F1F0-ATPase (ATP synthase) activities in platelets and brain from patients with Alzheimer's disease. Neurobiol Aging. 2002;23:371–376. doi: 10.1016/s0197-4580(01)00314-1. [DOI] [PubMed] [Google Scholar]
- 22.Bowling AC. Mutisya EM. Walker LC. Price DL. Cork LC. Beal MH. Age-dependent impairment of mitochondrial function in primate brain. J Neurochem. 1993;60:1964–1967. doi: 10.1111/j.1471-4159.1993.tb13430.x. [DOI] [PubMed] [Google Scholar]
- 23.Brand MD. The sites and topology of mitochondrial superoxide production. Exp Gerontol. 2010;45:466–472. doi: 10.1016/j.exger.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brown MR. Sullivan PG. Geddes JW. Synaptic mitochondria are more susceptible to Ca2+ overload than nonsynaptic mitochondria. J Biol Chem. 2006;28:11658–11668. doi: 10.1074/jbc.M510303200. [DOI] [PubMed] [Google Scholar]
- 25.Bubber P. Haroutunian V. Fisch G. Blass JP. Gibson GE. Mitochondrial abnormalities in Alzheimer brain: mechanistic implications. Ann Neurol. 2005;57:695–703. doi: 10.1002/ana.20474. [DOI] [PubMed] [Google Scholar]
- 26.Buchholz JN. Behringer EJ. Pottorf WJ. Pearce WJ. Vanterpool CK. Age-dependent changes in Ca2+ homeostasis in peripheral neurones: implications for changes in function. Aging Cell. 2007;6:285–296. doi: 10.1111/j.1474-9726.2007.00298.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Budimir A. Metal ions, Alzheimer's disease and chelation therapy. Acta Pharm. 2011;61:1–14. doi: 10.2478/v10007-011-0006-6. [DOI] [PubMed] [Google Scholar]
- 28.Cakatay U. Telci A. Kayalì R. Tekeli F. Akçay T. Sivas A. Relation of oxidative protein damage and nitrotyrosine levels in the aging rat brain. Exp Gerontol. 2001;36:221–229. doi: 10.1016/s0531-5565(00)00197-2. [DOI] [PubMed] [Google Scholar]
- 29.Camandola S. Mattson MP. Aberrant subcellular neuronal calcium regulation in aging and Alzheimer's disease. Biochim Biophys Acta. 2011;1813:965–973. doi: 10.1016/j.bbamcr.2010.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Canevari L. Clark JB. Bates TE. Beta-Amyloid fragment 25–35 selectively decreases complex IV activity in isolated mitochondria. FEBS Lett. 1999;457:131–134. doi: 10.1016/s0014-5793(99)01028-5. [DOI] [PubMed] [Google Scholar]
- 31.Caradonna S. Ladner R. Hansbury M. Kosciuk M. Lynch F. Muller S. Affinity purification and comparative analysis of two distinct human uracil-DNA glycosylases. Exp Cell Res. 1996;222:345–359. doi: 10.1006/excr.1996.0044. [DOI] [PubMed] [Google Scholar]
- 32.Cardoso SM. Proenca MT. Santos S. Santana I. Oliveira CR. Cytochrome c oxidase is decreased in Alzheimer's disease platelets. Neurobiol Aging. 2004;25:105–110. doi: 10.1016/s0197-4580(03)00033-2. [DOI] [PubMed] [Google Scholar]
- 33.Carreras MC. Franco MC. Peralta JG. Poderoso JJ. Nitric oxide, complex I, and the modulation of mitochondrial reactive species in biology and disease. Mol Aspects Med. 2004;25:125–139. doi: 10.1016/j.mam.2004.02.014. [DOI] [PubMed] [Google Scholar]
- 34.Castellani RJ. Rolston RK. Smith MA. Alzheimer disease. Dis Mon. 2010;56:484–546. doi: 10.1016/j.disamonth.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Celsi F. Pizzo P. Brini M. Leo S. Fotino C. Pinton P. Rizzuto R. Mitochondria, calcium and cell death: a deadly triad in neurodegeneration. Biochim Biophys Acta. 2009;1787:335–344. doi: 10.1016/j.bbabio.2009.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chakrabarti S. Munshi S. Banerjee K. Thakurta IG. Sinha M. Bagh MB. Mitochondrial dysfunction during brain aging: role of oxidative stress and modulation by antioxidant supplementation. Aging Dis. 2011;2:242–256. [PMC free article] [PubMed] [Google Scholar]
- 37.Chakraborty H. Ray SN. Chakrabarti S. Lipid peroxidation associated protein damage in rat brain crude synaptosomal fraction mediated by iron and ascorbate. Neurochem Int. 2001;39:311–317. doi: 10.1016/s0197-0186(00)00117-0. [DOI] [PubMed] [Google Scholar]
- 38.Chan DC. Mitochondria: dynamic organelles in disease, aging, and development. Cell. 2006;125:1241–1252. doi: 10.1016/j.cell.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 39.Chen D. Cao G. Hastings T. Feng Y. Pei W. O'Horo C. Chen J. Age-dependent decline of DNA repair activity for oxidative lesions in rat brain mitochondria. J Neurochem. 2002;81:1273–1284. doi: 10.1046/j.1471-4159.2002.00916.x. [DOI] [PubMed] [Google Scholar]
- 40.Chen H. Chan DC. Physiological functions of mitochondrial fusion. Ann NY Acad Sci. 2010;1201:21–25. doi: 10.1111/j.1749-6632.2010.05615.x. [DOI] [PubMed] [Google Scholar]
- 41.Cocco T. Sgobbo P. Clemente M, et al. Tissue-specific changes of mitochondrial functions in aged rats: effect of a long-term dietary treatment with N-acetylcysteine. Free Radic Biol Med. 2005;38:796–805. doi: 10.1016/j.freeradbiomed.2004.11.034. [DOI] [PubMed] [Google Scholar]
- 42.Cooke MS. Evans MD. Dizdaroglu M. Lunec J. Oxidative DNA damage: mechanisms, mutation, and disease. FASEB J. 2003;17:1195–1214. doi: 10.1096/fj.02-0752rev. [DOI] [PubMed] [Google Scholar]
- 43.Coskun PE. Beal MF. Wallace DC. Alzheimer’ s brains harbor somatic mtDNA control-region mutations that suppress mitochondrial transcription and replication. Proc Natl Acad Sci USA. 2004;101:10726–10731. doi: 10.1073/pnas.0403649101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Crompton M. The mitochondrial permeability transition pore and its role in cell death. Biochem J. 1999;341:233–249. [PMC free article] [PubMed] [Google Scholar]
- 45.Curti D. Rognoni F. Gasparini L, et al. Oxidative metabolism in cultured fibroblasts derived from sporadic Alzheimer's disease (AD) patients. Neurosci Lett. 1997;236:13–16. doi: 10.1016/s0304-3940(97)00741-6. [DOI] [PubMed] [Google Scholar]
- 46.de la Monte SM. Luong T. Neely TR. Robinson D. Wands JR. Mitochondrial DNA damage as a mechanism of cell loss in Alzheimer's disease. Lab Invest. 2000;80:1323–1335. doi: 10.1038/labinvest.3780140. [DOI] [PubMed] [Google Scholar]
- 47.de Souza-Pinto NC. Eide L. Hogue BA, et al. Repair of 8-oxodeoxyguanosine lesions in mitochondrial DNA depends on the oxoguanine dna glycosylase (OGG1) gene and 8-oxoguanine accumulates in the mitochondrial dna of OGG1-defective mice. Cancer Res. 2001;61:5378–5381. [PubMed] [Google Scholar]
- 48.De Stefani D. Raffaello A. Teardo E. Szabò I. Rizzuto R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature. 2011;476:336–340. doi: 10.1038/nature10230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Demple B. Sung JS. Molecular and biological roles of Ape1 protein in mammalian base excision repair. DNA Repair (Amst) 2005;4:1442–1449. doi: 10.1016/j.dnarep.2005.09.004. [DOI] [PubMed] [Google Scholar]
- 50.Detmer SA. Chan DC. Functions and dysfunctions of mitochondrial dynamics. Nat Rev Mol Cell Biol. 2007;8:870–879. doi: 10.1038/nrm2275. [DOI] [PubMed] [Google Scholar]
- 51.DiMauro S. Schon EA. Mitochondrial respiratory-chain diseases. N Engl J Med. 2003;348:2656–2668. doi: 10.1056/NEJMra022567. [DOI] [PubMed] [Google Scholar]
- 52.Dimroth P. Kaim G. Matthey U. Crucial role of the membrane potential for ATP synthesis by F(1)F(o) ATP synthases. J Exp Biol. 2000;203:51–59. doi: 10.1242/jeb.203.1.51. [DOI] [PubMed] [Google Scholar]
- 53.Dizdaroglu M. Kirkali G. Jaruga P. Formamidopyrimidines in DNA: mechanisms of formation, repair, and biological effects. Free Radic Biol Med. 2008;45:1610–1621. doi: 10.1016/j.freeradbiomed.2008.07.004. [DOI] [PubMed] [Google Scholar]
- 54.Dizdaroglu M. Base-excision repair of oxidative DNA damage by DNA glycosylases. Mutat Res. 2005;591:45–59. doi: 10.1016/j.mrfmmm.2005.01.033. [DOI] [PubMed] [Google Scholar]
- 55.Dumont M. Beal MF. Neuroprotective strategies involving ROS in Alzheimer disease. Free Radic Biol Med. 2011;51:1014–1026. doi: 10.1016/j.freeradbiomed.2010.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Evans JL. Goldfine ID. Maddux BA. Grodsky GM. Oxidative stress and stress-activated signaling pathways: a unifying hypothesis of type 2 diabetes. Endocr Rev. 2002;23:599–622. doi: 10.1210/er.2001-0039. [DOI] [PubMed] [Google Scholar]
- 57.Fang H. Chen M. Ding Y, et al. Imaging superoxide flash and metabolism-coupled mitochondrial permeability transition in living animals. Cell Res. 2011;21:1295–1304. doi: 10.1038/cr.2011.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ferrándiz ML. Martínez M. Juan ED. Díez A. Bustos G. Miquel J. Impairment of mitochondrial oxidative phosphorylation in the brain of aged mice. Brain Res. 1994;644:335–338. doi: 10.1016/0006-8993(94)91699-3. [DOI] [PubMed] [Google Scholar]
- 59.Filosto M. Scarpelli M. Cotelli MS, et al. The role of mitochondria in neurodegenerative diseases. J Neurol. 2011;258:1763–1774. doi: 10.1007/s00415-011-6104-z. [DOI] [PubMed] [Google Scholar]
- 60.Finefrock AE. Bush AI. Doraiswamy PM. Current status of metals as therapeutic targets. J am Geriatr Soc. 2003;51:1143–1148. doi: 10.1046/j.1532-5415.2003.51368.x. [DOI] [PubMed] [Google Scholar]
- 61.Floyd RA. Hensley K. Oxidative stress in brain aging. Implications for therapeutics of neurodegenerative diseases. Neurobiol Aging. 2002;23:795–807. doi: 10.1016/s0197-4580(02)00019-2. [DOI] [PubMed] [Google Scholar]
- 62.Fung H. Kow YW. Van Houten B, et al. Asbestos increases mammalian AP-endonuclease gene expression, protein levels, and enzyme activity in mesothelial cells. Cancer Res. 1998;58:189–194. [PubMed] [Google Scholar]
- 63.Gabbita SP. Lovell MA. Markesbery WR. Increased nuclear DNA oxidation in the brain in Alzheimer's disease. J Neurochem. 1998;71:2034–2040. doi: 10.1046/j.1471-4159.1998.71052034.x. [DOI] [PubMed] [Google Scholar]
- 64.Gallagher JJ. Finnegan ME. Grehan B. Dobson J. Collingwood JF. Lynch MA. Modest amyloid deposition is associated with iron dysregulation, microglial activation, and oxidative stress. J Alzheimers Dis. 2012;28:147–161. doi: 10.3233/JAD-2011-110614. [DOI] [PubMed] [Google Scholar]
- 65.Gao HM. Zhou H. Hong JS. NADPH oxidases: novel therapeutic targets for neurodegenerative diseases. Trends Pharmacol Sci. 2012;33:295–303. doi: 10.1016/j.tips.2012.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gemma C. Mesches MH. Sepesi B. Choo K. Holmes DB. Bickford PC. Diets enriched in foods with high antioxidant activity reverse age-induced decreases in cerebellar β-adrenergic function and increases in proinflammatory cytokines. J Neurosci. 2002;22:6114–6120. doi: 10.1523/JNEUROSCI.22-14-06114.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gilmer LK. Ansari MA. Roberts KN. Scheff SW. Age-related changes in mitochondrial respiration and oxidative damage in the cerebral cortex of the Fischer 344 rat. Mech Ageing Dev. 2010;131:133–143. doi: 10.1016/j.mad.2009.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gredilla R. Garm C. Holm R. Bohr VA. Stevnsner T. Differential age-related changes in mitochondrial DNA repair activities in mouse brain regions. Neurobiol Aging. 2010;31:993–1002. doi: 10.1016/j.neurobiolaging.2008.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gredilla R. Weissman L. Yang JL. Bohr VA. Stevnsner T. Mitochondrial base excision repair in mouse synaptosomes during normal aging and in a model of Alzheimer's disease. Neurobiol Aging. 2012;33:694–707. doi: 10.1016/j.neurobiolaging.2010.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gredilla R. DNA damage and base excision repair in mitochondria and their role in aging. J Aging Res. 2010;2011:257093. doi: 10.4061/2011/257093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Green K. Brand MD. Murphy MP. Prevention of mitochondrial oxidative damage as a therapeutic strategy in diabetes. Diabetes. 2004;53(Suppl 1):S110–S118. doi: 10.2337/diabetes.53.2007.s110. [DOI] [PubMed] [Google Scholar]
- 72.Guidi I. Galimberti D. Lonati S. Novembrino C, et al. Oxidative imbalance in patients with mild cognitive impairment and Alzheimer's disease. Neurobiol Aging. 2006;27:262–269. doi: 10.1016/j.neurobiolaging.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 73.Hamanaka RB. Chandel NS. Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes. Trends Biochem Sci. 2010;35:505–513. doi: 10.1016/j.tibs.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hansen AB. Griner NB. Anderson JP. Kujoth GC. Prolla TA. Loeb LA. Glick E. Mitochondrial DNA integrity is not dependent on DNA polymerase-beta activity. DNA Repair (Amst) 2006;5:71–79. doi: 10.1016/j.dnarep.2005.07.009. [DOI] [PubMed] [Google Scholar]
- 75.Harman D. Aging: a theory based on free radical and radiation chemistry. J Gerontol. 1956;11:298–300. doi: 10.1093/geronj/11.3.298. [DOI] [PubMed] [Google Scholar]
- 76.Harman D. The free radical theory of aging. Antioxid Redox Signal. 2003;5:57–56. doi: 10.1089/152308603770310202. [DOI] [PubMed] [Google Scholar]
- 77.Hazra TK. Izumi T. Boldogh I, et al. Identification and characterization of a human DNA glycosylase for repair of modified bases in oxidatively damaged DNA. Proc Natl Acad Sci U S A. 2002;99:3523–3528. doi: 10.1073/pnas.062053799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Head E. Liu J. Hagen TM. Muggenburg BA. Milgram NW. Ames BN. Cotman CW. Oxidative damage increases with age in a canine model of human brain aging. J Neurochem. 2002;82:375–381. doi: 10.1046/j.1471-4159.2002.00969.x. [DOI] [PubMed] [Google Scholar]
- 79.Hegde ML. Hegde PM. Holthauzen LM. Hazra TK. Rao KS. Mitra S. Specific Inhibition of NEIL-initiated repair of oxidized base damage in human genome by copper and iron: potential etiological linkage to neurodegenerative diseases. J Biol Chem. 2010;285:28812–28825. doi: 10.1074/jbc.M110.126664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hengartner MO. The biochemistry of apoptosis. Nature. 2000;407:770–776. doi: 10.1038/35037710. [DOI] [PubMed] [Google Scholar]
- 81.Hensley K. Hall N. Subramaniam R, et al. Brain regional correspondence between Alzheimer's disease histopathology and biomarkers of protein oxidation. J Neurochem. 1995;65:2146–2156. doi: 10.1046/j.1471-4159.1995.65052146.x. [DOI] [PubMed] [Google Scholar]
- 82.Hoeijmakers JH. Genome maintenance mechanisms for preventing cancer. Nature. 2001;411:366–374. doi: 10.1038/35077232. [DOI] [PubMed] [Google Scholar]
- 83.Howell N. Elson JL. Chinnery PF. Turnbull DM. mtDNA mutations and common neurodegenerative disorders. Trends Genet. 2005;11:583–586. doi: 10.1016/j.tig.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 84.Hu J. de Souza-Pinto NC. Haraguchi K, et al. Repair of formamidopyrimidines in DNA involves different glycosylases: role of the OGG1, NTH1, and NEIL1 enzymes. J Biol Chem. 2005;280:40544–40551. doi: 10.1074/jbc.M508772200. [DOI] [PubMed] [Google Scholar]
- 85.Huang X. Atwood CS. Hartshorn MA, et al. The A beta peptide of Alzheimer's disease directly produces hydrogen peroxide through metal ion reduction. Biochem. 1999;38:7609–7616. doi: 10.1021/bi990438f. [DOI] [PubMed] [Google Scholar]
- 86.Huang X. Moir RD. Tanzi RE. Bush AI. Rogers JT. Redox-active metals, oxidative stress, and Alzheimer's disease pathology. Ann N Y Acad Sci. 2004;1012:153–163. doi: 10.1196/annals.1306.012. [DOI] [PubMed] [Google Scholar]
- 87.Huffman JL. Sundheim O. Tainer JA. DNA base damage recognition and removal: new twists and grooves. Mutat Res. 2005;577:55–76. doi: 10.1016/j.mrfmmm.2005.03.012. [DOI] [PubMed] [Google Scholar]
- 88.Humpel C. Marksteiner J. Cerebrovascular damage as a cause for Alzheimer's disease. Curr Neurovasc Res. 2005;2:341–347. doi: 10.2174/156720205774322610. [DOI] [PubMed] [Google Scholar]
- 89.Ide H. Kotera M. Human DNA glycosylases involved in the repair of oxidatively damaged DNA. Biol Pharm Bull. 2004;27:480–485. doi: 10.1248/bpb.27.480. [DOI] [PubMed] [Google Scholar]
- 90.Imam SZ. Karahalil B. Hogue BA. Souza-Pinto NC. Bohr VA. Mitochondrial and nuclear DNA-repair capacity of various brain regions in mouse is altered in an age-dependent manner. Neurobiol Aging. 2006;27:1129–1136. doi: 10.1016/j.neurobiolaging.2005.06.002. [DOI] [PubMed] [Google Scholar]
- 91.Jeppesen DK. Bohr VA. Stevnsner T. DNA repair deficiency in neurodegeneration. Prog Neurobiol. 2011;94:166–200. doi: 10.1016/j.pneurobio.2011.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Jomova K. Valko M. Advances in metal-induced oxidative stress and human disease. Toxicol. 2011;283:65–87. doi: 10.1016/j.tox.2011.03.001. [DOI] [PubMed] [Google Scholar]
- 93.Kaguni LS. DNA polymerase gamma, the mitochondrial replicase. Annu Rev Biochem. 2004;73:293–320. doi: 10.1146/annurev.biochem.72.121801.161455. [DOI] [PubMed] [Google Scholar]
- 94.Karahalil B. de Souza-Pinto NC. Parsons JL. Elder RH. Bohr VA. Compromised incision of oxidized pyrimidines in liver mitochondria of mice deficient in NTH1 and OGG1 glycosylases. J Biol Chem. 2003;278:33701–33707. doi: 10.1074/jbc.M301617200. [DOI] [PubMed] [Google Scholar]
- 95.Kasai H. Nishimura S. Hydroxylation of deoxyguanosine at the C-8 position by ascorbic acid and other reducing agents. Nucleic Acids Res. 1984;12:2137–2145. doi: 10.1093/nar/12.4.2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kish SJ. Bergeron C. Rajput A. Brain cytochrome oxidase in Alzheimer's disease. J Neurochem. 1992;59:776–779. doi: 10.1111/j.1471-4159.1992.tb09439.x. [DOI] [PubMed] [Google Scholar]
- 97.Klungland A. Rosewell I. Hollenbach S. Larsen E. Daly G. Epe B. Seeberg E. Lindahl T. Barnes DE. Accumulation of premutagenic DNA lesions in mice defective in removal of oxidative base damage. Proc Natl Acad Sci U S A. 1999;96:13300–13305. doi: 10.1073/pnas.96.23.13300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kraytsberg Y. Kudryavtseva E. McKee AC. Geula C. Kowall NW. Khrapko K. Mitochondrial DNA deletions are abundant and cause functional impairment in aged human substantia nigra neurons. Nat Genet. 2006;38:518–520. doi: 10.1038/ng1778. [DOI] [PubMed] [Google Scholar]
- 99.Krokan HE. Nilsen H. Skorpen F. Otterlei M. Slupphaug G. Base excision repair of DNA in mammalian cells. FEBS Lett. 2000;476:73–77. doi: 10.1016/s0014-5793(00)01674-4. [DOI] [PubMed] [Google Scholar]
- 100.Kruman II. Wersto RP. Cardozo-Pelaez F. Smilenov L. Chan SL. Chrest FJ. Emokpae R., Jr Gorospe M. Mattson MP. Cell cycle activation linked to neuronal cell death initiated by DNA damage. Neuron. 2004;41:549–561. doi: 10.1016/s0896-6273(04)00017-0. [DOI] [PubMed] [Google Scholar]
- 101.Lauritzen KH. Cheng C. Wiksen H. Bergersen LH. Klungland A. Mitochondrial DNA toxicity compromises mitochondrial dynamics and induces hippocampal antioxidant defenses. DNA Repair (Amst) 2011;10:639–653. doi: 10.1016/j.dnarep.2011.04.011. [DOI] [PubMed] [Google Scholar]
- 102.Lauritzen KH. Moldestad O. Eide L, et al. Mitochondrial DNA toxicity in forebrain neurons causes apoptosis, neurodegeneration, and impaired behavior. Mol Cell Biol. 2010;30:1357–1367. doi: 10.1128/MCB.01149-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lavados M. Guillon M. Mujica MC. Rojo LE. Fuentes P. Maccioni RB. Mild cognitive impairment and Alzheimer patients display different levels of redox-active CSF iron. J Alzheimers Dis. 2008;13:225–232. doi: 10.3233/jad-2008-13211. [DOI] [PubMed] [Google Scholar]
- 104.Linnane AW. Marzuki S. Ozawa T. Tanaka M. Mitochondrial DNA mutations as an important contributor to ageing and degenerative diseases. Lancet. 1989;1:642–645. doi: 10.1016/s0140-6736(89)92145-4. [DOI] [PubMed] [Google Scholar]
- 105.Liu P. Qian L. Sung JS, et al. Removal of oxidative DNA damage via FEN1-dependent long-patch base excision repair in human cell mitochondria. Mol Cell Biol. 2008;28:4975–4987. doi: 10.1128/MCB.00457-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lovell MA. Ehmann WD. Butler SM. Markesbery WR. Elevated thiobarbituric acid-reactive substances and antioxidant enzyme activity in the brain in Alzheimer's disease. Neurology. 1995;45:1594–1601. doi: 10.1212/wnl.45.8.1594. [DOI] [PubMed] [Google Scholar]
- 107.Lovell MA. Ehmann WD. Mattson MP. Markesbery WR. Elevated 4-hydroxynonenal in ventricular fluid in Alzheimer's disease. Neurobiol Aging. 1997;18:457–461. doi: 10.1016/s0197-4580(97)00108-5. [DOI] [PubMed] [Google Scholar]
- 108.Lovell MA. Gabbita SP. Markesbery WR. Increased DNA oxidation and decreassed levels of repair products in Alzheimer's disease ventricular CSF. J Neurochem. 1999;72:771–776. doi: 10.1046/j.1471-4159.1999.0720771.x. [DOI] [PubMed] [Google Scholar]
- 109.Lu J. Wang K. Rodova M, et al. Polymorphic variation in cytochrome oxidase subunit genes. J Alzheimers Dis. 2010;21:141–154. doi: 10.3233/JAD-2010-100123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Luczaj W. Skrzydlewska E. DNA damage caused by lipid peroxidation products. Cell Mol Biol Lett. 2003;8:391–413. [PubMed] [Google Scholar]
- 111.Lustbader JW. Cirilli M. Lin C, et al. Abeta to mitochondrial toxicity in Alzheimer's disease. Science. 2004;304:448–452. doi: 10.1126/science.1091230. [DOI] [PubMed] [Google Scholar]
- 112.Lyras L. Cairns NJ. Jenner A. Jenner P. Halliwell B. An assessment of oxidative damage to proteins, lipids, and DNA in brain from patients with Alzheimer's disease. J Neurochem. 1997;68:2061–2069. doi: 10.1046/j.1471-4159.1997.68052061.x. [DOI] [PubMed] [Google Scholar]
- 113.Maki H. Origins of spontaneous mutations: specificity and directionality of base substitution, frameshift, and sequence-substitution mutageneses. Annu Rev Genet. 2002;36:279–303. doi: 10.1146/annurev.genet.36.042602.094806. [DOI] [PubMed] [Google Scholar]
- 114.Malins DC. Hellstrom KE. Anderson KM. Johnson PM. Vinson MA. Antioxidant-induced changes in oxidized DNA. Proc Natl Acad Sci U S A. 2002;99:5937–5941. doi: 10.1073/pnas.082111199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Mancuso M. Calsolaro V. Orsucci D. Carlesi C. Choub A. Piazza S. Siciliano G. Mitochondria, cognitive impairment, and Alzheimer's disease. Int J Alzheimers Dis. 2009;2009:951548. doi: 10.4061/2009/951548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Mannella CA. Structural diversity of mitochondria: functional implications. Ann N Y Acad Sci. 2008;1147:171–179. doi: 10.1196/annals.1427.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Mao P. Reddy PH. Aging and amyloid beta-induced oxidative DNA damage and mitochondrial dysfunction in Alzheimer's disease: implications for early intervention and therapeutics. Biochim Biophys Acta. 2011;1812:1359–1370. doi: 10.1016/j.bbadis.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Marcus DL. Thomas K. Rodriguez C, et al. Increased peroxidation and reduced antioxidant enzyme activity in Alzheimer's disease. Exp Neurol. 1998;150:40–44. doi: 10.1006/exnr.1997.6750. [DOI] [PubMed] [Google Scholar]
- 119.Markesbery WR. Lovell MA. Four-hydroxynonenal, a product of lipid peroxidation, is increased in the brain in Alzheimer's disease. Neurobiol Aging. 1998;19:33–36. doi: 10.1016/s0197-4580(98)00009-8. [DOI] [PubMed] [Google Scholar]
- 120.Markesbery WR. Oxidative stress hypothesis in Alzheimer's disease. Free Radic Biol Med. 1997;23:134–147. doi: 10.1016/s0891-5849(96)00629-6. [DOI] [PubMed] [Google Scholar]
- 121.Mastrogiacomo F. Bergeron C. Kish SJ. Brain alpha-ketoglutarate dehydrogenase complex activity in Alzheimer's disease. J Neurochem. 1993;61:2007–2014. doi: 10.1111/j.1471-4159.1993.tb07436.x. [DOI] [PubMed] [Google Scholar]
- 122.Mattson MP. Metal-catalyzed disruption of membrane protein and lipid signaling in the pathogenesis of neurodegenerative disorders. Ann N Y Acad Sci. 2004;1012:37–50. doi: 10.1196/annals.1306.004. [DOI] [PubMed] [Google Scholar]
- 123.Mecocci P. Beal MF. Cecchetti R, et al. Mitochondrial membrane fluidity and oxidative damage to mitochondrial DNA in aged and AD human brain. Mol Chem Neuropathol. 1997;31:53–64. doi: 10.1007/BF02815160. [DOI] [PubMed] [Google Scholar]
- 124.Mecocci P. MacGarvey U. Beal MF. Oxidative damage to mitochondrial DNA is increased in Alzheimer's disease. Ann Neurol. 1994;36:747–751. doi: 10.1002/ana.410360510. [DOI] [PubMed] [Google Scholar]
- 125.Melov S. Schneider JA. Day BJ, et al. A novel neurological phenotype in mice lacking mitochondrial manganese superoxide dismutase. Nat Genet. 1998;18:159–163. doi: 10.1038/ng0298-159. [DOI] [PubMed] [Google Scholar]
- 126.Milton RH, et al. CLIC1 function is required for beta-amyloid induced generation of reactive oxygen species by microglia. J Neurosci. 2008;28:11488–11499. doi: 10.1523/JNEUROSCI.2431-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Moreira PI. Carvalho C. Zhu X. Smith MA. Perry G. Mitochondrial dysfunction is a trigger of Alzheimer's disease pathophysiology. Biochim Biophys Acta. 2010;1802:2–10. doi: 10.1016/j.bbadis.2009.10.006. [DOI] [PubMed] [Google Scholar]
- 128.Moreira PI. Nunomura A. Nakamura M, et al. Nucleic acid oxidation in Alzheimer disease. Free Radic Biol Med. 2008;44:1493–1505. doi: 10.1016/j.freeradbiomed.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 129.Moreira PI. Zhu X. Wang X, et al. Mitochondria: a therapeutic target in neurodegeneration. Biochim Biophys Acta. 2010;1802:212–220. doi: 10.1016/j.bbadis.2009.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Moreira PI. Alzheimer's disease and diabetes: an integrative view of the role of mitochondria, oxidative stress, and insulin. J Alzheimers Dis. 2012;30:199–215. doi: 10.3233/JAD-2011-111127. [DOI] [PubMed] [Google Scholar]
- 131.Moreira PI. Duarte AI. Santos MS. Rego AC. Oliveira CR. An integrative view of the role of oxidative stress, mitochondria and insulin in Alzheimer's disease. J Alzheimers Dis. 2009;16:741–761. doi: 10.3233/JAD-2009-0972. [DOI] [PubMed] [Google Scholar]
- 132.Morland I. Rolseth V. Luna L. Rognes T. Bjoras M. Seeberg E. Human DNA glycosylases of the bacterial Fpg/MutM superfamily: an alternative pathway for the repair of 8-oxoguanine and other oxidation products in DNA. Nucleic Acids Res. 2002;30:4926–4936. doi: 10.1093/nar/gkf618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417:1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Naga KK. Sullivan PG. Geddes JW. High cyclophilin D content of synaptic mitochondria results in increased vulnerability to permeability transition. J Neurosci. 2007;27:7469–7475. doi: 10.1523/JNEUROSCI.0646-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Neiman M. Taylor DR. The causes of mutation accumulation in mitochondrial genomes. Proc Biol Sci. 2009;276:1201–1209. doi: 10.1098/rspb.2008.1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Nilsen H. Otterlei M. Haug T, et al. Nuclear and mitochondrial uracil-DNA glycosylases are generated by alternative splicing and transcription from different positions in the UNG gene. Nucleic Acids Res. 1997;25:750–755. doi: 10.1093/nar/25.4.750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Nishioka K. Ohtsubo T. Oda H. Fujiwara T. Kang D. Sugimachi K. Nakabeppu Y. Expression and differential intracellular localization of two major forms of human 8-oxoguanine DNA glycosylase encoded by alternatively spliced OGG1 mRNAs. Mol Biol Cell. 1999;10:1637–1652. doi: 10.1091/mbc.10.5.1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Nonaka I. Mitochondrial diseases. Curr Opin Neurol Neurosurg. 1992;5:622–632. [PubMed] [Google Scholar]
- 139.Nunomura A. Honda K. Takeda A. Hirai K. Zhu X. Smith MA. Perry G. Oxidative damage to RNA in neurodegenerative diseases. J Biomed Biotechnol. 2006;2006:82323. doi: 10.1155/JBB/2006/82323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Nunomura A. Moreira PI. Castellani RJ. Lee HG. Zhu X. Smith MA. Perry G. Oxidative damage to RNA in aging and neurodegenerative disorders. Neurotox Res. 2012;22:231–248. doi: 10.1007/s12640-012-9331-x. [DOI] [PubMed] [Google Scholar]
- 141.Nunomura A. Perry G. Aliev G. Hirai K. Takeda A. Balraj EK, et al. Oxidative damage is the earliest event in Alzheimer's disease. J Neuropathol Exp Neurol. 2001;60:759–767. doi: 10.1093/jnen/60.8.759. [DOI] [PubMed] [Google Scholar]
- 142.Nunomura A. Perry G. Pappolla MA. Wade R. Hirai K. Chiba S, et al. RNA oxidation is a prominent feature of vulnerable neurons in Alzheimer's disease. J Neurosci. 1999;19:1959–1964. doi: 10.1523/JNEUROSCI.19-06-01959.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.O'Connor TR. Boiteux S. Laval J. Ring-opened 7-methylguanine residues in DNA are a block to in vitro DNA synthesis. Nucleic Acids Res. 1988;16:5879–5894. doi: 10.1093/nar/16.13.5879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Ojaimi J. Masters CL. Opeskin K. Mckelvie P. Byrne E. Mitochondrial respiratory chain activity in the human brain as a function of age. Mech Ageing Dev. 1999;111:39–47. doi: 10.1016/s0047-6374(99)00071-8. [DOI] [PubMed] [Google Scholar]
- 145.Park L. Zhou P. Pitstick R, et al. Nox2-derived radicals contribute to neurovascularand behavioral dysfunction in mice overexpressing the amyloid precursor protein. Proc Natl Acad Sci USA. 2008;105:1347–1352. doi: 10.1073/pnas.0711568105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Parker WD., Jr Filley CM. Parks JK. Cytochrome oxidase deficiency in Alzheimer's disease. Neurol. 1990;40:1302–1303. doi: 10.1212/wnl.40.8.1302. [DOI] [PubMed] [Google Scholar]
- 147.Patten DA. Germain M. Kelly MA. Slack RS. Reactive oxygen species: stuck in the middle of neurodegeneration. J Alzheimer's Dis. 2010;20:S357–S367. doi: 10.3233/JAD-2010-100498. [DOI] [PubMed] [Google Scholar]
- 148.Praticò D. Lee VMY. Trojanowski JQ. Rokach J. Fitzgerald GA. Increased F2-isoprostanes in Alzheimer's disease: evidence for enhanced lipid peroxidation in vivo. FASEB J. 1998;12:1777–1783. doi: 10.1096/fasebj.12.15.1777. [DOI] [PubMed] [Google Scholar]
- 149.Querfurth HW. LaFerla FM. Alzheimer's disease. N Engl J Med. 2010;362:329–344. doi: 10.1056/NEJMra0909142. [DOI] [PubMed] [Google Scholar]
- 150.Rasola A. Bernardi P. Mitochondrial permeability transition in Ca(2+)-dependent apoptosis and necrosis. Cell Calcium. 2011;50:222–233. doi: 10.1016/j.ceca.2011.04.007. [DOI] [PubMed] [Google Scholar]
- 151.Rivkees SA. Kelley MR. Expression of a multifunctional DNA repair enzyme gene, apurinic/apyrimidinic endonuclease (APE; Ref-1) in the suprachiasmatic, supraoptic and paraventricular nuclei. Brain Res. 1994;666:137–142. doi: 10.1016/0006-8993(94)90296-8. [DOI] [PubMed] [Google Scholar]
- 152.Rizzuto R. Bernardi P. Pozzan T. Mitochondria as all-round players of the calcium game. J Physiol. 2000;529:37–47. doi: 10.1111/j.1469-7793.2000.00037.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Rizzuto R. Pinton P. Brini M. Chiesa A. Filippin L. Pozzan T. Mitochondria as biosensors of calcium microdomains. Cell Calcium. 1999;26:193–199. doi: 10.1054/ceca.1999.0076. [DOI] [PubMed] [Google Scholar]
- 154.Sabatini BL. Maravall M. Svoboda K. Ca(2+) signaling in dendritic spines. Curr Opin Neurobiol. 2001;11:349–356. doi: 10.1016/s0959-4388(00)00218-x. [DOI] [PubMed] [Google Scholar]
- 155.Santos RX. Correia SC. Wang X, et al. Alzheimer's disease: diverse aspects of mitochondrial malfunctioning. Int J Clin Exp Pathol. 2010;3:570–581. [PMC free article] [PubMed] [Google Scholar]
- 156.Santos RX. Correia SC. Zhu X, et al. Nuclear and mitochondrial DNA oxidation in Alzheimer's disease. Free Radic Res. 2012;6:565–576. doi: 10.3109/10715762.2011.648188. [DOI] [PubMed] [Google Scholar]
- 157.Sayre LM. Perry G. Harris PL. Liu Y. Schubert KA. Smith MA. In situ oxidative catalysis by neurofibrillary tangles and senile plaques in Alzheimer's disease: a central role for bound transition metals. J Neurochem. 2000;74:270–279. doi: 10.1046/j.1471-4159.2000.0740270.x. [DOI] [PubMed] [Google Scholar]
- 158.Sayre LM. Zelasko DA. Harris PL, et al. 4-Hydroxynonenal-derived advanced lipid peroxidation end products are increased in Alzheimer's disease. J Neurochem. 1997;68:2092–2097. doi: 10.1046/j.1471-4159.1997.68052092.x. [DOI] [PubMed] [Google Scholar]
- 159.Scherz-Shouval R. Elazar Z. Regulation of autophagy by ROS: physiology and pathology. Trends Biochem Sci. 2011;36:30–98. doi: 10.1016/j.tibs.2010.07.007. [DOI] [PubMed] [Google Scholar]
- 160.Sen T. Sen N. Jana S. Khan FH. Chatterjee U. Chakrabarti S. Depolarization and cardiolipin depletion in aged rat brain mitochondria: relationship with oxidative stress and electron transport chain activity. Neurochem Int. 2007;50:719–722. doi: 10.1016/j.neuint.2007.01.007. [DOI] [PubMed] [Google Scholar]
- 161.Shao C. Xiong S. Li GM, et al. Altered 8-oxoguanine glycosylase in mild cognitive impairment and late-stage Alzheimer's disease brain. Free Radic Biol Med. 2008;45:813–819. doi: 10.1016/j.freeradbiomed.2008.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Sheu KF. Kim YT. Blass JP, et al. An immunochemical study of the pyruvate dehydrogenase deficit in Alzheimer's disease brain. Ann Neurol. 1985;17:444–449. doi: 10.1002/ana.410170505. [DOI] [PubMed] [Google Scholar]
- 163.Shigenaga MK. Hagen TM. Ames BN. Oxidative damage and mitochondrial decay in aging. Proc Natl Acad Sci U S A. 1994;91:10771–10778. doi: 10.1073/pnas.91.23.10771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Simon DK. Lin MT. Zheng L, et al. Somatic mitochondrial DNA mutations in cortex and substantia nigra in aging and Parkinson's disease. Neurobiol Aging. 2004;25:71–81. doi: 10.1016/s0197-4580(03)00037-x. [DOI] [PubMed] [Google Scholar]
- 165.Sinclair DA. Toward a unified theory of caloric restriction and longevity regulation. Mech Ageing Dev. 2005;126:987–1002. doi: 10.1016/j.mad.2005.03.019. [DOI] [PubMed] [Google Scholar]
- 166.Smith CD. Carney JM. Starke-Reed PE, et al. Excess brain protein oxidation and enzyme dysfunction in normal aging and in Alzheimer disease. Proc Natl Acad Sci USA. 1991;88:10540–10543. doi: 10.1073/pnas.88.23.10540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Smith MA. Richey Harris PL. Sayre LM. Beckman JS. Perry G. Widespread peroxynitrite-mediated damage in Alzheimer's disease. J Neurosci. 1997;17:2653–2657. doi: 10.1523/JNEUROSCI.17-08-02653.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Smith MA. Zhu X. Tabaton M, et al. Increased iron and free radical generation in preclinical Alzheimer disease and mild cognitive impairment. J Alzheimers Dis. 2010;19:363–372. doi: 10.3233/JAD-2010-1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Soderling TR. CaM-kinases: modulators of synaptic plasticity. Curr Opin Neurobiol. 2000;10:375–380. doi: 10.1016/s0959-4388(00)00090-8. [DOI] [PubMed] [Google Scholar]
- 170.Sondheimer N. Glatz CE. Tirone JE. Deardorff MA. Krieger AM. Hakonarson H. Neutral mitochondrial heteroplasmy and the influence of aging. Hum Mol Genet. 2011;20:1653–1659. doi: 10.1093/hmg/ddr043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Starkov AA. Fiskum G. Chinopoulos C, et al. Mitochondrial α-ketoglutarate dehydrogenase complex generates reactive oxygen species. J Neurosci. 2004;24:7779–7788. doi: 10.1523/JNEUROSCI.1899-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Steenken S. Purine bases, nucleosides, and nucleotides: aqueous solution redox chemistry and transformation reactions of their radical cations and e- and OH adducts. Chem Rev. 1989;89:503–520. [Google Scholar]
- 173.Stierum RH. Croteau DL. Bohr VA. Purification and characterization of a mitochondrial thymine glycol endonuclease from rat liver. J Biol Chem. 1999;274:7128–7136. doi: 10.1074/jbc.274.11.7128. [DOI] [PubMed] [Google Scholar]
- 174.Stierum RH. Dianov GL. Bohr VA. Single-nucleotide patch base excision repair of uracil in DNA by mitochondrial protein extracts. Nucleic Acids Res. 1999;27:3712–3719. doi: 10.1093/nar/27.18.3712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Stowe DF. Camara AK. Mitochondrial reactive oxygen species production in excitable cells: modulators of mitochondrial and cell function. Antioxid Redox Signal. 2009;11:1373–1414. doi: 10.1089/ars.2008.2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Straface E. Matarrese P. Gambardella L, et al. Oxidative imbalance and cathepsin D changes as peripheral blood biomarkers of Alzheimer disease: A pilot study. FEBS Lett. 2005;579:2759–2766. doi: 10.1016/j.febslet.2005.03.094. [DOI] [PubMed] [Google Scholar]
- 177.Subba Rao K. Mechanisms of disease: DNA repair defects and neurological disease. Nat Clin Pract Neurol. 2007;3:162–172. doi: 10.1038/ncpneuro0448. [DOI] [PubMed] [Google Scholar]
- 178.Suzuki YJ. Forman HJ. Sevanian A. Oxidants as stimulators of signal transduction. Free Radic Biol Med. 1997;22:269–285. doi: 10.1016/s0891-5849(96)00275-4. [DOI] [PubMed] [Google Scholar]
- 179.Swerdlow RH. Khan SM. A “mitochondrial cascade hypothesis” for sporadic Alzheimer's disease. Med Hypotheses. 2004;63:8–20. doi: 10.1016/j.mehy.2003.12.045. [DOI] [PubMed] [Google Scholar]
- 180.Swerdlow RH. Khan SM. The Alzheimer's disease mitochondrial cascade hypothesis: an update. Exp Neurol. 2009;218:308–315. doi: 10.1016/j.expneurol.2009.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Swerdlow RH. Brain aging, Alzheimer's disease, and mitochondria. Biochim Biophys Acta. 2011;1812:1630–1639. doi: 10.1016/j.bbadis.2011.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Szabo C. Multiple pathways of peroxynitrite cytotoxicity. Toxicol Lett. 2003;140–141:105–112. doi: 10.1016/s0378-4274(02)00507-6. [DOI] [PubMed] [Google Scholar]
- 183.Szczesny B. Bhakat KK. Mitra S. Boldogh I. Age-dependent modulation of DNA repair enzymes by covalent modification and subcellular distribution. Mech Ageing Dev. 2004;125:755–765. doi: 10.1016/j.mad.2004.07.005. [DOI] [PubMed] [Google Scholar]
- 184.Szczesny B. Tann AW. Longley MJ. Copeland WC. Mitra S. Long patch base excision repair in mammalian mitochondrial genomes. J Biol Chem. 2008;283:26349–26356. doi: 10.1074/jbc.M803491200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Takao M. Aburatani H. Kobayashi K. Yasui A. Mitochondrial targeting of human DNA glycosylases for repair of oxidative DNA damage. Nucleic Acids Res. 1998;26:2917–2922. doi: 10.1093/nar/26.12.2917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Takuma K. Fang F. Zhang W, et al. RAGE-mediated signaling contributes to intraneuronal transport of amyloid-beta and neuronal dysfunction. Proc Natl Acad Scu USA. 2009;106:20021–20026. doi: 10.1073/pnas.0905686106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Takuma K. Yao J. Huang J, et al. ABAD enhances Abeta-induced cell stress via mitochondrial dysfunction. FASEB J. 2005;19:597–598. doi: 10.1096/fj.04-2582fje. [DOI] [PubMed] [Google Scholar]
- 188.Tang S. Huang T. Characterization of mitochondrial DNA heteroplasmy using a parallel sequencing system. Biotechniques. 2010;48:287–296. doi: 10.2144/000113389. [DOI] [PubMed] [Google Scholar]
- 189.Taylor RC. Cullen SP. Martin SJ. Apoptosis: controlled demolition at the cellular level. Nat Rev Mol Cell Biol. 2008;9:231–241. doi: 10.1038/nrm2312. [DOI] [PubMed] [Google Scholar]
- 190.Torreilles F. Salman-Tabcheh S. Guerin M. Torreilles J. Neurodegenerative disorders: the role of peroxynitrite. Brain Res Rev. 1999;30:153–163. doi: 10.1016/s0165-0173(99)00014-4. [DOI] [PubMed] [Google Scholar]
- 191.Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol. 2003;552:335–344. doi: 10.1113/jphysiol.2003.049478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Valko M. Leibfritz D. Moncol J. Cronin MT. Mazur M. Telser J. Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol. 2007;39:44–84. doi: 10.1016/j.biocel.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 193.Valla J. Schneider L. Niedzielko T, et al. Impaired platelet mitochondrial activity in Alzheimer's disease and mild cognitive impairment. Mitochondrion. 2006;6:323–330. doi: 10.1016/j.mito.2006.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Virag L. Szabo E. Gergely P. Szabo C. Peroxynitrite-induced cytotoxicity: mechanism and opportunities for intervention. Toxicol Lett. 2003;140–141:113–124. doi: 10.1016/s0378-4274(02)00508-8. [DOI] [PubMed] [Google Scholar]
- 195.Wallace DC. Stugard C. Murdock D. Schurr T. Brown MD. Ancient mtDNA sequences in the human nuclear genome: a potential source of errors in identifying pathogenic mutations. Proc Natl Acad Sci USA. 1997;94:14900–14905. doi: 10.1073/pnas.94.26.14900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Wallace DC. Animal models for mitochondrial disease. Methods Mol Biol. 2002;197:3–54. doi: 10.1385/1-59259-284-8:003. [DOI] [PubMed] [Google Scholar]
- 197.Wang J. Xiong S. Xie C. Markesbery WR. Lovell MA. Increased oxidative damage in nuclear and mitochondrial DNA in Alzheimer's disease. J Neurochem. 2005;93:953–962. doi: 10.1111/j.1471-4159.2005.03053.x. [DOI] [PubMed] [Google Scholar]
- 198.Wang W. Fang H. Groom L, et al. Superoxide Flashes in Single Mitochondria. Cell. 2008;134:279–290. doi: 10.1016/j.cell.2008.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Weissman L. de Souza-Pinto NC. Stevnsner T. Bohr VA. DNA repair, mitochondria, and neurodegeneration. Neurosci. 2007;145:1318–1329. doi: 10.1016/j.neuroscience.2006.08.061. [DOI] [PubMed] [Google Scholar]
- 200.Wilkinson B, et al. Fibrillar beta-amyloid-stimulated intracellular signaling cascades require Vav for induction of respiratory burst and phagocytosis in monocytes and microglia. J Biol Chem. 2006;281:20842–20850. doi: 10.1074/jbc.M600627200. [DOI] [PubMed] [Google Scholar]
- 201.Wilson DM., III Barsky D. The major human abasic endonuclease: formation, consequences and repair of abasic lesions in DNA. Mutat Res. 2001;485:283–307. doi: 10.1016/s0921-8777(01)00063-5. [DOI] [PubMed] [Google Scholar]
- 202.Wilson TM. Rivkees SA. Deutsch WA. Kelley MR. Differential expression of the apurinic/apyrimidinic endonuclease (APE/ref-1) multifunctional DNA base excision repair gene during fetal development and in adult rat brain and testis. Mutat Res. 1996;362:237–248. doi: 10.1016/0921-8777(95)00053-4. [DOI] [PubMed] [Google Scholar]
- 203.Wojda U. Salinska E. Kuznicki J. Calcium ions in neuronal degeneration. IUBMB Life. 2008;60:575–590. doi: 10.1002/iub.91. [DOI] [PubMed] [Google Scholar]
- 204.Yakes FM. Van Houten B. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc Natl Acad Sci USA. 1997;94:514–519. doi: 10.1073/pnas.94.2.514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Yang JL. Weissman L. Bohr VA. Mattson MP. Mitochondrial DNA damage and repair in neurodegenerative disorders. DNA Repair (Amst) 2008;7:1110–1120. doi: 10.1016/j.dnarep.2008.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Yao Y. Zhukareva V. Sung S, et al. Enhanced brain levels of 8,12-iso-iPF2α-VI differentiate AD from frontotemporal dementia. Neurology. 2003;61:475–478. doi: 10.1212/01.wnl.0000070185.02546.5d. [DOI] [PubMed] [Google Scholar]
- 207.Zheng L. Zhou M. Guo Z, et al. Human DNA2 is a mitochondrial nuclease/helicase for efficient processing of DNA replication and repair intermediates. Mol Cell. 2008;32:325–336. doi: 10.1016/j.molcel.2008.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Zimmermann H. Neurotransmitter release. FEBS Lett. 1990;268:394–399. doi: 10.1016/0014-5793(90)81292-v. [DOI] [PubMed] [Google Scholar]
- 209.Zoratti M. Szabo I. The mitochondrial permeability transition. Biochim Biophys Acta. 1995;1241:139–176. doi: 10.1016/0304-4157(95)00003-a. [DOI] [PubMed] [Google Scholar]
- 210.Zucker RS. Calcium- and activity-dependent synaptic plasticity. Curr Opin Neurobiol. 1999;9:305–313. doi: 10.1016/s0959-4388(99)80045-2. [DOI] [PubMed] [Google Scholar]