

Abstract
Dithiocarbamates are a class of sulfur-based metal-chelating compounds with various applications in medicine. We reported previously that certain members of dithiocarbamates, such as diethyldithiocarbamate, disulfiram (DSF) and pyrrolidine dithiocarbamate (PDTC), were able to bind with tumor cellular copper to inhibit tumor growth through the inhibition of proteasome activity and induction of cancer cell apoptosis. Since the DSF is an irreversible inhibitor of aldehyde dehydrogenase (ALDH), its ALDH-inhibitory activity might potentially affect its usefulness as an anti-cancer drug. For the purpose of selecting potent anti-cancer compounds that are not ALDH inhibitors and mapping out preliminary structure–activity relationship trends for these novel compounds, we synthesized a series of PDTC analogues and chose three novel compounds to study their ALDH-inhibitory activity, proteasome-inhibitory activity as well as the cancer cell apoptosis-inducing activity. The results showed that compared to DSF, compound 9 has less ALDH inhibition activity, and the in vitro results also proved the positive effects of 9-Cu in proteasome inhibition and apoptosis induction in breast cancer cells, suggesting that 9 as a lead compound could be developed into a novel proteasome inhibitor anti-cancer drug.
Keywords: ALDH, Dithiocarbamates, Cancer, Proteasome inhibitor, Disulfiram
1. Introduction
In multi-cellular organisms, apoptosis is a conserved cellular death program [1]. It plays an important role in tissue homeostasis, development, and defense against infections and mutations. Genes controlling the apoptotic process are suppressed or over expressed in many human diseases, especially in cancer [2,3].
Copper is an essential trace element that can affect many biological pathways in human body [4,5]. It can activate some critical proteins such as Cu–Zn superoxide dismutase, tyrosinase, ceruloplasmin and cytochrome oxidase, which are important in fundamental biological pathways [6]. Therefore, the copper concentration in human body is tightly regulated [7]. However, the concentration of copper in cancerous tissues, such as breast, prostate, lung, and brain, is higher than that of the normal tissues [8–11]. In the serum of breast cancer patients, the level of copper can reach 1.67 μg/ml. This copper level is much higher than the healthy controls (0.98 μg/ml) [12]. For now, there is no explanation for this cancer-associated copper elevation. It was speculated that this accumulation might play a vital role in angiogenesis, a process critical for tumor growth [13,14]. Copper can stimulate the production of cytokine, degradation of extracellular matrix, proliferation and migration of endothelial cell. In recent years, the methods of controlling the growth of tumor by eliminating the copper in serum of human body have been developed [15]. Tetrathiomolybdate (TM), which has the ability of copper-chelating was effective at inhibiting the growth of breast, prostate and lung tumors in various mouse models [16]. However, cancer kept developing in patients before the concentration of copper was lowered also suggesting that just passively chelating free copper is not enough to eliminate tumors.
The ubiquitin–proteasome pathway is essential for many fundamental cellular processes, such as cell cycle, apoptosis, angiogenesis and differentiation [17]. This pathway contributes to the pathological state of several human diseases including cancer, in which some regulatory proteins are either stabilized due to decreased degradation or lost due to accelerated degradation [18]. The 20S proteasome, the core of 26S proteasome complex, contains multiple peptidase activities (including the chymotrypsin-like, trypsin-like and peptidylglutamyl peptide hydrolyzing-like/PGPH-like) [19]. It has been reported that all three types of activities contributed significantly to protein breakdown and their relative importance varied widely with the substrate [20]. However, only the inhibition of chymotrypsin-like but not other proteasomal activities is a strong stimulus that induces apoptosis [21,22].
Small molecule proteasome inhibitors can be developed as anti-cancer agents. These inhibitors can block proteasome function without, or slightly affecting other normal biological processes in the cell. The fact that cancer cells are more sensitive to the proteasome inhibitors than normal or untransformed cells makes these small molecules even more attractive potential anti-cancer drugs [22–24].
Dithiocarbamates are a class of copper-chelating compounds with various applications in medicine for the treatment of bacterial and fungal infections, and possible treatment of AIDS [25,26]. One of the dithiocarbamates, pyrrolidine dithiocarbamate (PDTC), is a synthetic antioxidant that has a potential to inhibit NFκB activation [27,28]. We have previously found that PDTC formed a complex with copper and that PDTC-copper complex inhibited cell proliferation and induces apoptosis, and ubiquitin–proteasome pathway inhibition in cultured breast and prostate cancer cells [29].
Aldehyde dehydrogenases (ALDH) are NADP+ (NAD+) dependent enzymes. They catalyze the oxidation of endogenous and exogenous aldehydes [30,31]. The cytosolic (ALDH1) and mitochondrial isozymes (ALDH2) are the most abundant in human body. Disulfiram (DSF), a member of the dithiocarbamate family, is the first drug that is developed for the treatment of alcoholism by inhibiting the ALDH1 activity. DSF bound tumor cellular copper and induced apoptotic cell death in human breast cancer cells in vitro and in vivo through proteasome inhibition [32]. Thus, DSF may have an important useful role in the treatment of human cancers. However, the ALDH inhibitor property of DSF may decrease the concentration that function as drug in the treatment of cancer. Thus, discovering novel cancer inhibitory compounds without the inhibition of ALDH activity is essential. Here we report the cancer inhibitory activity of potential non-ALDH binding PDTC analogues in human breast cancer cells through the inhibition of proteasomal chymotrypsin-like activity.
2. Materials and methods
2.1. Reagents
CuCl2, disulfuram (DSF), 3-[4,5-dimethyltiazol-2-yl]-2.5-diphenyl-tetrazolium bromide (MTT), dimethylsulfoxide (DMSO) and other chemicals were purchased from Sigma–Aldrich (St. Louis, MO, USA). Dulbecco’s modified Eagle’s medium (DMEM)/F12, fetal bovine serum, horse serum, sodium bicarbonate, N-(2-hydroxyethyl) piperazine-N’-ethanesulfonic acid (HEPES) buffer solution, penicillin, and streptomycin were purchased from Invitrogen (Carlsbad, CA, USA). Fluorogenic peptide substrate (Suc-LLVY-AMC) for the proteasomal chymotrypsin-like activity was from Calbiochem (San Diego, CA, USA). Mouse monoclonal antibody against human poly(ADP-ribose) polymerase (PARP) was purchased from Biomol International (Plymouth Meeting, PA). Mouse monoclonal antibodies against Bax (B-9), goat polyclonal antibody against actin (C-11) and secondary antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Mouse monoclonal antibody against ubiquitin (P4D1) was from Cell Signaling Technology Inc. (Danvers, MA, USA), Mouse monoclonal antibody against NOXA was from Abcam Inc. (Cambridge, UK) and mouse polyclonal antibody against actin was from BIO-RAD (CA, USA).The BU-ANNEXIN V-FITC Apoptosis Detection Kit was from Biouniquer Technology (Biouniquer Technology CO., Ltd). Apo-ONE® Homogeneous Caspase-3/7 Assay Kit was from Promega (Promega Biotech CO., Ltd). All the chemicals used for synthesis of PDTC analogues were purchased from ACROS. The water used in this study was purified by reverse osmosis on a Milli-Ro followed by ion-exchange (Cascada, USA). The copper solution was prepared by dissolving CuCl2 in DMSO.
2.2. Synthesis of PDTC analogues
Dialkyl-substituted dithiocarbamate analogues were prepared by the one-step reaction of secondary amine with carbon disulfide in sodium hydroxide and methanol [33] (see Scheme 1). The mixture was heated to 40 °C and stirred well for 30–120 min, and the methanol was then removed by distillation. Crude products were further purified by recrystallization.
Scheme 1.
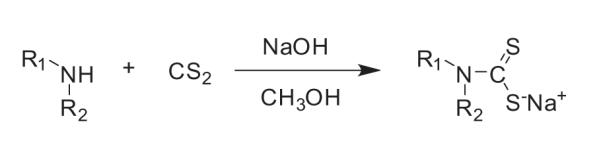
Synthesis of PDTC analogues.
2.3. Measurement of binding constants of PDTC analogues and copper
Binding constant between a PDTC analogue and CuCl2 was determined by UV spectroscopy at 25 °C. The concentration of PDTC analogue solution was 100 μM and the concentration of the CuCl2 was increased stepwise from 5 to 250 μM.
2.4. Computational docking
We chose the structure of human mitochondrial aldehyde dehydrogenase (ALDH2) and sheep liver ALDH1 from the website http://www.pdb.org. The PDB IDs are 2vle and 1bxs respectively. The docking software is Discovery Studio and Material Studio. Uses a CHARMm-based molecular dynamics scheme to dock ligands into a receptor binding site. Random ligand conformations are generated using high-temperature MD. The conformations are then translated into the binding site. Candidate poses are then created using random rigid-body rotations followed by simulated annealing. A final minimization is then used to refine the ligand poses. Many of these steps are optional [34].
2.5. Cell proliferation assay
The MTT assay was used to measure the effects of the compound-copper mixtures on breast cancer cell proliferation. Cells were plated in a 96-well plate and grown to 70–80% confluency, followed by the addition of each compound-copper mixture at the concentrations indicated. After incubation at 37 °C for 24 h, inhibition of cell proliferation was measured using MTT assay.
2.6. Cell cultures and whole cell extract preparation
Human breast cancer MDA-MB-231 cells were maintained in DMEM/F12 supplemented with 5% (v/v) fetal bovine serum, 100 units/ml of penicillin, and 100 μg/ml of streptomycin. Human breast cancer DCIS cells were maintained in DMEM/F12 supplemented with 5% (v/v) horse bovine serum, 0.029 M sodium bicarbonate, 10 mM HEPES buffer solution, 100 units/ml of penicillin, and 100 μg/ml of streptomycin at 37 °C in a humidified incubator with an atmosphere of 5% CO2. MCF10DCIS.com is a clonal breast cancer cell line, derived from in vivo passages of MCF10AT (tumorigenic) in nude mice by researchers in the Barbara Ann Karmanos Cancer Institute at Wayne State University and the reference [35]. Cells were treated with compounds, mixtures, or with an equivalent volume of solvent as indicated in the figure legends. Whole cell extracts were prepared as described previously [36]. Briefly, cells were washed twice with phosphate buffered saline and homogenized in a lysis buffer (50 mM tris(hydroxymethyl)ami-nomethane–HCl, pH 8.0, 150 mM NaCl, 0.5% NP40, Protease Inhibitor Cocktails). After they had been rocked for 20 min at 4 °C, the mixtures were centrifuged at 12,000 g for 15 min and the supernatants were collected as whole cell extracts.
2.7. In vitro proteasome activity assay
The cell-free chymotrypsin-like activity of the proteasome was determined by measuring the release of the AMC groups from a substrate as previously described [36]. Breast cancer cells were grown to 70–80% confluency, treated with PTDC analogues, CuCl2, or the 1:1 mixture as indicated, harvested, and then used for whole cell extract preparation. Proteasomal chymotrypsin-like activity was assayed as previously described [29]. Briefly, cell lysates (7.5 μg) were incubated with 40 μM fluorogenic substrate for proteasomal chymotrypsin-like activity for 2 h at 37 °C in 100 μL of assay buffer 50 μM tris(hydroxymethyl)aminometh-ane–HCl, pH 7.5. After incubation, production of hydrolyzed 7-amino-4-methylcoumarin groups was measured using a Victor3 multilabel counter (Perkin-Elmer, Boston, MA, USA). Statistical analysis was performed using SigmaPlot 8.0.
2.8. Western blot analysis
Breast cancer MDA-MB-231 and DCIS cells were exposed to PDTC analogues, CuCl2, or 1:1 mixture, followed by preparation of whole cell extracts. Cell lysates (50 μg) were separated by electrophoresis on 10–15% sodium dodecyl sulfate polyacrylamide gels and transferred onto a nitrocellulose membrane. The membranes were probed to determine levels of ubiquitinated proteins, PARP cleavage, the pro-apoptosis protein Bax and the induction of NOXA. The horseradish peroxidase-conjugated antimouse or antigoat antibodies were used as secondary antibodies, followed by visualization using an enhanced chemiluminescence kit (Amersham Biosciences, Piscataway, NJ, USA).
2.9. Apoptosis assay
The percentage of cells undergoing apoptosis was determined by Annexin V staining by using the BU-ANNEXIN V-FITC apoptosis detection kit (Biouniquer Technology CO., Ltd). 3 × 105 MDA-MB-231 cells were seeded in 60 mm plastic tissue culture dishes and incubated overnight. Cells were then treated for 24 h with 1.4 μM 9-Cu mixture or vehicle in the presence or absence of 20 μM caspase inhibitor Z-VAD-FMK. DSF-Cu mixture-treated cells were measured as positive control. Cells were harvested and washed in ice-cold PBS buffer and then incubated with FITC-labeled Annexin V for 15 min, PI was added to distinguish necrotic cells (Annexin V−/PI+) from early apoptotic cells (Annexin V+/PI−) and late apoptotic cells (Annexin V+/PI+). Flow cytometric analysis (FACSCalibur, BD Immunocytometry Systems, San Jose, CA) was performed to determine the percentage of apoptotic cells in each sample.
2.10. Caspase activity assay
Caspase 3/7 assay (Apo-ONE® Homogeneous Caspase-3/7 Assay, Promega Biotech CO., Ltd) was performed according to the manufacturers’ recommendation. 1.5 × 104 cells per well were plated in 96-well tissue culture plates and incubated overnight. Cells were then treated with 1.4 μM 9-Cu mixture or DMSO vehicle in the presence or absence of 20 μM Z-VAD-FMK caspase inhibitor for 24 h. DSF-Cu mixture-treated cells as positive control. The fluorescent caspases-3/7 substrate Z-DEVD-R110 was added to each well. Contents were mixed by gentle shaking at 300 r.p.m. for 5 min, and the plates incubated at room temperature in the dark for 18 h. Samples were read using a Victor3 multilabel counter (PerkinElmer, Boston, MA, USA) at 485 nm (excitation)/535 nm (emission). Statistical analysis was performed using SigmaPlot 8.0.
3. Results
3.1. Synthesis and characterization of PDTC analogues
Nine secondary amines (Fig. 1A) were used for synthesis of PDTC analogues. Since these compounds have different nucleophilicity, the reactions were carried out under different conditions. Aliphatic amines could easily react with CS2 in NaOH at room temperature with a yield of 90%. Secondary amines containing aromatic groups were less reactive and were treated at higher temperature (50 °C) and yielded about 50% of final product. Products were characterized by NMR and LC/MS.
Fig. 1.

Chemical structures of compound 1–9 and the Anti-proliferative effect of the copper mixtures with different DSF analogues. (A) Chemical structures of compound 1–9. (B, C) the Anti-proliferative effect of the copper mixtures with different DSF analogues. DCIS (B) and MDA-MB-231 (C) cells were treated with the copper mixtures of nine PDTC analogues at different concentrations as indicated. After 24 h, the cell viability was measured by MTT assay as described in Section 2. DMSO was used as negative control and DSF as positive control.
3.2. Mixtures of 9 with copper inhibits proliferation of human breast cancer MDA-MB-231 and DCIS Cells
In order to discover anti-cancer PDTC analogues without ALDH inhibition activity, we first determined anti-proliferative effects of compounds 1–9 in highly metastatic MDA-MB-231 and malignant MCF10DCIS.com (DCIS) breast cancer cells. We found that the mixtures of Cu and compound 9 had strong growth inhibition activity, causing 94% and 99% inhibition in DCIS cells and MDA-MB-231 cells respectively at 1.0 μM (Fig. 1B). Mixture 1-Cu was slightly less potent, causing 94% and 53% inhibition in DCIS cells and MDA-MB-231cells respectively at 2.5 μM. The Cu mixtures of other compounds, Cu or compound alone showed weaker or no growth inhibition at the concentrations tested. These suggested that the copper-dependent growth-inhibitory effect of PDTC analogues is highly related to their chemical structures (Fig. 1B). Results showed that R1 should be a phenyl group with one or two carbon spacing, while R2 must be small (methyl group) (see Fig. 1A).
3.3. Compound 9 is not likely ALDH inhibitor as judged by docking studies
In order to investigate the potential ALDH inhibition by these compounds, we constructed two computational docking models of human ALDH from the structure of sheep liver ALDH1 (PDB ID 1bxs) and mitochondrial aldehyde dehydrogenase ALDH2 (PDB ID 2vle). ALDH2 is a tetrameric enzyme that comprising 500 amino acid residues [37]. Each subunit consists of three domains and one of them is the catalytic domain. Inside the catalytic domain, amino acid residue Cys302 has been identified as the key group [38] to interact with the –SH group in inhibitors [39] and forms a covalent bond. After ligand docking, we compared the distance between the thiol group on compound 1, 5, 9, DSF and that on Cys302 (Table 1). In ALDH2, the separations of the –SH group of 9 from the sulfur atom of Cys302 is 4.166 Å while this distance for DSF is 3.053 Å (Fig. 2A). In ALDH1, the distance is 4.198 Å and 3.469 Å for 9 and DSF respectively (Fig. 2B). Computer docking results suggested that compound 9 is less likely to inhibit ALDH and, therefore, a plausible anti-cancer lead compound. Its mechanism of action was then thoroughly investigated.
Table 1.
The distance between the thiol on compound DSF, 1, 5, 9 and thiol on Cys302.
| Compound | Distance in ALDH1 (Å) | Distance in ALDH2 (Å) |
|---|---|---|
| DSF | 3.469 | 3.053 |
| 1 | 3.311 | 4.514 |
| 5 | 3.478 | 3.525 |
| 9 | 4.198 | 4.166 |
Fig. 2.

Interactions between ALDH and compounds. (A) Details of the region of ALDH2 around the compounds, showing the hydrogen-bonding distance formed between the -SH of compound and Cys302. The side chain of the cysteine residues Cys302 is shown. (B) Details of the Cys302 of ALDH1 binding distance of the compound binding site. The backbone of ALDH is shown as a line model with the side chain of the Cys302 residues forming contact with compounds. The structures of compounds are shown as ball-and-stick model. The different atoms are marked as different colors: S (yellow), N (blue), C (gray), H (white), O (red). (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.).
3.4. Mixture 9-Cu could induce cancer cell apoptosis in a concentration- and time-dependent manner
To further investigate the mechanisms that caused the anti-proliferative effect of PDTC analogues, we further studied cellular effects induced by compounds 1, 5 and 9. After treatment, cell condensation and rounding were detected in the cells treated with 1-Cu and 9-Cu, but not in cells treated with 5-Cu mixture even at concentration of 2.0 μM (Fig. S1A and B). A dose- and time-dependent production of cell apoptosis-specific PARP cleavage fragment p89 and p65 were detected in MDA-MB-231 cells after 9-Cu treatment (Fig. 4A and B). The accumulation of PARP cleavage fragment p89 was detected after treatment for 2 h in MDA-MB-321 cells, and increased with time (Fig. 4A–C). PARP cleavage fragment p89 and p65 were also detected in DCIS in a dose-dependent manner (Fig. 4A). This further supports caspase-3 and calpain activation. Furthermore, accumulation of pro-apoptotic protein Bax was detected in cells treated with 9-Cu mixtures (Fig. 4A–C).
Fig. 4.

Induction of apoptosis in cancer cells by 9-copper mixtures. (A) Dose-dependent poly(ADP-ribose) polymerase (PARP) cleavage and Bax accumulation in DCIS cells treated with 1, 5 and 9-Cu mixtures at increasing concentrations for 24 h. (B) Dose-dependent poly(ADP-ribose) polymerase (PARP) cleavage and Bax accumulation in MDA-MB-231 cells treated with 1, 5 and 9-Cu mixtures at increasing concentrations for 24 h. (C) Time-dependent poly(ADP-ribose) polymerase (PARP) cleavage and Bax accumulation in MDA-MB-231 cells treated with 2.5 μM of 5 and 9-Cu mixtures for up to 24 h. (D) Induction of apoptosis in MDA-MB-231 cells after treatment with 1.4 μM of DSF-Cu mixture, 9-Cu mixture or vehicle in the presence and absence of Z-VAD-FMK inhibitor for 24 h by Annexin V/PI staining (also see Fig. S2). (E) Detection of caspase-3/7 activity in MDA-MB-231 cells after treatment with 1.4 μM of DSF-Cu mixture, 9-Cu mixture or vehicle in the presence and absence of Z-VAD-FMK for 24 h using the Apo-ONE® homogeneous Caspase-3/7 Assay.
The appearance of phosphatidyl-serine on the extracellular side of the cell membrane was quantified by Annexin V/PI staining. In cells treated by 9-Cu, apoptosis was induced as detected by Annexin V-FITC/PI positivity (Fig. 4D and Fig. S2). The percentages of apoptotic cells (both early stage and late stage) were 57.3 ± 2.2% and 46.2 ± 5.2% respectively upon treatment with 1.4 μM of DSF-Cu mixture or 9-Cu mixture for 24 h (Fig. 4D). In the presence of 20 μM caspase inhibitor Z-VAD-FMK, the percentages of apoptotic cells (both early stage and late stage) were 13.7 ± 0.8% and 16.4 ± 0.7% respectively upon treatment with DSF-Cu mixture or 9-Cu mixture (Fig. 4D). The results demonstrated that the activation of caspase-3 might play a crucial role in 9-copper mixture-induced apoptosis.
To further discern the mechanism by which 9-Cu induces the breast cancer cell apoptosis, we measured the activation of caspase3/7 activity in MDA-MB-231 cells treated with 1.4 μM of 9-Cu mixture or vehicle in the presence or absence of 20 μM caspase inhibitor Z-VAD-FMK. Cells treated with 1.4 μM of DSF-Cu mixture were used as positive control (Fig. 4E). The caspase 3/7 activity of MDA-MB-231 treated with DSF-Cu and 9-Cu mixtures are 61.6% and 60.6% respectively, corresponding to at least 6-fold increase of caspase 3/7 activity compared with that of cells treated with vehicle (Fig. 4E). In contrast caspase 3/7 activity was obviously inhibited when Z-VAD-FMK was also added (Fig. 4E), showing nearly 2–3 folds decrease of caspase activity. These results further demonstrated that caspase-3/7 was involved in the 9-Cu mixture-induced apoptosis, which was consistent with the results by Annexin V/PI staining asssay.
3.5. Proteasome inhibition by 9-Cu mixture
Previous reports have proved that selective inhibition of cancer cell proteasomes blocked the cancer cell’s metabolic regulators and induced cancer cell apoptosis [22,23,40,41]. To further investigate whether 9-Cu mixture could inhibit the proteasome activity, we first performed the cell-free chymotrypsin-like activity assay in vitro (Fig. 5). The results showed that the 9-Cu mixture inhibited the chymotrypsin-like activity in a concentration-dependent manner (Fig. 5A and C). In contrast, 1-Cu and 5-Cu did not cause chymotrypsin-like activity inhibition at the concentrations used. However, the accumulation of ubiquitinated proteins was detected by Western blot analysis in the cells treated with 9-Cu and 1-Cu but not in cells treated with 5-Cu (Fig. 5B and D).
Fig. 5.

Inhibition of proteasomal chymotrypsin-like activity by 1-Cu, 5-Cu and 9-Cu mixtures. (A) The DCIS cells were treated with 0.5, 1.0 and 2.0 μM of the indicated mixtures for 24 h, followed by the measurement of proteasomal CT-like activity. (B) Western blot analysis for the accumulation of ubiquitinated proteins and NOXA induction in the extracts prepared from the DCIS cells for 24 h. (C) The MDA-MB-231 cells were treated with 0.5, 1.0 and 2.0 μM of the indicated mixtures for 24 h, followed by the measurement of proteasomal CT-like activity. (D) Western blot analysis for the accumulation of ubiquitinated proteins and NOXA induction in the extracts prepared from the MDA-MB-231 cells for 24 h. (E) The MDA-MB-231 cells were exposed to a 2.5 μM mixture of 9-Cu and 5-Cu for the indicated times, followed by measurement of proteasomal CT-like activity. (F) Western blot analysis using specific antibodies to ubiquitinated proteins and NOXA in the extracts prepared form the MDA-MB-231 cells for the indicated times.
To further ascertain the proteasome-inhibitory activity of 9-Cu, MDA-MB-231 cells were treated with 2.5 μM of 9-Cu and 5-Cu mixture for up to 24 h. We found that about 39% inhibition of the proteasomal chymotrypsin-like activity was achieved by 9-Cu mixture after 2 h of treatment, 46% inhibition after 4 h, 65% inhibition after 8 h, 90% inhibition after 16 h. Proteasome inhibition was also confirmed by the accumulation of ubiquitinated proteins (Fig. 5E and F).
Since NOXA is ubiquitinated and degraded through the proteasome [42], its expression can be an indirect marker of proteasome inhibition, thus we analyzed the expression of NOXA. Our result showed that exposure of breast cancer cells to 9-Cu induced a concentration- and time-dependent accumulation of NOXA, which correlated well to the accumulation of the poly-ubiquitinated proteins (Fig. 5). In contrast, there is no such effect in cells treated with 5-Cu mixture, compound 5 and 9 or CuCl2 alone, suggesting that proteasome inhibition by 9-Cu might be reason for caspase 3 activation and cell apoptosis induction.
4. Discussion
High levels of copper are found in serum and tumors of cancer patients [8–11]. The dramatic differences in Cu accumulation between normal and tumor tissues, and the requirement of Cu in tumor angiogenesis suggest that targeting cellular Cu can be a strategy for the treatment of breast caner. Previously, Cu depletion by TM has been suggested as a means to control angiogenesis and inhibit the tumor growth [16,43,44]. In clinical trials, TM treatment successfully blocked angiogenesis. However, the TM-copper complex did not have the ability to inhibit the proteasome and failed to kill cultured cancer cells [32].
The copper-binding compounds DSF and PDTC analogues have a wide range of bioactivities. Previously studies suggest that DSF is able to bind copper forming a complex with proteasome-inhibitory activity and suppress proliferation of cultured cancer cells [32].
The ubiquitin/proteasome-dependent pathway plays an essential role in up-regulation of proliferation, down-regulation of apoptosis, promotion of angiogenesis, and development of drug resistance in human tumor cells [21,45,46]. Proteasome inhibitors can be employed as potential novel anti-cancer drugs [46–48]. PS-341 (Bortezomib, Velcade), a potent and selective boronic acid proteasome inhibitor, is currently being developed for treating hematological malignant and solid tumors. Clinical trial data confirm the antitumor activity of PS-341. However, some associated side effects were observed [47,48], suggesting the need to discover novel proteasome inhibitors with no, or significantly reduced, toxicity. Copper chelators would be more effective in cancer inhibitor by both copper elimination and proteasome inhibition.
We and others have recently reported organic Cu-containing compounds, including DSF-Cu and PDTC-Cu as a novel class of proteasome inhibitors and apoptosis inducers [32,49,50]. However, DSF is an irreversible inhibitor of aldehyde dehydrogenase (ALDH) in clinical alcoholism treatment. Its anti-cancer activity may be potentially deactivated by ALDH. To develop potent anti-cancer compounds that are not ALDH inhibitors, we synthesized a series of PDTC analogues and studied their ALDH-independent anti-cancer activities.
Cu mixtures of compounds 1 and 9 were identified as potent agents that induced cell death in human breast cancer cells (Fig. 1B). In docking studies using structures of ALDH1 and ALDH2 as templates, we found that it is less likely that compound 9 can form covalent bond with Cys302 in both ALDH1 and ALDH2 (Fig. 2A and B). With potent anti-cancer activity and no potentially ALDH inhibition, 9 can be a promising lead compound for the treatment of breast cancer.
The cancer cell inhibitory effect of compound 9 was associated with its copper binding (Fig. 3) and apoptosis induction effect in DCIS and MDA-MB-231 cells. The 9-Cu mixture significantly reduced chymotrypsin-like activity (Fig. 5) and accumulation of ubiquitinated proteins (Fig. 5) in a dose- and time-dependent manner in both cellular and cell-free assays suggesting that proteasome might be the target of this anti-cancer agent.
Fig. 3.
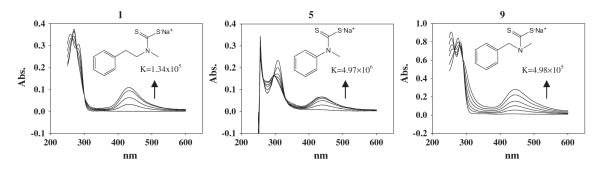
Copper complex formation and the binding of compound with copper. The UV spectra of complexes formed between PDTC analogues 1, 5 and 9 and copper and their binding constants. The concentration of PDTC analogue was 100 μM. The absorption band of the complex at 430 nm increases as Cu concentrations were 10, 20, 30, 40, 50 and 60 μM.
Furthermore, our results (Fig. 4D and E) showed that Z-VAD-FMK attenuated the activition of caspase-3 in 9-copper mixture-treated MDA-MB-231 cells, demonstrating that caspase-3 activation is a key mechanism of 9-copper mixture-induced apoptosis.
In this study, we identified 9-Cu as a promising agent in proteasome inhibition and apoptosis induction in breast cancer cells with no potential ALDH inhibition. The combination of anti-angiogenesis and proteasome inhibition is an effective approach that may result in direct damage to cancer tissues by simultaneous Cu elimination, and tumor cell apoptosis induction through the de novo formation of Cu-based proteasome inhibitors within tumors.
Supplementary Material
Acknowledgments
This work is supported by Grants from the National Natural Science Foundation of China (20902054 to S.M.Z. and 90913006 to B.Y.), Karmanos Cancer Institute 2009 Pilot project funding (to Q.P.D.) and the National Cancer Institute (1R01CA120009, 3R01CA120009-04S1, 1R21CA139386-01, to Q.P.D.), the American Lebanese Syrian Associated Charities (ALSAC), and St. Jude Children’s Research Hospital. We thank Yanwei Li for technical assistance in the computational docking analysis.
Footnotes
Conflict of interest None declared.
Appendix A. Supplementary material Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.canlet. 2010.09.010.
References
- [1].Jacobson M, Weil M, Raff M. Programmed cell death review in animal development. Cell. 1997;88:347–354. doi: 10.1016/s0092-8674(00)81873-5. [DOI] [PubMed] [Google Scholar]
- [2].Thatte U, Dahanukar S. Apoptosis: clinical relevance and pharmacological manipulation. Drugs. 1997;54:511–532. doi: 10.2165/00003495-199754040-00002. [DOI] [PubMed] [Google Scholar]
- [3].Song Z, Steller H. Death by design: mechanism and control of apoptosis. Trends Cell Biol. 1999;9:49–52. [PubMed] [Google Scholar]
- [4].Gregoriadis G, Apostolidis N, Romanos A, Paradellis T. A comparative study of trace elements in normal and cancerous colorectal tissues. Cancer. 1983;52:508–519. doi: 10.1002/1097-0142(19830801)52:3<508::aid-cncr2820520322>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- [5].O’dell B. Biochemistry of copper. Med. Clin. N. Am. 1976;60:687–703. doi: 10.1016/s0025-7125(16)31853-3. [DOI] [PubMed] [Google Scholar]
- [6].Tapiero H, Townsend D, Tew K. Trace elements in human physiology and pathology, Copper. Biomed. Pharmacother. 2003;57:386–398. doi: 10.1016/s0753-3322(03)00012-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Labbe S, Thiele D. Pipes and wiring: the regulation of copper uptake and distribution in yeast. Trends Microbiol. 1999;7:500–505. doi: 10.1016/s0966-842x(99)01638-8. [DOI] [PubMed] [Google Scholar]
- [8].Diez M, Arroyo M, Cerdan F, Munoz M, Martin M, Balibrea J. Serum and tissue trace metal levels in lung cancer. Oncology. 1989;46:230–235. doi: 10.1159/000226722. [DOI] [PubMed] [Google Scholar]
- [9].Geraki K, Farquharson M, Bradley D. Concentrations of Fe, Cu and Zn in breast tissue: a synchrotron XRF study. Phys. Med. Biol. 2002;47:2327–2339. doi: 10.1088/0031-9155/47/13/310. [DOI] [PubMed] [Google Scholar]
- [10].Yoshida D, Ikeda Y, Nakazawa S. Quantitative analysis of copper, zinc and copper/zinc ratio in selected human brain tumors. J. Neuro-Oncol. 1993;16:109–115. doi: 10.1007/BF01324697. [DOI] [PubMed] [Google Scholar]
- [11].Nayak S, Bhat V, Upadhyay D, Udupa S. Copper and ceruloplasmin status in serum of prostate and colon cancer patients. Indian J. Physiol. Pharmacol. 2003;47:108–110. [PubMed] [Google Scholar]
- [12].Gupta SK, Shukla VK, Vaidya MP, Roy SK, Gupta S. Serum trace elements and Cu/Zn ratio in breast cancer patients. J. Surg. Res. 1991;46:178–181. doi: 10.1002/jso.2930460311. [DOI] [PubMed] [Google Scholar]
- [13].Brewer G. Copper control as an antiangiogenic anticancer therapy: lessons from treating Wilson’s disease. Exp. Biol. Med. 2001;226:665–673. doi: 10.1177/153537020222600712. [DOI] [PubMed] [Google Scholar]
- [14].Eatock M, Schtzlein A, Kaye S. Tumour vasculature as a target for anticancer therapy. Cancer Treat. Rev. 2000;26:191–204. doi: 10.1053/ctrv.1999.0158. [DOI] [PubMed] [Google Scholar]
- [15].Adsule S, Barve V, Ahmed F, Dou Q, Padhye S, Sarkar F. Novel Schiff base copper complexes of quinoline-2 carboxaldehyde as proteasome inhibitors in human prostate cancer cells. J. Med. Chem. 2006;49:7242–7246. doi: 10.1021/jm060712l. [DOI] [PubMed] [Google Scholar]
- [16].Brewer G. The use of copper-lowering therapy with tetrathiomolybdate in medicine. Expert Opin. Investig. Drugs. 2009;18:89–97. doi: 10.1517/13543780802621859. [DOI] [PubMed] [Google Scholar]
- [17].Orlowski R, Dees E. The role of the ubiquitination–proteasome pathway in breast cancer: applying drugs that affect the ubiquitin–proteasome pathway to the therapy of breast cancer. Breast Cancer Res. 2002;5:1–7. doi: 10.1186/bcr460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Ciechanover A, Everett R, Orr A, Preston C, Arnold I, Pfeiffer K, Neupert W, Stuart R, Schgger H, Wouters F. The ubiquitin–proteasome pathway: on protein death and cell life FREE. EMBO J. 1998;17:7151–7160. doi: 10.1093/emboj/17.24.7151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Seemuller E, Lupas A, Stock D, Lowe J, Huber R, Baumeister W. Proteasome from thermoplasma acidophilum: a threonine protease. Science. 1995;268:579–582. doi: 10.1126/science.7725107. [DOI] [PubMed] [Google Scholar]
- [20].Kisselev AF, Callard A, Goldberg AL. Importance of the different proteolytic sites of the proteasome and the efficacy of inhibitors varies with the protein substrate. J. Biol. Chem. 2006;281:8582–8590. doi: 10.1074/jbc.M509043200. [DOI] [PubMed] [Google Scholar]
- [21].Lopes U, Erhardt P, Yao R, Cooper G. P53-dependent induction of apoptosis by proteasome inhibitors. J. Biol. Chem. 1997;272:12893–12896. doi: 10.1074/jbc.272.20.12893. [DOI] [PubMed] [Google Scholar]
- [22].Adams J. Potential for proteasome inhibition in the treatment of cancer. Drug Discov. Today. 2003;8:307–315. doi: 10.1016/s1359-6446(03)02647-3. [DOI] [PubMed] [Google Scholar]
- [23].Almond J, Cohen G. The proteasome: a novel target for cancer chemotherapy. Leukemia. 2002;16:433–443. doi: 10.1038/sj.leu.2402417. [DOI] [PubMed] [Google Scholar]
- [24].Dou Q, Li B. Proteasome inhibitors as potential novel anticancer agents. Drug Resist. Update. 1999;2:215–223. doi: 10.1054/drup.1999.0095. [DOI] [PubMed] [Google Scholar]
- [25].Schreck R, Meier B, Mannel D, Droge W, Baeuerle P. Dithiocarbamates as potent inhibitors of nuclear factor kappa B activation in intact cells. J. Exp. Med. 1992;175:1181–1194. doi: 10.1084/jem.175.5.1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Malaguarnera L, Pilastro M, DiMarco R, Scifo C, Renis M, Mazzarino M, Messina A. Cell death in human acute myelogenous leukemic cells induced by pyrrolidinedithiocarbamate. Apoptosis. 2003;8:539–545. doi: 10.1023/a:1025550726803. [DOI] [PubMed] [Google Scholar]
- [27].Parodi F, Mao D, Ennis T, Bartoli M, Thompson R. Suppression of experimental abdominal aortic aneurysms in mice by treatment with pyrrolidine dithiocarbamate, an antioxidant inhibitor of nuclear factor-κB. J. Vasc. Surg. 2005;41:479–489. doi: 10.1016/j.jvs.2004.12.030. [DOI] [PubMed] [Google Scholar]
- [28].Zhang Y. Cancer-preventive isothiocyanates: measurement of human exposure and mechanism of action. Mutat. Res. – Fund. Mol. Mech. Mutagen. 2004;555:173–190. doi: 10.1016/j.mrfmmm.2004.04.017. [DOI] [PubMed] [Google Scholar]
- [29].Daniel K, Orlu S, Cui Q, Miller F, Dou Q. Clioquinol and pyrrolidine dithiocarbamate complex with copper to form proteasome inhibitors and apoptosis inducers in human breast cancer cells. Breast Cancer Res. 2005;7:897–908. doi: 10.1186/bcr1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Goedde H. Werner, Agarwal D. Pharmacogenetics of aldehyde dehydrogenase (ALDH) Pharmacol. Ther. 1989;45:345–371. doi: 10.1016/0163-7258(90)90071-9. [DOI] [PubMed] [Google Scholar]
- [31].Weiner H, Wang X. Aldehyde dehydrogenase and acetaldehyde metabolism. Alcohol Alcoholism. 1994;2:141–145. [PubMed] [Google Scholar]
- [32].Chen D, Cui Q, Yang H, Dou Q. Disulfiram, a clinically used anti-alcoholism drug and copper-binding agent, induces apoptotic cell death in breast cancer cultures and xenografts via inhibition of the proteasome activity. Cancer Res. 2006;66:10425–10433. doi: 10.1158/0008-5472.CAN-06-2126. [DOI] [PubMed] [Google Scholar]
- [33].Clifford AM, Lichty JG. The reaction of diazonium salts on sodium alkyl dithiocarbamates. J. Am. Chem. Soc. 1932;54:1163–1166. [Google Scholar]
- [34].Wu G, Robertson D, Brooks C, Vieth M. Detailed analysis of grid-based molecular docking: a case study of CDOCKER-A CHARMm-based MD docking algorithm. J. Comput. Chem. 2003;24:1549–1562. doi: 10.1002/jcc.10306. [DOI] [PubMed] [Google Scholar]
- [35].Miller F, Santner S, Tait L, Dawson P. MCF10DCIS.Com xenograft model of human comedo ductal carcinoma in situ. J. Natl. Cancer Inst. 2000;92:1185–1186. doi: 10.1093/jnci/92.14.1185a. [DOI] [PubMed] [Google Scholar]
- [36].An B, Goldfarb RH, Siman R, Dou QP. Novel dipeptidyl proteasome inhibitors overcome Bcl-2 protective function and selectively accumulate the cyclin-dependent kinase inhibitor p27 and induce apoptosis in transformed, but not normal, human fibroblasts. Cell Death Differ. 1998;5:1062–1075. doi: 10.1038/sj.cdd.4400436. [DOI] [PubMed] [Google Scholar]
- [37].Ni L, Zhou J, Hurley T, Weiner H. Human liver mitochondrial aldehyde dehydrogenase: three-dimensional structure and the restoration of solubility and activity of chimeric forms. PRS. 1999;8:2784–2790. doi: 10.1110/ps.8.12.2784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Weiner H, Farres J, Rout U, Wang X, Zheng C. Site directed mutagenesis to probe for active site components of liver mitochondrial aldehyde dehydrogenase. Adv. Exp. Med. Biol. 1995;372:1–7. doi: 10.1007/978-1-4615-1965-2_1. [DOI] [PubMed] [Google Scholar]
- [39].Shen M, Johnson K, Mays D, Lipsky J, Naylor S. Determination of in vivo adducts of disulfiram with mitochondrial aldehyde dehydrogenase. Biochem. Pharmacol. 2001;61:537–545. doi: 10.1016/s0006-2952(00)00586-4. [DOI] [PubMed] [Google Scholar]
- [40].Adams J. The development of proteasome inhibitors as anticancer drugs. Cancer Cell. 2004;5:417–421. doi: 10.1016/s1535-6108(04)00120-5. [DOI] [PubMed] [Google Scholar]
- [41].Adams J. The proteasome: a suitable antineoplastic target. Nat. Rev. Cancer. 2004;4:349–360. doi: 10.1038/nrc1361. [DOI] [PubMed] [Google Scholar]
- [42].Baou M, Kohlhaas SL, Butterworth M, Vogler M, Dinsdale D, Walewska R, Majid A, Eldering E, Dyer MJS, Cohen GM. Role of NOXA and its ubiquitination in proteasome inhibitor-induced apoptosis in chronic lymphocytic leukemia cells. Haematology. 2010;95:1510–1518. doi: 10.3324/haematol.2010.022368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Brewer GJ, Dick RD, Grover DK, LeClaire V, Tseng M, Wicha M, Pienta K, Redman BG, Jahan T, Sondak VK. Treatment of metastatic cancer with tetrathiomolybdate, an anticopper, antiangiogenic agent: phase I study. Clin. Cancer Res. 2000;6:1–10. [PubMed] [Google Scholar]
- [44].Redman BG, Esper P, Pan Q, Dunn RL, Hussain HK, Chenevert T, Brewer GJ, Merajver SD. Phase II trial of tetrathiomolybdate in patients with advanced kidney cancer. Clin. Cancer Res. 2003;9:1666–1671. [PubMed] [Google Scholar]
- [45].Daniel KG, Harbach RH, Guida WC, Dou QP. Copper storage diseases: Menkes, Wilsons, and cancer. Front. Biosci. 2004;9:2652–2662. doi: 10.2741/1424. [DOI] [PubMed] [Google Scholar]
- [46].Tanimoto Y, Onishi Y, Hashimoto S, Kizaki H. Peptidyl aldehyde inhibitors of proteasome induce apoptosis rapidly in mouse lymphoma RVC cells. J. Biochem. 1997;121:542–549. doi: 10.1093/oxfordjournals.jbchem.a021620. [DOI] [PubMed] [Google Scholar]
- [47].Adams J. Preclinical and clinical evaluation of proteasome inhibitor PS-341 for the treatment of cancer. Curr. Opin. Chem. Biol. 2002;6:493–500. doi: 10.1016/s1367-5931(02)00343-5. [DOI] [PubMed] [Google Scholar]
- [48].Dou QP, Goldfarb RH. Bortezomib (millennium pharmaceuticals) IDrugs. 2002;5:828–834. [PubMed] [Google Scholar]
- [49].Chen SH, Lin JK, Liang YC, Pan MH, Liu SH, Lin-Shiau SY. Involvement of activating transcription factors JNK, NF-κB, and AP-1 in apoptosis induced by pyrrolidine dithiocarbamate/Cu complex. Eur. J. Pharmacol. 2008;594:9–17. doi: 10.1016/j.ejphar.2008.07.024. [DOI] [PubMed] [Google Scholar]
- [50].Yu Z, Wang F, Milacic V, Li X, Cui QC, Zhang B, Yan B, Dou QP. Evaluation of copper-dependent proteasome-inhibitory and apoptosis-inducing activities of novel pyrrolidine dithiocarbamate analogues. Int. J. Mol. Med. 2007;20:919–925. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.