

Abstract
Glycosaminoglycans (GAGs) are complex polysaccharides composed of hexosamine-containing disaccharide repeating units. The three most studied classes of GAGs, heparin/heparan sulfate, hyaluronan and chondroitin/dermatan sulfate, are essential macromolecules. GAGs isolated from animal and microbial sources have been utilized therapeutically, but naturally occurring GAGs are extremely heterogeneous limiting further development of these agents. These molecules pose difficult targets to construct by classical organic syntheses due to the long chain lengths and complex patterns of modification by sulfation and epimerization. Chemoenzymatic synthesis, a process that employs exquisite enzyme catalysts and various defined precursors (e.g. uridine 5′-diphosphosphate-sugar donors, sulfate donors, acceptors and oxazoline precursors), promises to deliver homogeneous GAGs. This review covers both theoretical and practical issues of GAG oligosaccharide and polysaccharide preparation as single molecular entities and in library formats. Even at this early stage of technology development, nearly monodisperse GAGs can be made with either natural or artificial structures.
Keywords: chondroitin, epimerase, glycosaminoglycan, glycosyltransferase, heparan sulfate, heparin, hyaluronan or hyaluronic acid, libraries, microfluidics, oligosaccharide, polysaccharide, sulfatase, sulfotransferase, synthase, UDP-sugar
Introduction
Glycosaminoglycans in animals
Carbohydrates play many roles in biology, but the glycosaminoglycans (GAGs) of animals are truly exceptional polymers with a plethora of chemical structures and bioactivities (Sugahara et al. 2003; Toole 2004; Bishop et al. 2007). The three most abundant classes of GAG polymers in vertebrates are (i) heparan sulfate (HS) and heparin, (ii) hyaluronan (HA) and (iii) chondroitin sulfate (CS) and dermatan sulfate (DS). HS is found throughout the animal kingdom including the primitive coelenterates (e.g. Hydra); thus, it appears that the most complicated GAG is also the most primordial (DeAngelis 2002). Unsulfated chondroitin exists in nematodes (e.g. Caenorhabditis), whereas poorly sulfated CS is found in arthropods (e.g. Drosophila) and more extensively sulfated CS in most animals. HA appears to be the most recent GAG to evolve in animals and is only found in chordates (e.g. Amphioxus and higher). Most, if not all, mammalian cells or their surrounding extracellular matrix possesses at least one of the types of the GAG molecule, but in many cases, multiple GAGs co-exist. Genetic knockout or knockdown studies in various organisms from worms to mice demonstrate that the GAGs are crucial for development/organogenesis and thus appear to be essential for multicellular life.
Natural GAG and glycoconjugate structures
The GAG chains of animals are polysaccharides composed of repeating disaccharide building blocks constructed from a hexosamine and an uronic acid residue (Table I). Keratan sulfate (KS) is another member of the GAG family, but due to its distinctive structure, lack of uronic acid, relatively low abundance in the body and less well-known biological roles, KS will not be covered in this review. In vertebrates, the backbones of HS and CS (heparosan and chondroitin, respectively) are modified by sulfation, but HA is not sulfated naturally. In more ancient animals, sulfation on chondroitin does not always occur. In more evolutionarily modern organisms, some of the HS and CS backbones are further modified by C5-epi, resulting in the conversion of D-glucuronic acid (GlcA) to l-iduronic acid (IdoA). It is well-known that the level of sulfation and epimerization varies from slight to extreme depending on the organism, tissue, developmental stage and health status; this diversity as well as heterogeneity makes GAG analysis and synthesis challenging (Laremore et al. 2009).
Table I.
Animal GAGs and their major backbone structures
| GAG | Major backbone repeat structures |
|---|---|
| Hyaluronan, hyaluronic acid (HA) | [-4-GlcAβ-1-3-GlcNAc-β1-]n |
| Chondroitin | [-4-GlcAβ-1-3-GalNAc-β1-]n |
| CS | [-4-GlcA2Xβ-1-3-GalNAc4X6X-β1-]n |
| DS | [-4-IdoAα or GlcA2X β–1-3-GalNAc4X6X -β1-]n |
| Heparosan | [-4-GlcAβ1-4-GlcNAc-α1-]n |
| HS | [-4-GlcAβ or IdoA2Xα-1-4-GlcNY3X6X-α1-]n |
| Heparin | [-4-IdoAα or GlcA2Xβ/-1-4-GlcNY3X6X-α1-]n |
GlcA, glucuronic acid; IdoA, iduronic acid; GalNAc, N-acetylgalactosamine; GlcNAc, N-acetylglucosamine; GlcN, glucosamine; Y = Ac, SO3−, or H; X = OH or SO3−. With the exception of HA, sulfo groups can be found at various O-positions in the sugar ring. KS, with a non-acidic [-3Galβ1-4GlcNAcβ1]n backbone, is not listed.
All three GAGs can be found attached to proteins during their duty cycle in the body. For example, HA is initially biosynthesized without a protein-based primer or core, but in some tissues and fluids, a fraction of the HA is covalently attached to inter-α-trypsin inhibitor forming a HA–SHAP complex and it gains a new function (Zhuo et al. 2006). In contrast, both CS and HS are always built in the Golgi on a tetrasaccharide linkage region attached to various secreted or membrane-bound core proteins. Heparin, one of the most widely used drugs in hospitals, is actually a heavily modified form of HS that is released from its core protein and stored intracellularly in mast cell granules (Linhardt 2003).
Due to the abundance of hydroxyl and carboxyl groups as well as in many cases, sulfo groups, GAGs exhibit a hydrophilic, cation-binding physiochemical nature. The resulting hydrated and anionic GAG structure, especially in the case of HA, is an important factor for creating various anatomical structures including the eyes, joints, heart and myriad intricate tissues. Probably more important are the biological interactions of GAGs with various protein-based receptors, adhesions and fibrous elements. In many cases, cell–cell or cell–matrix adhesion in mammals relies on the GAGs. In addition to serving as anchors, GAGs, especially HS and HA, have very important roles in signaling. For example, GAG-stimulated pathways are used to control cellular behavior including proliferation, differentiation and motility.
Many different oligosaccharide sequences differing in sulfate and IdoA content and distribution are present in the sulfated GAGs, giving them the potential to bind numerous protein ligands and effectors in a selective manner. Organizing and maintaining the trillions of human cells, present in ∼200 different cell types, GAGs could certainly make use of such coding. The hypothesis that GAGs have a sulfate code that might be deciphered is a topic of great interest (Bülow and Hobert 2006; Kreuger et al. 2006; Ly et al. 2011; Li, Ly, et al. 2012). Understanding this “code” requires access to pure and chemically defined GAGs or GAG fragments.
Experimental issues and caveats with natural GAG extracts
GAGs are often extracted from animal tissues or prepared through microbial fermentation (Linhardt and Gunay 1999; DeAngelis et al. 2002; Widner et al. 2005; Wang et al. 2010; Bhaskar et al. 2012). Industries operate at the ton scale for heparin (porcine intestine), HA (rooster comb; Streptococcus or recombinant Bacillus bacteria) and CS (bovine or shark cartilage) production (Bhaskar et al. 2012). GAG biosynthesis in vivo results in a variety of sizes and modification patterns; thus, there will also always be some variability in GAGs obtained from natural sources. Furthermore, seasonal variations, environmental factors, feedstocks as well as the intrinsic differences between animal breeds add to the variability of naturally occurring GAGs. Typically, large-scale physicochemical separation technologies cannot resolve completely intact GAGs; thus, most commercial preparations are quite often heterogeneous with respect to size and/or sulfation. In some cases, due to the similar nature of GAGs, cross-contamination of HS and the various forms of CS and DS can also occur. One expensive and generally effective methodology utilizes selective GAG degrading enzymes (e.g. heparin lyases or chondroitin lyases) to remove the undesirable GAG impurities from the target GAG. This approach, however, is not useful for the large-scale (kilogram to ton) production of pure GAGs. Another issue is that other potentially bioactive entities (i.e. virus, prions or growth modulating factors) may remain associated with the GAG chains in extracts; thus, extremely careful preparation and analysis is required for any naturally derived extract including recombinant vertebrate systems.
On the industrial scale, since a large population of animals is required, the supply chain is immense and globally interconnected and has been subject to willful contamination as seen in the 2008 Chinese heparin crisis (Liu et al. 2009). Somewhere in the heparin production process, presumably after the extraction of crude heparin from tissue at the slaughterhouse and before the manufacture of the active pharmaceutical ingredient (pharmacopeial heparin) using certified Good Manufacturing Processes in a pharmaceutical company, the heparin was adulterated with a chemically modified oversulfated CS (Guerrini et al. 2008). This contaminated heparin went undetected because of limitations in the pharmacopeial assays used at the time and entered the market place resulting in serious adverse side effects associated with the death of over 100 patients. This worldwide crisis has led to more stringent control of the manufacture and testing of heparin, but animal sourced GAGs will always pose a risk because their naturally complex structure makes assessing the presence of unknown contaminants difficult and because of the vastly different level of regulations for the food chain and the drug chain.
To avoid these kinds of problems, there is a need to produce GAGs synthetically. One early success story is Arixtra, a sulfated pentasaccharide that binds and activates antithrombin III and is the active sequence in therapeutic heparin. However, the synthesis of this relatively simple pentasaccharide required dozens of chemical steps. The chemical synthesis of oligosaccharides longer than five sugar units with selectivity and reasonable yield is virtually impossible using conventional methods based on orthogonal blocking groups and selective protection and deprotection. In addition, chemical synthesis often results in the undesired formation of toxic byproducts and waste solvents that may contaminate the product and the environment.
This review discusses progress over the last decade in preparing free GAG chains using chemoenzymatic synthesis. Enzymes, the best chemists on the planet, play a key role in preparing these long and complex polymers in an efficient, reliable and greener fashion. Furthermore, this approach simplifies the analysis of the products and provides a platform for generating oligosaccharide libraries.
Strategic planning and methodologies
Nature of the GAG targets
GAGs in the body range from tens to thousands of disaccharide units in length depending on the tissue, age and health conditions. The issue of chain-length polydispersity (the breadth of the polymer size distribution) is important because the physical nature [viscosity and occupancy of three-dimensional (3D) space] as well as biological activities (defined by the ability of proteins to bind) of GAGs change depending on the size. The absence of sharply defined regulation for polysaccharide size control in vivo has always been an issue for the glycobiologist to grapple. In contrast, nucleic acid and protein biosyntheses are template driven and utilize origins/promoters and terminators or start and stop codons, respectively, to afford defined macromolecular products.
In addition, for HS and CS, it is well established that the sulfation pattern is very important for binding to target proteins (Capila and Linhardt 2002; Lindahl and Li 2009). Perhaps the most vexing and challenging issue is that sulfated GAG biosynthesis in any cell-based system always results in heterogeneous products. Multiple enzymes are operating in the Golgi apparatus as the glycoconjugates translocate to the cell surface; therefore, complete sulfation does not always occur resulting in the biosynthesis of a variety of GAG species. Is this observed GAG variety a complex code or a result of cellular limitations? Or is there an inter-relationship between these two possibilities that the human body has exploited after ∼400–600 million years of evolution?
Chemoenzymatic synthesis of GAGs: attributes and hurdles
Chemoenzymatic syntheses and clever processing strategies have now allowed the preparation of more defined GAGs and GAG oligosaccharides as well as libraries of diversity and purity greater than possible by extracting polysaccharides from natural sources alone or by directed synthesis (Pervin et al. 1995; Hileman et al. 1997; Yang et al. 2000; Liu et al. 2010; Xu et al. 2011; Chai et al. 2012). As described above, large complex GAGs are not very amenable to current organic synthesis methods; the repetitive steps of protection, activation, coupling and de-protection are quite laborious and often require much optimization and customization for each step. In addition, the control of product formation is less than ideal; therefore, many separation steps may be needed to remove undesirable isomers (especially the incorrect anomers) as well as the unavoidable failure products that add cost and decrease overall yields. In contrast, enzymes have exquisite stereoselectivity (α- vs β-glycosidic linkages) and regio-selectivity (position on sugar ring, e.g. C2, C3 etc.) for connecting monosaccharide units. Most glycosyltransferases (GTases) rarely appear to make a mistake with a natural substrate, but by design enzymes can be “fooled” into using unnatural substrates [see Uridine 5′-diphosphosphate (UDP)-sugar analogs section].
Fortunately, many animal-derived and many microbial-derived GAG polymerizing and modifying enzymes have been identified, expressed and utilized for reactions in vitro (Table II). The backbones of GAG chains can be efficiently and controllably synthesized in vitro (as discussed in detail later) using the relevant GTases to add the monosaccharide units from UDP-sugar donors onto an acceptor or primer sugar (Figure 1). The bifunctional GTases catalyzing GAG biosynthesis are termed synthases (or polymerases) and are among the most useful of enzymes for the synthesis of polysaccharides in vitro.
Table II.
Useful GAG chemoenzymatic reactions
| Reaction | Catalyst examples |
|---|---|
| Oligosaccharide synthesis: | |
| I. Acceptor + UDP-GlcA → GlcA-acceptor | Mutant PmHAS or PmHS1 or PmHS2 |
| II. Acceptor + UDP-HexNAc → HexNAc-acceptor | Mutant PmHAS; KfiA |
| III. Acceptor + UDP-R analogs → R-acceptor | PmHAS; PmHS2; KfiA |
| Polysaccharide synthesis: | |
| IV. n R׳-oxazoline → GAGa | Testicular hyaluronidase |
| V. Acceptor + UDP-GlcA + UDP-HexNAc → GAG-acceptorb | PmHAS; PmHS1; PmCS; KfoC |
| VI. Acceptor + UDP-R + UDP-R” → GAG[R/R”]-acceptorb | PmHAS; PmHS2; PmHS1/2 Chimeras |
| Chimeric GAG formation via transglycosylation: | |
| VII. GAG1 + GAG2 → GAG1-GAG2c | Testicular hyaluronidase |
| Backbone modification:d | |
| VIII. GAG → N-S-GAG | GlcNAc N-deacetylase/N-STasee |
| IX. GAG(GlcA) → GAG(IdoA) | Uronyl C5-epimerase |
| X. N-GAG → N,O-S-GAG | OSTs |
| XI. N,O-S-GAG → N-S-GAG | GAG sulfatases |
| Backbone transfer/activation: | |
| XII. GAG-protein → GAG-R’” | Patnopecten xylosidase |
The general reaction schemes and examples of the relevant enzymes are summarized. See Figure 2 for a schematic overview. R’, R” = unnatural sugar analog; R’” = another protein or alkyne.
aSee Figure 3 for details. In addition, more than one type of oxazoline intermediate can be co-polymerized to form hybrid GAGs.
bAn exogenous acceptor is not absolutely required for V or VI because UDP-GlcNAc can serve as the initial acceptor (the resulting reducing terminal UDP on the nascent GAG chain is labile). Also, chimeric HA/chondroitin GAGs can be made by this method as well depending on the choice of acceptor, donor and synthase.
cNote that chain fragmentation of the donor GAG occurs to afford extension of the recipient acceptor GAG; only a simplified reaction overview is shown here with the target chimeric GAG depicted.
dThe HS and CS modification enzymes do not cross-react with the non-cognate GAG (i.e. distinct STases, sulfatases and epimerases are required for each GAG class).
eThis reaction can also be done chemically with the HS containing-GlcN[TFA] analog (reactions III or VI) via deprotection with base and chemical sulfonation.
Fig. 1.

GAG elongation reactions. GTases, including the GAG synthases, can transfer an uronic acid (top) or a hexosamine (bottom) from a UDP-sugar donor to an acceptor in vivo or in vitro. In this example, GlcA or GlcNAc, respectively, is transferred to a nascent heparosan chain.
One area that is still being elucidated is the control of the sulfotransferases (STases) that transfer sulfo groups from the donor 3′-phosphoadenosine-5′-phosphosulfate (PAPS) to the GAG chain and the control of epimerases that convert GlcA into IdoA. There are several types of N- and O-Stases (OSTs), enzymes that add a sulfo group to the amine of a glucosamine (GlcN) residue in HS or heparin or to a hydroxyl of a saccharide residue, respectively. In many cases, there are multiple isozymes with distinct substrate specificities within each STases class (Esko and Selleck 2002; Pavão et al. 2006; Peterson et al. 2009; Liu et al. 2012; Sheng et al. 2012). The OST isozymes exhibit selectivity based on the sulfation state of the substrate. Basically, putting on the first sulfo group on the GAG chain affects the efficiency and placement of subsequent sulfation added to the same or proximal monosaccharide by other STases. For example, 2-O-ST requires the prior introduction of an N-sulfo group into the adjacent GlcN residue. Sulfatases may be employed to remove certain sulfo groups as well.
The HS and chondroitin C5-epimerases (C5-epi; note two distinct enzyme classes that only work on a single GAG type) also exhibit complex specificity (Pacheco et al. 2009; Sheng et al. 2012). The epimerization of HS, for example, requires the presence of N-sulfo groups, and the rules for C5-epi acting on CS epimerization are currently being elucidated (Sheng et al. 2012). Therefore, if harnessed correctly and precisely, many target GAG polymers are possible using chemoenzymatic synthesis.
Overview of chemoenzymatic GAG synthesis strategies and challenges
The use of enzymes in vitro to produce or modify sugar polymers by a variety of reactions (listed in Table II) has been reported in a range of synthetic modes. As depicted in Figure 2, these modes include: (a) a semi-synthetic approach where the GAG backbone polymer produced in vivo is extracted from an organism and then modified in vitro (reactions I–III, V–XII), (b) swapping portions of existing GAG chains through transglycosylation reactions in vitro (reaction VII), (c) building GAG chains with degradation enzymes and transition state intermediates in vitro (reaction IV) or (d) building GAG chains with synthases/GTases and UDP-sugars in vitro (reactions I–III, V, VI). In all four formats, after sugar backbone isolation or synthesis, modifications of the chain such as the addition/removal of sulfo groups and/or isomerization of uronic acid moieties are pursued utilizing recombinant enzymes typically cloned from vertebrates (reactions VIII and XI).
Fig. 2.

Schematic of general GAG chemoenzymatic synthesis formats. Various chemoenzymatic synthetic modes allow the enzymatic transformation and/or production in vitro (arrows) of GAG oligosaccharides and polysaccharides (black or white rectangles). In (A), a semi-synthetic approach where the GAG backbone polymer produced in vivo is extracted from an organism and then modified with enzymes (in this example, STase and epimerase to add sulfo groups, S, and create IdoA, I) is employed. In (B), testicular hyaluronidase swaps portions of existing GAG chains (donor, black, and acceptor, white) through transglycosylation reactions to create a series of related hybrid chains with extensions of different sizes in the population. In (C), testicular hyaluronidase builds GAG chains by polymerizing oxazoline disaccharides (box-oxa) to create a series of polymers with a wide size distribution. In (D), GAG synthases/GTases transfer a single sugar or thousands of sugars from UDP-sugar donors (box-UDP) depending on the strategy employed. Three examples are shown here (from top to bottom): (1) uncontrolled polymerization resulting in a series of products with a wide size distribution, (2) single sugar extension of an acceptor (white rectangle) as shown in Figure 1 or (3) acceptor-mediated synchronized extension to form polymers with a narrow sized distribution (note: final product size is controlled by acceptor/donor stoichiometry). These approaches can be performed in series or combined or repetitively repeated to produce a wide range of products, e.g. the GAG backbone products of B–D can be modified as in A etc. For the specific reactions and useful catalysts, see Table II.
The semi-synthetic approach (a) employs naturally occurring polydisperse polysaccharides; thus, there are cost savings on purified UDP-sugars and enzymes, but the polymer size and composition control are sacrificed (as well as the potential for carryover of impurities). Typically, the GAG chain is isolated either in a uniformly unmodified state from a microbial source or in a complex heterogeneous state from an animal tissue before treatment with various modification enzymes (see later sections for examples). Domain swapping through transglycosylation (b) may also use such “natural” GAGs as feedstocks.
When building GAGs with synthases/GTases and UDP-sugars (Figure 1) (d) the GAG chain synthesis reaction may be carried out in one of two preferred formats depending on the target size: (i) step-wise elongation (i.e. one sugar unit at a time) of oligosaccharides in vitro (reactions I–III; Figure 2, mode D.2) or (ii) assembling a GAG polysaccharide in a synchronized polymerization reaction in vitro (reactions V and VI; Figure 2, mode D.3). Both of these formats yield more defined products with narrow size distributions (monodisperse or nearly so) and potentially more controllable compositions than in semi-synthesis of a GAG polymer produced in vivo (a), in domain swapping through transglycosylation (b) or in building GAGs using degradation enzymes and transition state intermediates (c). However, the use of synthases/GTases and UDP-sugars (d) is considerably more expensive as it requires purified components.
A further benefit of building GAGs using either degradation enzymes and transition state intermediates (c) or the use of synthases/GTases and UDP-sugars (d) is that the monosaccharide units in the GAGs produced may be either the naturally occurring sugars or chemically synthesized unnatural analogs possessing either new functionality (e.g. a tag or reactive group) or a potential biological modifier moiety (i.e. a group that prevents or enhances binding, signaling, cleavage etc.) (reaction III or VI).
In any of these formats, a particularly useful strategy (described in greater detail later) is to prepare a library of similar modified GAGs in parallel where separate combinatorial syntheses each utilize an alternate order of addition or the exact choice of the various GTases, STases, epimerase, sulfatases etc. Each member of the library can then be tested for bioactivities with proteins or in cultured cells or in animals and also analyzed to define unique chemical structures responsible for activity. Although the exact catalysts employed (down to the specific isoforms) and the order of addition directs the synthesis, precise control is not often possible when long polysaccharides are subjected to modification reactions. Even if overall disaccharide compositions are similar, their linear arrangement can differ; the potential for various domain structures with different lengths, percentages of modified or unmodified units or orientation (i.e. reducing vs non-reducing terminus) adds another level of complexity to chemoenzymatic synthesis.
After synthesis, it is also critical to verify the molecular structure of the GAG product. However, it is currently beyond analytical capabilities to establish the exact structure of intact GAG chains larger than ∼5000 Da (equivalent to ∼20 sugars) due to their complex physical–chemical properties and their highly variable repeating disaccharide structure (Wang et al. 2010). In contrast, rapid, inexpensive and rather definitive polypeptide or DNA sequencing is performed routinely; even the sequencing of N-glycans is now possible in a number of specialized laboratories. Typically, a GAG polymer must be fragmented (using a bottom-up approach) to perform its disaccharide compositional analysis and oligosaccharide mapping. Recent top-down GAG sequencing methodologies relying on tandem mass spectroscopy have been used to determine the sequence of intact GAG chains of short lengths (e.g. 25–43 monosaccharide units; Ly et al. 2011).
Production and use of natural structure and chimeric backbones
Unsulfated GAGs, HA, chondroitin and heparosan (Table I), can be prepared through microbial fermentation (DeAngelis et al. 2002; Widner et al. 2005; Wang et al. 2010). This in vivo approach using bacteria is currently limited to the production of the natural GAG backbone, and furthermore, it is difficult to prepare GAG backbones of defined molecular weight. Production optimization, through genetic manipulations or by varying fermentation conditions, has been reviewed elsewhere (Cimini et al. 2012; DeAngelis 2012). Several groups have focused on the native Escherichia coli K5 heparosan as the HS/heparinoid starting material that is then modified by various methods (described later in “Modification”) as in reactions VIII-XI (Kuberan et al. 2003; Li, Sheng, et al. 2012). Another interesting recent report for preparing natural GAG backbones is the molecular cloning of the E. coli heparosan production KfiA-D genes (encoding three proteins of the GTase complex and the UDP-glucose dehydrogenase for making UDP-GlcA) into a laboratory strain of E. coli lacking the transporter proteins that are normally employed to secrete capsular polymers. Therefore, these recombinant bacteria make the heparosan polymer intracellularly (Barreteau et al. 2012). The same group also co-expressed a heparosan-degrading enzyme (a lyase) in some constructs to produce short oligosaccharides that would be a useful feedstock for the HS modification enzymes described later.
With respect to making natural GAG polymers in vitro, much progress has been made using two entirely distinct classes of carbohydrate-active enzyme tools. The first class, the GTases (or various mutated versions), build GAGs through the use of UDP-sugar precursors (Figures 1 and 2) that emulate the cellular carbohydrate biosynthetic pathways. For these methods, certain bacterial GTase enzymes have been much more useful than the non-homologous vertebrate counterparts due to better expression, higher stability and other intrinsic properties. Such catalytic workhorses include the Pasteurella HA, chondroitin or heparosan synthases and their homologs, and Escherichia KfiA [transfers α-N-acetylglucosamine (α-GlcNAc) unit of heparosan; Table II] that have been useful for making HA, chondroitin or heparosan polymers (DeAngelis et al. 2003; Jing and DeAngelis 2004; Sismey-Ragatz et al. 2007; Liu et al. 2010). These microbial enzymes add sugars to the non-reducing terminus of acceptors (Figure 1), the same directionality as the vertebrate enzymes. The Streptococcus HA synthases, however, which operate through reducing end addition (the opposite direction as above), are not very useful catalysts in vitro due to their inability to extend exogenously added sugar chains. Surprisingly, the corresponding recombinant E. coli K5 GlcA-Tase enzyme, KfiC, has not been useful as a catalyst in vitro due to expression problems when expressed independently of KfiA and B (Sugiura et al. 2010).
The second class of useful catalysts is a natural HA and CS degrading enzyme, testicular hyaluronidase (HAase; EC 3.2.1.35). This enzyme can be used in the “reverse” direction to make polymers either by transglycosylation (a disaccharide unit is added to the chain terminus rather than the hydrolysis of the chain by a water molecule), swapping domains of existing GAGs (Chen et al. 2009; Kakizaki et al. 2012; Table II, reaction VII) or by the polymerization of disaccharide oxazoline transition state intermediates (Ochiai et al. 2007; Figure 3; Table II, reaction IV). Unfortunately, there is no known comparable enzyme with the ability to make heparosan or HS by either of these approaches.
Fig. 3.
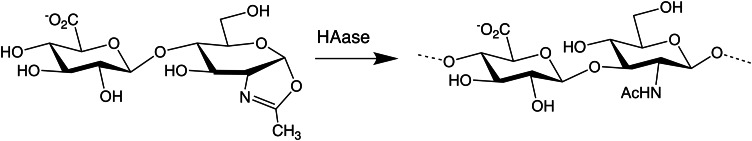
Hyaluronidase-mediated GAG polymerization reaction. The creation of a GAG chain (HA in this example) by repetitive ligation of oxazoline disaccharides by the HA-degrading enzyme is depicted. The cyclic oxazoline group mimics the transition state intermediate where the acetyl group participates in the glycosidic bond cleavage during normal GAG degradation. Unsulfated HA or unsulfated chondroitin or 4-S chondroitin disaccharide intermediates, but not 6-S, may be employed in the “reverse” reaction to synthesize GAG polymers.
Domain swapping through transglycosylation has been used to generate unnatural chimeric HA–CS or CS–HA GAGs, where typically each reaction yields a series of oligosaccharide products with very similar structures (i.e. not a specific targeted product; reaction VII). A variety of GAG polymers composed of HA and/or chondroitin/CS have been reported (Endo and Kakizaki 2012). DS, however, was not useful as an acceptor or a donor GAG for transglycosylation; thus, the IdoA appears to be blocking the reaction.
In a very useful example of using the HA degradation enzyme and transition state intermediates to build GAGs (Figure 3; Table II, reaction IV), 4-O-sulfo-N-acetylgalactosamine (GalNAc) oxazoline intermediates were polymerized with testicular HAase to produce a pure chondroitin-4-sulfate (CS-A) polymer with molecular weights in the range of 104 (Fujikawa et al. 2005). Such a defined material is not found in nature, particularly in common animal sources (e.g. shark, bovine, porcine) where sulfo groups are present on a variable fraction of the 2-, 4- and 6-O-positions within a given CS chain. Unfortunately, the 6-O-sulfo GalNAc oxazoline cannot currently be used produce the chondroitin-6-sulfate (CS-C) polymer in a similar fashion. While chimeric HA–CS molecules can be made by domain swapping through transglycosylation (Figure 2B), or building GAGs using degradation enzymes and transition state intermediates (c), the synthetic control with respect to size distribution is not always possible. These approaches do have the advantage of introducing O-sulfo chondroitin in unique ways not yet possible using synthases/GTases and UDP-sugars (d).
As examples of exquisite synthetic control utilizing GAG GTases, two approaches using synthases/GTases and UDP-sugars (d) have been employed to create either (i) defined oligosaccharides (Table II, reactions I-III) or (ii) quasi-monodisperse polysaccharides (reactions V and VI) (Figure 1). For the goal of making long polymers (e.g. ∼10 to ∼3000 kDa) in a rapid fashion, the raw polymerizing ability of bifunctional synthases, such as PmHAS, PmCS or PmHS1, is useful. However, for shorter chains (i.e. oligosaccharides less than 20–25 monosaccharide units), precise size control needs more careful planning and/or the use of monofunctional GTases such as synthase mutants or a naturally occurring enzyme such as KfiA.
One strategy to harness the synthase for oligosaccharide synthesis is to employ a pair of monofunctional synthase mutants in an alternating stepwise fashion (e.g. reactions I then II and so on; Figure 2, D.2) to elongate a GAG acceptor (DeAngelis et al. 2003; Kane et al. 2006; Chavaroche et al. 2012). Basically, Pasteurella synthases contain two relatively independent GTases in one polypeptide chain; mutation of one active site (e.g. changing the critical DXD motif to NXN) typically does not affect the catalysis at the other site. A particularly useful format is to immobilize the enzyme on a bead to facilitate the removal of the catalyst after a step is finished, because if both enzyme components, the GlcA-Tase and the N-acetylhexosamine (HexNAc)-Tase, are present with both types of UDP-sugars simultaneously, then runaway polymerization will form undesirable longer products. HA chains up to 20 monosaccharides in length have been made by repetitive extension of a tetrasaccharide acceptor using this stepwise approach (DeAngelis et al. 2003). This monofunctional mutant method allows the reaction mixture to contain both UDP-sugars and be continuously processed without GAG intermediate purification steps after each sugar addition, a considerable savings in both labor and time.
Alternatively, in a second oligosaccharide synthesis strategy, any GAG GTase may be used for stepwise extensions (Figure 1) with the following two provisos: only one UDP-sugar (e.g. UDP-GlcA for reaction I) is added at a time with the enzyme and each GAG oligosaccharide intermediate is purified before proceeding to the next elongation step with the second UDP-sugar (in this case, UDP-HexNAc for reaction II) and the next GTase. Heparosan chains of up to 21 monosaccharide units have been made using KfiA or PmHS2 (as the source of GlcA-Tase) in alternating steps (Liu et al. 2010; Xu et al. 2011, 2012). In a third strategy, the acceptor may be immobilized on a surface [e.g. microplate well, nanoparticles in a microfluidic channel etc. (Martin et al. 2009; Powell et al. 2010)] and the reaction mix containing a single GTase and a single UDP-sugar that is exchanged at each step; this synthesis method is ideally suited for the library or array format (Park et al. 2008; Powell et al. 2010; Rogers and Hsieh-Wilson 2012).
Synchronized extension reactions (Table II, reactions V and VI; Figure 2, D.3) have been employed with success to produce nearly monodisperse size-defined polysaccharides (chains greater than 20–25 monosaccharide units). Basically, some non-processive GTase enzymes (e.g. PmHAS, PmCS and PmHS1 synthases) elongate acceptors much faster than they initiate new chains de novo. Therefore, in reactions with acceptor, all the GAG chains are elongated in parallel; thus, the extended polymers all possess very similar lengths or quasi-monodisperse size distributions. In contrast, for reactions without the acceptor (Figure 2, D.1), the new GAG chain is formed through the coupling of the two UDP-sugars in a slow process; the first such chains to be initiated will have longer extension time compared with the various chains started later in the reaction. Thus, such asynchronous extensions yield polysaccharide products with a wide size distribution. The acceptor/donor ratio is varied to control the specific size obtained. Polymers with polydispersities of 1.01–1.2 (close to the monodisperse value of 1) with sizes from ∼20 to 2000 kDa for HA (Jing and DeAngelis 2004) and 20 to 800 kDa for heparosan (Sismey-Ragatz et al. 2007) have been obtained. In contrast, natural GAG polymers have much higher polydispersities (1.3–2.5). With respect to scalability, these synchronized reactions have been performed at the multigram level for commercial purposes (SelectHA™ of Hyalose, LLC; HEPtune™ of Caisson Biotech, LLC).
As an example of a novel chimeric polymer made using synthases/GTases and UDP-sugars (d), the Pasteurella HA synthase will assemble block polymers of HA-unsulfated chondroitin or HA–CS attached together; these structures combine the two GAG components of aggrecan proteoglycan assemblies without involving a core protein (Tracy et al. 2007). Similarly, the chondroitin synthases will make chondroitin-HA or chondroitin-CS polymers. The basis for these syntheses is that the two enzymes have discrete UDP-sugar donor specificity, but they can extend non-cognate GAG acceptors (i.e. either synthase can elongate HA or chondroitin acceptors as the HexNAc C4 position is not key for acceptor substrate recognition). The size of the extension is controlled by stoichiometry of the GAG acceptor and the UDP-sugars employed. As mentioned earlier, the alternative method to create chimeric GAGs, transglycosylation (Table II, reaction VII), is somewhat less controllable.
UDP-sugar analogs
Monosaccharides, in the form of UDP-sugar, are donors in the polysaccharide biosynthetic pathway, being transferred to a glycosyl acceptor by a GTase or a synthase (Watkins 1986). Natural and unnatural GAGs can be prepared by using recombinant GAG synthases and chemically modified UDP-sugar nucleotides (reaction III or VI; Figure 1) (Sismey-Ragatz et al. 2007). A major current, but fading, limitation to the enzymatic preparation of GAG polysaccharides and oligosaccharides is the availability and cost of sugar nucleotides and their analogs and derivatives due to the difficulty of their syntheses (Karst and Linhardt 2003). Although they can be prepared in several ways, a purely chemical approach requires a long and tedious synthesis and yields of UDP-donor are generally low and the production of kilogram amounts is probably not commercially viable. Chemical synthesis is particularly important in preparing unnatural UDP-sugars such as UDP-IdoA (Weïwer et al. 2008). Chemoenzymatic synthesis offers an alternative for the synthesis of natural UDP-sugars and UDP-sugars containing some minor modifications (Figure 4). Furthermore, chemoenzymatic synthesis eliminates the use of protection and deprotection steps required for chemical synthesis, circumventing the difficulties inherent to the formation of a pyrophosphate bond.
Fig. 4.
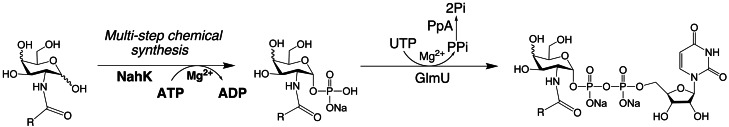
Enzymatic synthesis of UDP-GlcNAc and UDP-GalNAc analogs. A variety of UDP-sugar precursor analogs can be produced. Two alternative strategies are employed to create the 1-phosphorylated monosaccharide: chemical synthesis (Masuko et al. 2012; italics) or enzymatic synthesis (Chen et al. 2011; bold; NahK = N-acetylhexosamine 1-kinase). Then UDP is exchanged for the phosphate by GlmU to create the UDP-sugar. PpA (pyrophosphatase) drives the reaction by cleaving pyrophosphate, PPi, to inorganic phosphate, Pi. Donor analogs are then useful substrates for production of the GAG chain by synthases or GTases. The new functional group, R, can alter the GAG's chemical reactivity, biological response and/or chemophysical properties. In addition to the C2 position of the HexNAc (shown here), C6 has been relatively amenable to modification.
Wang et al. reported the enzymatic synthesis of UDP-GlcNAc/UDP-GalNAc analogs using recombinant E. coli N-acetylglucosamine-1-phosphate uridyltransferase (GlmU; Guan et al. 2009). However, low yields were reported for N-modified GalNAc-1-phosphate analogs, a problem that was later resolved using recombinant human UDP-GalNAc pyrophosphorylase (AGX1) for the synthesis of UDP-GalNAc analogs (Guan et al. 2010). Chen et al. (2011) reported a one-pot three-enzyme synthesis of UDP-GlcNAc derivatives using a HexNAc-1-kinase and GlmU but only a few derivatives were successfully synthesized (Figure 4). Alternatively, eight different GalNAc-1-phosphate analogs have recently been synthesized (Figure 4) and their recognition by the GlmU enzyme has been studied (Masuko et al. 2012).
Out of the eight HexNAc-1-phosphate analogs tested, six were accepted as substrates by GlmU, affording the corresponding UDP-sugar nucleotides in high yields. GlmU accepted neither 2-deoxy-2-azidoglucose (GlcN3)-1-phosphate nor GlcN-1-phosphate, so these two additional UDP-sugars required chemical synthesis. GlcNAc-1-phosphate and N-trifluoroacetylglucosamine (GlcN[TFA])-1-phosphate were excellent substrates, yielding more than 70% conversion. Since GalNAc-1-phosphate was accepted by GlmU, the 4-OH group appears not to play a critical role in enzyme recognition. Both GlcN-alkyne-1-phosphate and GalN-alkyne-1-phosphate gave moderate yields of UDP-sugars using GlmU. Based on these observations, it appears that the amide bond on the 2-amino functional group is essential for substrate recognition by GlmU but the configuration at C4 is not important.
However, even if one class of enzymes can make such analogs, other catalysts such as the GTases are not assured of having the same promiscuity. Therefore, these novel UDP-sugar nucleotide analogs were then tested for incorporation by various GAG synthases in vitro. PmHAS and PmCS produce HA and chondroitin polysaccharides, respectively (Jing and DeAngelis 2003). PmHS1 and PmHS2 are two homologous isozymes that both can polymerize heparosan, the precursor of HS and heparin (Sismey-Ragatz et al. 2007). The hexosamine analogs were tested in a chain polymerization assay using radioactively labeled UDP-[3H]GlcA; a radioactive signal results only if the hexosamine is co-polymerized into the disaccharide repeats of a GAG product (Jing and DeAngelis 2000; Sismey-Ragatz et al. 2007; Masuko et al. 2012).
PmCS used both UDP-GalNAc and UDP-GalN-alkyne. PmHAS utilized UDP-GlcNAc, UDP-GlcN-alkyne, UDP-GlcN[TFA], UDP-GlcN-alkene and UDP-GlcN-N-tert-butoxycarbonyl ([tBoc]). PmHS1 utilized UDP-GlcNAc, UDP-GlcN-alkyne, UDP-GlcN-alkene and UDP-GlcN-[tBoc]. PmHS2 utilized UDP-GlcNAc, UDP-GlcN-alkyne, UDP-GlcN[TFA], UDP-GlcN-alkyne, UDP-GlcN-fluorenylmethyloxycarbonyl ([Fmoc]) and UDP-GlcN-[tBoc] (Masuko et al. 2012). Unfortunately, neither PmHS1 nor PmHS2 nor PmCS accepted UDP-IdoA as a donor (Weïwer et al. 2008); thus, the C5-epi will continue to be needed to make natural HS or DS GAGs.
Production of GAG analog backbones
Two approaches have been utilized to prepare novel polymers with unnatural structures unlike animal-derived GAGs: the use of known GAG GTases with UDP-sugar analogs that substitute for a natural sugar unit (Table II, reactions III and VI), and novel synthases that polymerize non-animal GAG structures. In the first case, sugar analogs (discussed in the previous section) have been tested that (i) increase the hydrophobicity of the normal HexNAc unit (e.g. GlcN-propanoyl or GlcN-butanoyl with one or two extra CH2 groups, respectively), (ii) add a new chemical functionality (e.g. 6-azido-GlcNAc, GlcN-propyne etc.) or (iii) allow selective deprotection to reveal a new functionality (e.g. GlcN[TFA] to GlcN using base treatment). In summary, some synthases can use certain UDP-sugar analogs as both a donor (to add onto a growing chain) and an acceptor (to serve as a site for extension). Surprisingly, reagents with substitutions at the 6-OH or the 2-N positions of the HexNAc can be accommodated by multiple GTase enzymes.
A particularly interesting case is the UDP-GlcN[TFA] analog; the Pasteurella PmHAS, PmHS2 and the E. coli KfiA can utilize this analog quite well. The TFA protecting group can be removed selectively by mild volatile base treatment, without removal of any natural acetyl groups or cleavage of the GAG chain. The new amine in a GAG chain is useful for attaching amine-reactive molecules (e.g. NHS-esters, isothiocyanates) or for N-sulfonation by either an enzyme (reaction VIII) or a simple chemical reagent. Therefore, the careful placement of a GlcN[TFA] unit in an oligosaccharide using reaction III (Liu et al. 2010) or a polysaccharide by reaction VI (Sismey-Ragatz et al. 2007; Otto et al. 2012) will direct selective N-sulfo modification. In the case of HS biosynthesis, the introduction of an N-sulfo group is the first step of heparosan modification, thus allowing the glycochemist to emulate nature.
A technical issue on this front, however, is that the native enzymes are sometimes not ideal. Specifically, PmHS2 uses the TFA analog quite well, but this enzyme does not make monodisperse polymers and is prone to starting chains de novo rather than building exclusively on existing chains. In contrast, PmHS1 does not use the TFA analog with high efficiency, but makes narrow size distribution heparosan chains and prefers to elongate existing chains. Therefore, a panel of PmHS1/PmHS2 chimeric enzymes was generated and screened for both analog use and acceptor elongation activity (Otto et al. 2012). A new catalyst with the combination of useful attributes from both synthases was shown to be useful in forming heparosan with defined blocks or segments of GlcA-GlcNAc and GlcA-GlcN[TFA] repeats. Goals for such synthetic HS precursors include determining how the initial N-sulfate domain controls/influences the pattern of epimerization and/or the introduction of O-sulfo groups.
In a separate pursuit to expand the GAG toolbox, bioinformatic screens for homologs/analogs of known synthases seeks to find catalysts with alternative transferase activities. A novel GAG producing enzyme, CtTS, was identified that produces the heparosan analog, testosteronan (Otto et al. 2011). This new polymer has α-linked rather than the β-linked GlcA found in heparosan (Figure 5). Some HS-binding proteins may tolerate the testosteronan backbone, whereas others will not; therefore, alternative selectivity may be observed due to its alternative glycosidic conformation.
Fig. 5.

Comparison of unmodified heparosan, epimerized heparosan and testosteronan (top to bottom). The differences in the structures of the various GAG polymers may allow for recognition by certain HS-binding proteins, but not others, thus opening possibilities for discovery of more selective ligands.
Modification of GAG backbones with STases, epimerases and sulfatases
The biosynthesis of heparin and HS in animals is achieved by a similar group of enzymes, while certain specialized enzymes might be required for the complete biosynthesis of heparin. The pathway involves multiple enzymes, including HS polymerase, epimerase and STases (Figure 6). The pathway covers the synthesis of a linkage region tetrasaccharide, backbone elongation and backbone modification. In some cases, sulfatases then remodel the HS chain by removing certain O-sulfo groups. Although CS chains are started similarly with the same linkage tetrasaccharide, the biosynthetic pathways then diverge using distinct elongation and modification enzymes.
Fig. 6.

The biosynthetic pathway of HS. To maintain clarity, the steps involved in the synthesis of the backbone have been excluded. The modifications require a series of specialized enzymes, including N-deacetylase/N-STase, C5-epimerase, 2-OST, 6-OST and 3-OST. The modification sites at each step are boxed. All of these transformations have been reproduced in vitro via chemoenzymatic synthesis.
In nature, the sulfated GAGs assemble while attached to a core protein of a proteoglycan via a linkage region (GlcA-Gal-Gal-Xyl), which requires four enzymes for its assembly. The first GlcNAc is added by EXTL3, then the HS polymerase (EXT complexes) adds alternating GlcA and GlcNAc units generating an unsulfated and unepimerized HS backbone. The backbone is then modified by N-deacetylase/N-STase, C5-epi (converting GlcA to IdoA), 2-OST, 6-OST and 3-OST to produce the fully modified HS. In contrast, CS is initiated by adding a β-linked GalNAc to the linkage tetrasaccharide followed by elongation with the unsulfated GlcA-GalNAc backbone via chondroitin polymerases (not homologous to EXTs nor bacterial synthases). The chain is modified by various chondroitin-specific STases (4-, 6- and/or 4,6-OSTs) depending on the tissue and developmental state. To create DS, two C5-epi enzymes (distinct from the HS-acting enzyme) convert elected GlcA residues to IdoA residues to afford DS.
An abundant source of enzymes for carrying out the chemoenzymatic modification of HS and heparin are recombinant vertebrate proteins expressed in E. coli, insect cells and yeast (Figure 6). The use of Kluyveromyces yeast to make key modification enzymes in readily purified, secreted soluble forms facilitates what was once a laborious process (Zhou et al. 2011). In the same report, a method to prepare PAPS, the sulfo donor that all STases require, in a much more economical fashion (∼1/5000 of previous costs) was described. These HS-active enzymes are currently used to modify the polysaccharides and oligosaccharides in vitro to synthesize products with different sulfation patterns. The applied use of the comparable recombinant CS-active enzymes has recently been used to make a variety of 4-, 6- or 4,6-O-sulfo-chondroitin chains from an unsulfated backbone made with the E. coli K4 chondroitin polymerase (Sugiura et al. 2012).
Defined heptasaccharide analogs of Arixtra®, the synthetic drug prepared laboriously using organic chemistry, were prepared by stepwise chemoenzymatic synthesis by Table II, Reactions I–III (Figure 2, D.2) followed by reactions VIII–X (Xu et al. 2011). First, a disaccharide acceptor was extended stepwise by incubation with KfiA and a UDP-hexosamine (either UDP-GlcNAc or UDP-GlcN[TFA]) then by PmHS2 and UDP-GlcA; the process was repeated to make the GAG backbone. Strategic positioning of the TFA-containing units directed the precise installation of the N-sulfo groups after basic deprotection and N-STase treatment. The oligosaccharides were treated either during or after completion of the backbone synthesis process with epimerase and OSTs (order of addition 2-, 6-, 3-OST). These 10- or 12-step processes gave ∼45 or ∼37% yields, respectively, and the products possessed very similar anticoagulant potencies to Arixtra®.
Another superb example by the same group was the chemoenzymatic synthesis of oligosaccharides up to 21-mers containing the binding sites for both antithrombin III (AT) and thrombin separated by a short linker (Xu et al. 2012). While not structurally homogenous, due to the incomplete action of the GAG modification enzymes, C5-epi and 2-OST, the 21-mer product had the desirable anticoagulant activity.
Another recent advance was reported where the anticoagulant properties of naturally occurring heparin were improved through remodeling with recombinant human sulfatase (Sulf2) to remove specific 6-O-sulfo groups (Table II, reaction XI) involved in platelet factor 4 binding, which did not affect binding of antithrombin III (Pempe et al. 2012). This modification should reduce the detrimental side effect of heparin-induced thrombocytopenia. Another promising target area is non-anticoagulant heparins; basically, animal-derived heparin has many activities with potential for use in cancer and inflammation, but these leads cannot be fully exploited because of bleeding complications that would be associated with most current preparations. Thus, while semi-synthetic methodologies (a) may be useful for a safer and more controlled supply chain, in the long term, building GAG chains with synthases/GTases and UDP-sugars (d) may provide the safest products.
Synthesis of neoproteoglycans
In further steps toward the synthesis of artificial glycoconjugates, methods have been reported with utility to construct proteoglycan mimics by attaching natural GAG chains to other macromolecules. One interesting example of the semi-synthetic approach involves isolating then functionalizing a CS chain from a proteoglycan through a transglycosylation reaction (Table II, reaction XII; Yamaguchi et al. 2010). Basically, the Patnopecten endo-β-xylosidase cuts the GAG chain at the linker tetrasaccharide, but when cleaving in the presence of propargyl alcohol (containing a alkyne functionality), this reagent is added to the reducing end of the CS chain instead of a water molecule. The released GAG is suitable for “click” chemistry with azido groups present on another molecule.
The same group in another report described employing transglycosylation with the same xylosidase to reconstruct neoglycoproteins; the GAG chain was added to a suitable acceptor peptide lacking a GAG chain (Endo and Kakizaki 2012). The same report also described the use of a trimmed decorin proteoglycan as the acceptor for adding on new GAG chains with HAase using transglycosylation (Figure 2B; Table II, reaction VII). Although it is difficult to control precisely the GAG portions of these novel products, such strides are promising first steps for re-creating or re-modeling proteoglycans to assess the biological roles of both the GAG and the core protein molecules.
Production of GAG libraries
Since the first carbohydrate microarray appeared in the literature in 2002 (Wang et al. 2002), such microarrays have been promising tools to probe carbohydrate–protein interactions, particularly in the developing field of glycomics (Wang 2003; Noti et al. 2006; Stevens et al. 2006; Ban and Mrksich 2008; Liang et al. 2008; Park et al. 2008). Over a thousand spots on a microarray chip can be conveniently monitored using very small amounts of both ligand and analyte to elucidate the specific antibodies useful in the diagnosis of diseases, to identify inhibitors of carbohydrate–protein interactions and to study events involving carbohydrate–cellular interactions (Feizi et al. 2003; Flitsch and Ulijn 2003; Wang 2003; de Paz, Noti, et al. 2006; de Paz and Seeberger, 2006; de Paz, Spillmann, et al. 2006;Noti et al. 2006; Stevens et al. 2006; Zhi et al. 2006; de Paz et al. 2007; Ban and Mrksich 2008; Liang et al. 2008). Despite the importance of measuring binding events on carbohydrate microarrays, this technology has not been extensively applied to probe protein binding to GAGs because of their heterogeneity. 2D surface chemistries for site-specific and covalent immobilization generally result in low signal intensity and substantial non-specific binding of target proteins because of an insufficient number of binding sites and the presence of surface-protein interactions. Chip design using polymers such as hydrogels or dendrimers provide a 3D structure with more binding sites required for signal amplification (Seurynck-Servoss et al. 2007). Specific interactions between heparin and AT were observed on a poly-l-lysine-based 3D HepGlyChip providing enhanced signal to noise ratio and minimized non-specific binding of AT (Park et al. 2008).
Microfluidics and lab-on-a-chip technologies enable reactions on the micro scale and the nano scale, reducing reagent consumption and analysis time, increasing reaction control and throughput, and providing opportunities for full automation (Srinivasan et al. 2004). Two types of microfluidic systems have been developed: (i) channel microfluidics, which involves fluid flow in patterned channels; and (ii) digital microfluidics, wherein open droplet movement occurs through the process of electrowetting on a 2D grid-like platform. Digital microfluidics has gained popularity by eliminating many of the constraints associated with fixed channels (Wheeler 2008) and allowing individual droplets in a biochemical array to be addressed. Some previous applications of digital microfluidics include glucose and other enzyme-based assays, preparation of protein samples for matrix-assisted laser desorption/ionization mass spectrometry, polymerase chain reaction and cell-based assays (Srinivasan et al. 2004; Chang et al. 2006; Moon et al. 2006; Barbulovic-Nad et al. 2008; Miller and Wheeler 2008; Kwon et al. 2012).
Digital microfluidics chips consist of an array of electrodes coated with an insulator followed by a hydrophobic layer (Figure 7). Droplet movement in digital microfluidics is driven by electrowetting, the ability of a surface to tune its wettability by the application of electrical pulses. To operate a digital microfluidic device, a droplet of fluid is placed over one electrode and then a voltage is applied to an adjacent electrode, causing the insulator above that electrode to become charged. This process makes the destination electrode more hydrophilic causing the droplet to move (Wheeler 2008). This wettability of the surface is reversible, thus allowing the droplet to be moved to an adjacent electrode of choice. In this manner, sample-containing droplets may be transported, mixed and separated on the chip.
Fig. 7.

Digital microfluidic chip used to perform enzymatic reactions. The top panel is a magnified picture of a digital microfluidic chip of gold electrodes, buffer reservoirs and wires on a glass substrate coated with Teflon. A glass coverslip coated with a transparent and conductive layer coated with a thin layer of Teflon is laid on top of a spacer on this digital microfluidic chip (bottom panel). The gap between the digital microfluidic chip electrode and the glass coverslip is filled with internal silicone oil (this serves to minimize the evaporation of these nanoliter-sized droplets. Samples in aqueous buffer containing Golgi enzymes, UDP-sugars and PAPS are placed on buffer reservoirs through holes in the coverslip and are held in position by applying a voltage to the reservoirs through a connecting wire (Martin et al. 2009). When a voltage is applied to a buffer reservoir or electrode-square the hydrophobicity of the Teflon layer is overcome through electrowetting making the aqueous droplet adhere to the newly hydrophilic surface. Droplets can be split off from the reservoir droplet and moved to the adjacent electrode-square by applying voltages. Once the droplet is transferred to an electrode it can be similarly moved to the adjacent electrode, merged with another droplet or split into two droplets on separate electrode-squares. Thus, enzymes can be added to substrates and reactions can be conducted with both temporal and spatial control.
HS was modified by 3-OST-1 using digital microfluidics to afford an HS with an increased affinity for the anticoagulant protein AT (Martin et al. 2009). This research represents a first step towards the construction of an artificial Golgi organelle that may serve as a test-bed to better understand how the natural Golgi controls the biosynthesis of GAGs.
Summary and future directions
In the last decade, various groups have made great strides in producing GAGs with structures identical or similar to natural molecules in vitro. With respect to molecular weight control of the GAG backbone, scientists have bested the natural products as demonstrated by the creation of nearly monodisperse artificial preparations. For HS, the positioning of N-sulfo groups can be done very precisely within a GAG chain in vitro. But the lack of precise control in introducing O-sulfo groups and epimerizing uronic acid residues in vitro can still humble scientists trying to prepare lab-made GAGs. Despite these challenges, some artificial GAGs are still more defined than the heterogeneous GAGs extracted from animal sources. Perhaps microfluidic-based syntheses will allow further refinement of chemoenzymatic methods and result in higher efficiencies and/or better control of sequential backbone modifications.
The forthcoming availability of defined GAGs promises to help both basic and applied science in making discoveries towards structure/function relationships of GAGs. The availability of synthetic GAGs should help sort out the role of sulfated domain structures in fine-tuning the biological outputs. The availability of such designer GAGs, produced in vitro, promises more consistent and selective new therapeutic products and a better-secured supply chain. We look forward to the next decade of new developments in synthetic control to improve scale-up, allowing broader exploration and access of these critically important biopolymers.
In the distant future, metabolic engineering may be used to construct GAGs in cultured animal or microbial cells, allowing a single cell line to produce the products with desired structures. The production of GAGs using a metabolic engineering approach is no easy task. Even seemingly simple manipulation, such as switching the production of HS to heparin in cultured Chinese hamster ovary (CHO) cells, poses an incredible challenge (Baik et al. 2012). For example, the overexpression of NDST-2 and 3-OST-1 in CHO cells cannot yet yield polysaccharides that resemble heparin, suggesting gaps in our fundamental understanding of control in GAG biosynthesis. Yeast cells, also containing a Golgi, have been engineered to prepare humanized glycoproteins (Wildt and Gerngross 2005). Ultimately, it might even be possible to engineer sulfated GAGs in bacteria (Wang et al. 2011). Despite those challenges, a cleverly designed metabolic engineering approach might one day provide a unique solution to simplify the multi-enzyme synthesis of GAGs.
Funding
We also thank funding agencies including the National Institutes of Health (R01GM38060, GM090127, HL096972, HL062244, HL62244), the National Science Foundation and the Oklahoma Center for Advancement of Science & Technology.
Conflict of interest
None declared.
Abbreviations
AT, antithrombin III; CHO, Chinese hamster ovary; CS, chondroitin sulfate; DS, dermatan sulfate; [Fmoc], fluorenylmethyloxycarbonyl; GAG, glycosaminoglycan; GalNAc, N-acetylgalactosamine; GlcA, glucuronic acid; GlmU, N-acetylglucosamine-1-phosphate uridyltransferase; GlcN, glucosamine; GlcN3, 2-deoxy-2-azidoglucose; GlcNAc, N-acetylglucosamine; GTase, glycosyltransferase; HA, hyaluronan or hyaluronic acid; HAase, hyaluronidase; HexNAc, N-acetylhexosamine; HS, heparan sulfate; IdoA, iduronic acid; KS, Keratan sulfate; OST, O-STase; PAPS, 3′-phosphoadenosine-5′-phosphosulfate; STase, sulfotransferase; Tase, transferase; [tBoc], N-tert-butoxycarbonyl; [TFA], trifluoroacetyl; UDP, uridine 5′-diphosphosphate.
Acknowledgements
We thank our colleagues in the field for numerous observations and exciting discussions on GAG chemistry and glycobiology.
References
- Baik JY, Gasimli L, Yang B, Datta P, Zhang F, Glass CA, Esko JD, Linhardt RJ, Sharfstein S. Metabolic engineering of Chinese hamster ovary cells: Towards a bioengineered heparin. Metabol Engineer. 2012;14:81–90. doi: 10.1016/j.ymben.2012.01.008. doi:10.1016/j.ymben.2012.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ban L, Mrksich M. On-chip synthesis and label-free assays of oligosaccharide arrays. Angew Chem Int Ed. 2008;47:3396–3399. doi: 10.1002/anie.200704998. doi:10.1002/anie.200704998. [DOI] [PubMed] [Google Scholar]
- Barbulovic-Nad I, Yang H, Park PS, Wheeler AR. Digital microfluidics for cell-based assays. Lab Chip. 2008;8:519–526. doi: 10.1039/b717759c. doi:10.1039/b717759c. [DOI] [PubMed] [Google Scholar]
- Barreteau H, Richard E, Drouillard S, Samain E, Priem B. Production of intracellular heparosan and derived oligosaccharides by lyase expression in metabolically engineered E. coli K-12. Carbohydr Res. 2012;360:19–24. doi: 10.1016/j.carres.2012.07.013. doi:10.1016/j.carres.2012.07.013. [DOI] [PubMed] [Google Scholar]
- Bhaskar U, Sterner E, Hickey AM, Onishi A, Zhang F, Dordick JS, Linhardt RJ. Engineering of routes to heparin and related polysaccharides. Applied Microbiol Biotech. 2012;93:1–16. doi: 10.1007/s00253-011-3641-4. doi:10.1007/s00253-011-3641-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop JR, Schuksz M, Esko JD. Heparan sulphate proteoglycans fine-tune mammalian physiology. Nature. 2007;446:1030–1037. doi: 10.1038/nature05817. doi:10.1038/nature05817. [DOI] [PubMed] [Google Scholar]
- Bülow HE, Hobert O. The molecular diversity of glycosaminoglycans shapes animal development. Annu Rev Cell Dev Biol. 2006;22:375–407. doi: 10.1146/annurev.cellbio.22.010605.093433. doi:10.1146/annurev.cellbio.22.010605.093433. [DOI] [PubMed] [Google Scholar]
- Capila I, Linhardt RJ. Heparin-protein interactions. Angew Chem Int Ed. 2002;41:390–412. doi: 10.1002/1521-3773(20020201)41:3<390::aid-anie390>3.0.co;2-b. doi:10.1002/1521-3773(20020201)41:3<390::AID-ANIE390>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- Chai C, Solakyildirim K, Yang B, Beaudet JM, Weyer A, Linhardt RJ, Zhang F. Semi-synthesis of chondroitin sulfate-E from chondroitin sulfate-A. Carbohydr Polymers. 2012;87:822–829. doi: 10.1016/j.carbpol.2011.08.075. doi:10.1016/j.carbpol.2011.08.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang YH, Lee GB, Huang FC, Chen YY, Lin JL. Integrated polymerase chain reaction chips utilizing digital microfluidics. Biomed Microdevices. 2006;8:215–225. doi: 10.1007/s10544-006-8171-y. doi:10.1007/s10544-006-8171-y. [DOI] [PubMed] [Google Scholar]
- Chavaroche AA, van den Broek LA, Boeriu C, Eggink G. Synthesis of heparosan oligosaccharides by Pasteurella multocida PmHS2 single-action transferases. Appl Microbiol Biotechnol. 2012;95:1199–1210. doi: 10.1007/s00253-011-3813-2. doi:10.1007/s00253-011-3813-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F, Kakizaki I, Yamaguchi M, Kojima K, Takagaki K, Endo M. Novel products in hyaluronan digested by bovine testicular hyaluronidase. Glycoconj J. 2009;26:559–566. doi: 10.1007/s10719-008-9200-2. doi:10.1007/s10719-008-9200-2. [DOI] [PubMed] [Google Scholar]
- Chen Y, Thon V, Li Y, Yu H, Ding L, Lau K, Qu J, Hie L, Chen X. One-pot three-enzyme synthesis of UDP-GlcNAc derivatives. Chem Commun. 2011;47(38):10815–10817. doi: 10.1039/c1cc14034e. doi:10.1039/c1cc14034e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cimini D, Rosa MD, Schiraldi C. Production of glucuronic acid-based polysaccharides by microbial fermentation for biomedical applications. Biotechnol J. 2012;7:237–250. doi: 10.1002/biot.201100242. doi:10.1002/biot.201100242. [DOI] [PubMed] [Google Scholar]
- DeAngelis PL. Evolution of glycosaminoglycans and their glycosyltransferases: Implications for the extracellular matrices of animals and the capsules of pathogenic bacteria. Anat Rec. 2002;268:317–326. doi: 10.1002/ar.10163. doi:10.1002/ar.10163. [DOI] [PubMed] [Google Scholar]
- DeAngelis PL. Glycosaminoglycan polysaccharide biosynthesis and production: Today and tomorrow. Appl Microbiol Biotechnol. 2012;94:295–305. doi: 10.1007/s00253-011-3801-6. doi:10.1007/s00253-011-3801-6. [DOI] [PubMed] [Google Scholar]
- DeAngelis PL, Gunay NS, Toida T, Mao W-J, Linhardt RJ. Identification of the capsular polysaccharides of type D and F Pasteurella multocida as unmodified heparin and chondroitin, respectively. Carbohydr Res. 2002;337:1547–1552. doi: 10.1016/s0008-6215(02)00219-7. doi:10.1016/S0008-6215(02)00219-7. [DOI] [PubMed] [Google Scholar]
- DeAngelis PL, Oatman LC, Gay DF. Rapid chemoenzymatic synthesis of monodisperse hyaluronan oligosaccharides with immobilized enzyme reactors. J Biol Chem. 2003;278:35199–35203. doi: 10.1074/jbc.M306431200. doi:10.1074/jbc.M306431200. [DOI] [PubMed] [Google Scholar]
- de Paz JL, Moseman EA, Noti C, Polito L, von Andrian UH, Seeberger PH. Profiling heparin-chemokine interactions using synthetic tools. ACS Chem Biol. 2007;2:735–744. doi: 10.1021/cb700159m. doi:10.1021/cb700159m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Paz JL, Noti C, Seeberger PH. Microarrays of synthetic heparin oligosaccharides. J Am Chem Soc. 2006;128:2766–2767. doi: 10.1021/ja057584v. doi:10.1021/ja057584v. [DOI] [PubMed] [Google Scholar]
- de Paz JL, Seeberger PH. Recent advances in carbohydrate microarrays. QSAR Comb Sci. 2006;25:1027–1032. doi:10.1002/qsar.200640081. [Google Scholar]
- de Paz JL, Spillmann D, Seeberger PH. Microarrays of heparin oligosaccharides obtained by nitrous acid depolymerization of isolated heparin. Chem Commun. 2006;29:3116–3118. doi: 10.1039/b605318a. doi:10.1039/b605318a. [DOI] [PubMed] [Google Scholar]
- Endo M, Kakizaki I. Synthesis of neoproteoglycans using the transglycosylation reaction as a reverse reaction of endo-glycosidases. Proc Jpn Acad Ser B Phys Biol Sci. 2012;88:327–344. doi: 10.2183/pjab.88.327. doi:10.2183/pjab.88.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esko JD, Selleck SB. Order out of chaos: Assembly of ligand binding sites in heparan sulfate. Annu Rev Biochem. 2002;71:435–471. doi: 10.1146/annurev.biochem.71.110601.135458. doi:10.1146/annurev.biochem.71.110601.135458. [DOI] [PubMed] [Google Scholar]
- Feizi T, Fazio F, Chai W, Wong CH. Carbohydrate microarrays—a new set of technologies at the frontiers of glycomics. Curr Opin Struct Biol. 2003;13:637–645. doi: 10.1016/j.sbi.2003.09.002. doi:10.1016/j.sbi.2003.09.002. [DOI] [PubMed] [Google Scholar]
- Flitsch SL, Ulijn RV. Sugars tied to the spot. Nature. 2003;421:219–220. doi: 10.1038/421219a. doi:10.1038/421219a. [DOI] [PubMed] [Google Scholar]
- Fujikawa S, Ohmae M, Kobayashi S. Enzymatic synthesis of chondroitin 4-sulfate with well-defined structure. Biomacromolecules. 2005;6:2935–2942. doi: 10.1021/bm050364p. doi:10.1021/bm050364p. [DOI] [PubMed] [Google Scholar]
- Guan W, Cai L, Fang J, Wu B, Wang PG. Enzymatic synthesis of UDP-GlcNAc/UDP-GalNAc analogs using N-acetylglucosamine 1-phosphate uridyltransferase (GlmU) Chem Commun. 2009;45:6976–6978. doi: 10.1039/b917573c. doi:10.1039/b917573c. [DOI] [PubMed] [Google Scholar]
- Guan W, Cai L, Wang PG. Highly efficient synthesis of UDP-GalNAc/GlcNAc analogues with promiscuous recombinant human UDP-GalNAc pyrophosphorylase AGX1. Chem Eur J. 2010;16(45):13343–13345. doi: 10.1002/chem.201002315. doi:10.1002/chem.201002315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrini M, Beccati D, Shriver Z, Naggi AM, Bisio A, Capila I, Lansing J, Guglieri S, Fraser B, Al-Hakim A, et al. Oversulfated chondroitin sulfate is a major contaminant in heparin associated with adverse clinical events. Nat Biotechnol. 2008;26:669–775. doi: 10.1038/nbt1407. doi:10.1038/nbt1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hileman RE, Smith AE, Toida T, Linhardt RJ. Preparation and structure of heparin lyase-derived heparan sulfate oligosaccharides. Glycobiology. 1997;7:231–239. doi: 10.1093/glycob/7.2.231. doi:10.1093/glycob/7.2.231. [DOI] [PubMed] [Google Scholar]
- Jing W, DeAngelis PL. Dissection of the two transferase activities of the Pasteurella multocida hyaluronan synthase: Two active sites exist in one polypeptide. Glycobiology. 2000;10:883–889. doi: 10.1093/glycob/10.9.883. doi:10.1093/glycob/10.9.883. [DOI] [PubMed] [Google Scholar]
- Jing W, DeAngelis PL. Enzymatic synthesis of UDP-GlcNAc/UDP-GalNAc analogs using N-acetylglucosamine 1-phosphate uridyltransferase (GlmU) Glycobiology. 2003;13:661–671. doi: 10.1093/glycob/cwg085. doi:10.1093/glycob/cwg085. [DOI] [PubMed] [Google Scholar]
- Jing W, DeAngelis PL. Synchronized chemoenzymatic synthesis of monodisperse hyaluronan polymers. J Biol Chem. 2004;279:42345–42349. doi: 10.1074/jbc.M402744200. doi:10.1074/jbc.M402744200. [DOI] [PubMed] [Google Scholar]
- Kakizaki I, Suto S, Tatara Y, Nakamura T, Endo M. Hyaluronan-chondroitin hybrid oligosaccharides as new life science research tools. Biochem Biophys Res Commun. 2012;423:344–349. doi: 10.1016/j.bbrc.2012.05.127. doi:10.1016/j.bbrc.2012.05.127. [DOI] [PubMed] [Google Scholar]
- Kane TA, White CL, DeAngelis PL. Functional characterization of PmHS1, a Pasteurella multocida heparosan synthase. J Biol Chem. 2006;281:33192–33197. doi: 10.1074/jbc.M606897200. doi:10.1074/jbc.M606897200. [DOI] [PubMed] [Google Scholar]
- Karst NA, Linhardt RJ. Recent chemical and enzymatic approaches to the synthesis of glycosaminoglycan oligosaccharides. Curr Med Chem. 2003;10:1993–2031. doi: 10.2174/0929867033456891. doi:10.2174/0929867033456891. [DOI] [PubMed] [Google Scholar]
- Kreuger J, Spillmann D, Li JP, Lindahl U. Interactions between heparan sulfate and proteins: The concept of specificity. J Cell Biol. 2006;174:323–327. doi: 10.1083/jcb.200604035. doi:10.1083/jcb.200604035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuberan B, Lech MZ, Beeler DL, Wu ZL, Rosenberg RD. Enzymatic synthesis of antithrombin III-binding heparan sulfate pentasaccharide. Nat Biotechnol. 2003;21:1343–1346. doi: 10.1038/nbt885. doi:10.1038/nbt885. [DOI] [PubMed] [Google Scholar]
- Kwon SJ, Lee KB, Solakyildirim K, Masuko S, Ly M, Zhang F, Li L, Dordick JS, Linhardt RJ. Ultrasensitive carbohydrate detection by Glyco-qPCR amplification and applications in glycobiology. Angew Chem. 2012;51:11800–11804. doi: 10.1002/anie.201205112. doi:10.1002/anie.201205112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laremore TN, Zhang F, Dordick JS, Liu J, Linhardt RJ. Recent progress and applications in glycosaminoglycan and heparin research. Curr Opin Chem Biol. 2009;13:633–640. doi: 10.1016/j.cbpa.2009.08.017. doi:10.1016/j.cbpa.2009.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Ly M, Linhardt RJ. Proteoglycan Sequence. Mol BioSyst. 2012;8:1613–1625. doi: 10.1039/c2mb25021g. doi:10.1039/c2mb25021g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Sheng J, Liu Y, Li J, Liu J, Wang F. Heparosan-derived heparan sulfate/heparin-like compounds: One kind of potential therapeutic agents. Med Res Rev. 2012 doi: 10.1002/med.21263. doi:10.1002/med.21263. [DOI] [PubMed] [Google Scholar]
- Liang PH, Wu CY, Greenberg WA, Wong CH. Glycan arrays: Biological and medical applications. Curr Opin Chem Biol. 2008;12:86–92. doi: 10.1016/j.cbpa.2008.01.031. doi:10.1016/j.cbpa.2008.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindahl U, Li JP. Interactions between heparan sulfate and proteins-design and functional implications. Int Rev Cell Mol Biol. 2009;276:105–159. doi: 10.1016/S1937-6448(09)76003-4. doi:10.1016/S1937-6448(09)76003-4. [DOI] [PubMed] [Google Scholar]
- Linhardt RJ. Heparin: Structure and activity. J Med Chem. 2003;46:2551–2554. doi: 10.1021/jm030176m. doi:10.1021/jm030176m. [DOI] [PubMed] [Google Scholar]
- Linhardt RJ, Gunay NS. Production and chemical processing of low molecular weight heparins. Semin Thromb Hemost. 1999;25:5–16. doi:10.1055/s-2007-996417. [PubMed] [Google Scholar]
- Liu J, Moon AF, Sheng J, Pedersen LC. Understanding the substrate specificity of the heparan sulfate sulfotransferases by an integrated biosynthetic and crystallographic approach. Curr Opin Struct Biol. 2012;22:550–557. doi: 10.1016/j.sbi.2012.07.004. doi:10.1016/j.sbi.2012.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu R, Xu Y, Chen M, Weïwer M, Bridges A, DeAngelis PL, Zhang Q, Linhardt RJ, Liu J. Chemoenzymatic design of heparan sulfate oligosaccharides. J Biol Chem. 2010;285:34240–34249. doi: 10.1074/jbc.M110.159152. doi:10.1074/jbc.M110.159152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Zhang Z, Linhardt RJ. Lessons learned from the contamination of heparin. Nat Product Reports. 2009;26:313–321. doi: 10.1039/b819896a. doi:10.1039/b819896a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ly M, Leach FE, III, Laremore TN, Toida T, Amster IJ, Linhardt RJ. The proteoglycan bikunin has a defined sequence. Nat Chem Biol. 2011;7:827–833. doi: 10.1038/nchembio.673. doi:10.1038/nchembio.673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin JG, Gupta M, Xu Y, Akella S, Liu J, Dordick JS, Linhardt RJ. Towards an artificial Golgi: Redesigning the biological activities of heparan sulfate on a digital microfluidic chip. J Am Chem Soc. 2009;131:11041–11048. doi: 10.1021/ja903038d. doi:10.1021/ja903038d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuko S, Bera S, Green DE, Weïwer M, DeAngelis PL, Linhardt RJ. Chemoenzymatic synthesis of UDP-GlcNAc and UDP-GalNAc analogs for the preparation of unnatural glycosaminoglycans. J Org Chem. 2012;77:1449–1456. doi: 10.1021/jo202322k. doi:10.1021/jo202322k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller EM, Wheeler AR. A digital microfluidic approach to homogeneous enzyme assays. Anal Chem. 2008;80:1614–1619. doi: 10.1021/ac702269d. doi:10.1021/ac702269d. [DOI] [PubMed] [Google Scholar]
- Moon H, Wheeler AR, Garrell RL, Loo JA, Kim CJ. An integrated digital microfluidic chip for multiplexed proteomic sample preparation and analysis by MALDI-MS. Lab Chip. 2006;6:1213–1219. doi: 10.1039/b601954d. doi:10.1039/b601954d. [DOI] [PubMed] [Google Scholar]
- Noti C, de Paz JL, Polito L, Seeberger PH. Preparation and use of microarrays containing synthetic heparin oligosaccharides for the rapid analysis of heparin-protein interactions. Chem Eur J. 2006;12:8664–8686. doi: 10.1002/chem.200601103. doi:10.1002/chem.200601103. [DOI] [PubMed] [Google Scholar]
- Ochiai H, Ohmae M, Mori T, Kobayashi S. Hyaluronidase-catalyzed copolymerization for the single-step synthesis of functionalized hyaluronan derivatives. Biomacromolecules. 2007;8:1327–1332. doi: 10.1021/bm061136d. doi:10.1021/bm061136d. [DOI] [PubMed] [Google Scholar]
- Otto NJ, Green DE, Masuko S, Mayer A, Tanner ME, Linhardt RJ, DeAngelis PL. Structure/function analysis of Pasteurella multocida heparosan synthases: Toward defining enzyme specificity and engineering novel catalysts. J Biol Chem. 2012;287:7203–7212. doi: 10.1074/jbc.M111.311704. doi:10.1074/jbc.M111.311704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto NJ, Solakyildirim K, Linhardt RJ, DeAngelis PL. Comamonas testosteronan synthase, a bifunctional glycosyltransferase that produces a unique heparosan polysaccharide analog. Glycobiology. 2011;21:1331–1340. doi: 10.1093/glycob/cwr072. doi:10.1093/glycob/cwr072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacheco B, Malmström A, Maccarana M. Two dermatan sulfate epimerases form iduronic acid domains in dermatan sulfate. J Biol Chem. 2009;284:9788–9795. doi: 10.1074/jbc.M809339200. doi:10.1074/jbc.M809339200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park TJ, Lee MY, Dordick JS, Linhardt RJ. Signal amplification of target protein on heparin glycan microarray. Anal Biochem. 2008;383:116–121. doi: 10.1016/j.ab.2008.07.037. doi:10.1016/j.ab.2008.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavão MS, Vilela-Silva AC, Mourão PA. Biosynthesis of chondroitin sulfate: From the early, precursor discoveries to nowadays, genetics approaches. Adv Pharmacol. 2006;53:117–140. doi: 10.1016/S1054-3589(05)53006-0. doi:10.1016/S1054-3589(05)53006-0. [DOI] [PubMed] [Google Scholar]
- Pempe EH, Burch TC, Law CJ, Liu J. Substrate specificity of 6-O-endosulfatase (Sulf-2) and its implications in synthesizing anticoagulant heparan sulfate. Glycobiology. 2012;22:1353–1362. doi: 10.1093/glycob/cws092. doi:10.1093/glycob/cws092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pervin A, Gallo C, Jandik KA, Han X-J, Linhardt RJ. Preparation and structural characterization of large heparin-derived oligosaccharides. Glycobiology. 1995;5:83–95. doi: 10.1093/glycob/5.1.83. doi:10.1093/glycob/5.1.83. [DOI] [PubMed] [Google Scholar]
- Peterson S, Frick A, Liu J. Design of biologically active heparan sulfate and heparin using an enzyme-based approach. Nat Prod Rep. 2009;26:610–627. doi: 10.1039/b803795g. doi:10.1039/b803795g. [DOI] [PubMed] [Google Scholar]
- Powell AK, Ahmed YA, Yates EA, Turnbull JE. Generating heparan sulfate saccharide libraries for glycomics applications. Nat Protoc. 2010;5:821–833. doi: 10.1038/nprot.2010.17. doi:10.1038/nprot.2010.17. [DOI] [PubMed] [Google Scholar]
- Rogers CJ, Hsieh-Wilson LC. Microarray method for the rapid detection of glycosaminoglycan-protein interactions. Methods Mol Biol. 2012;808:321–336. doi: 10.1007/978-1-61779-373-8_22. doi:10.1007/978-1-61779-373-8_22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seurynck-Servoss SL, White AM, Baird CL, Rodland KD, Zangar RC. Evaluation of surface chemistries for antibody microarrays. Anal Biochem. 2007;371:105–115. doi: 10.1016/j.ab.2007.07.010. doi:10.1016/j.ab.2007.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng J, Xu Y, Dulaney SB, Huang X, Liu J. Uncovering biphasic catalytic mode of C5-epimerase in heparan sulfate biosynthesis. J Biol Chem. 2012;287:20996–21002. doi: 10.1074/jbc.M112.359885. doi:10.1074/jbc.M112.359885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sismey-Ragatz AE, Green DE, Otto NJ, Rejzek M, Field RA, DeAngelis PL. Chemoenzymatic synthesis with distinct Pasteurella heparosan synthases: monodisperse polymers and unnatural structures. J Biol Chem. 2007;282:28321–28327. doi: 10.1074/jbc.M701599200. doi:10.1074/jbc.M701599200. [DOI] [PubMed] [Google Scholar]
- Srinivasan V, Pamula V, Fair R. An integrated digital microfluidic lab-on-a-chip for clinical diagnostics on human physiological fluids. Lab Chip. 2004;4:310–315. doi: 10.1039/b403341h. doi:10.1039/b403341h. [DOI] [PubMed] [Google Scholar]
- Stevens J, Blix O, Paulson C, Wilson IA. Glycan microarray technologies: Tools to survey host specificity of influenza viruses. Nat Rev Microbiol. 2006;4:857–864. doi: 10.1038/nrmicro1530. doi:10.1038/nrmicro1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugahara K, Mikami T, Uyama T, Mizuguchi S, Nomura K, Kitagawa H. Recent advances in the structural biology of chondroitin sulfate and dermatan sulfate. Curr Opin Struct Biol. 2003;13:612–620. doi: 10.1016/j.sbi.2003.09.011. doi:10.1016/j.sbi.2003.09.011. [DOI] [PubMed] [Google Scholar]
- Sugiura N, Baba Y, Kawaguchi Y, Iwatani T, Suzuki K, Kusakabe T, Yamagishi K, Kimata K, Kakuta Y, Watanabe H. Glucuronyltransferase activity of KfiC from Escherichia coli strain K5 requires association of KfiA: KfiC and KfiA are essential enzymes for production of K5 polysaccharide, N-acetylheparosan. J Biol Chem. 2010;285:1597–1606. doi: 10.1074/jbc.M109.023002. doi:10.1074/jbc.M109.023002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiura N, Shioiri T, Chiba M, Sato T, Narimatsu H, Kimata K, Watanabe H. Construction of a chondroitin sulfate library with defined structures and analysis of molecular interactions. J Biol Chem. 2012;287:43390–43400. doi: 10.1074/jbc.M112.412676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toole BP. Hyaluronan: From extracellular glue to pericellular cue. Nat Rev Cancer. 2004;4:528–539. doi: 10.1038/nrc1391. doi:10.1038/nrc1391. [DOI] [PubMed] [Google Scholar]
- Tracy BS, Avci FY, Linhardt RJ, DeAngelis PL. Acceptor specificity of the Pasteurella hyaluronan and chondroitin synthases and production of chimeric glycosaminoglycans. J Biol Chem. 2007;282:337–344. doi: 10.1074/jbc.M607569200. doi:10.1074/jbc.M607569200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D. Carbohydrate microarrays. Proteomics. 2003;3:2167–2175. doi: 10.1002/pmic.200300601. doi:10.1002/pmic.200300601. [DOI] [PubMed] [Google Scholar]
- Wang Z, Dordick JS, Linhardt RJ. E. coli K5 heparosan fermentation and improvement by genetic engineering. Bioeng Bugs. 2011;2:1–15. doi: 10.4161/bbug.2.1.14201. doi:10.4161/bbug.2.1.14315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Liu S, Trummer BJ, Deng C, Wang A. Carbohydrate microarrays for the recognition of cross-reactive molecular markers of microbes and host cells. Nate Biotechnol. 2002;20:275–281. doi: 10.1038/nbt0302-275. doi:10.1038/nbt0302-275. [DOI] [PubMed] [Google Scholar]
- Wang Z, Ly M, Zhang F, Zhong W, Suen A, Dordick JS, Linhardt RJ. E. coli K5 fermentation and the preparation of heparosan, a bioengineered heparin precursor. Biotechnol Bioeng. 2010;107:964–973. doi: 10.1002/bit.22898. doi:10.1002/bit.22898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins WM. Glycosyltransferases, early history, development and future prospects. Carbohydr Res. 1986;149:1–12. doi: 10.1016/s0008-6215(00)90364-1. doi:10.1016/S0008-6215(00)90364-1. [DOI] [PubMed] [Google Scholar]
- Weïwer M, Sherwood T, Green DE, Chen M, DeAngelis PL, Liu J, Linhardt RJ. Synthesis of uridine 5′-diphosphoiduronic acid: A potential substrate for the chemoenzymatic synthesis of heparin. J Org Chem. 2008;73:7631–7637. doi: 10.1021/jo801409c. doi:10.1021/jo801409c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler AR. Putting electrowetting to work. Science. 2008;322:539–540. doi: 10.1126/science.1165719. doi:10.1126/science.1165719. [DOI] [PubMed] [Google Scholar]
- Widner B, Behr R, Von Dollen S, Tang M, Heu T, Sloma A, Sternberg D, DeAngelis PL, Weigel PH, Brown S. Hyaluronic acid production in Bacillus subtilis. Appl Environ Microbiol. 2005;71:3747–3752. doi: 10.1128/AEM.71.7.3747-3752.2005. doi:10.1128/AEM.71.7.3747-3752.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wildt S, Gerngross TU. The humanization of N-glycosylation pathways in yeast. Nat Rev Microbiol. 2005;3:119–128. doi: 10.1038/nrmicro1087. doi:10.1038/nrmicro1087. [DOI] [PubMed] [Google Scholar]
- Xu Y, Masuko S, Takieddin M, Xu H, Liu R, Jing J, Mousa S, Linhardt RJ, Liu J. Chemoenzymatic synthesis of structurally homogeneous ultra-low molecular weight heparins. Science. 2011;334:498–501. doi: 10.1126/science.1207478. doi:10.1126/science.1207478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Pempe EH, Liu J. Chemoenzymatic synthesis of heparin oligosaccharides with both anti-factor Xa and anti-factor IIa activities. J Biol Chem. 2012;287:29054–29061. doi: 10.1074/jbc.M112.358523. doi:10.1074/jbc.M112.358523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi M, Takagaki K, Kojima K, Hayashi N, Chen F, Kakizaki I, Kon A, Endo M. Novel proteoglycan glycotechnology: Chemoenzymatic synthesis of chondroitin sulfate-containing molecules and its application. Glycoconj J. 2010;27:189–198. doi: 10.1007/s10719-009-9252-y. doi:10.1007/s10719-009-9252-y. [DOI] [PubMed] [Google Scholar]
- Yang HO, Gunay NS, Toida T, Kuberan B, Yu G, Kim YS, Linhardt RJ. Preparation and structural determination of dermatan sulfate-derived oligosaccharides. Glycobiology. 2000;10:1033–1040. doi: 10.1093/glycob/10.10.1033. doi:10.1093/glycob/10.10.1033. [DOI] [PubMed] [Google Scholar]
- Zhi ZI, Powell AK, Turnbull JE. Fabrication of carbohydrate microarrays on gold surfaces: Direct attachment of nonderivatized oligosaccharides to hydrazide-modified self-assembled monolayers. Anal Chem. 2006;78:4786–4793. doi: 10.1021/ac060084f. doi:10.1021/ac060084f. [DOI] [PubMed] [Google Scholar]
- Zhou X, Chandarajoti K, Pham TQ, Liu R, Liu J. Expression of heparan sulfate sulfotransferases in Kluyveromyces lactis and preparation of 3′-phosphoadenosine-5′-phosphosulfate. Glycobiology. 2011;21:771–780. doi: 10.1093/glycob/cwr001. doi:10.1093/glycob/cwr001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuo L, Kanamori A, Kannagi R, Itano N, Wu J, Hamaguchi M, Ishiguro N, Kimata K. SHAP potentiates the CD44-mediated leukocyte adhesion to the hyaluronan substratum. J Biol Chem. 2006;281:20303–20314. doi: 10.1074/jbc.M506703200. doi:10.1074/jbc.M506703200. [DOI] [PubMed] [Google Scholar]