

Abstract
Several neurodegerative diseases are caused by expansion of a trinucleotide repeat tract in a critical gene. The mechanism of repeat instability is not yet defined, but in mice it requires MutSβ, a complex of MSH2 and MSH3. We showed previously that transcription through a CAG repeat tract induces repeat instability in human cells via a pathway that requires the mismatch repair (MMR) components, MSH2 and MSH3, and the entire transcription-coupled nucleotide excision repair pathway (Lin et al., Nat. Struct. Mol. Biol. 13:189–190; Lin and Wilson, Mol. Cell. Biol. 27:6209–6217). Here, we examine the role of downstream MMR processing components on transcription-induced CAG instability, using our selection assay for repeat contraction. In contrast to knockdowns of MSH2 or MSH3, which reduce repeat contractions, we show that siRNA-mediated depletion of MLH1 or PMS2 increases contraction frequency. Knockdown of DNMT1, which has been identified as an MMR factor in genetic studies, also elevates the frequency of contraction. Simultaneous knockdowns of MLH1 or DNMT1 along with MSH2, XPA, or BRCA1, whose individual knockdowns each decrease CAG contraction, yield intermediate frequencies. In sharp contrast, double knockdown of MLH1 and DNMT1 additively increases the frequency of CAG contraction. These results show that MMR components can alter repeat stability in diverse ways, either enhancing or suppressing CAG contraction, and they provide insight into the influence of MMR components on transcription-induced CAG repeat instability.
Introduction
Microsatellite sequences, consisting of tandemly repeated units of one to a few nucleotides, are widely represented in eukaryotic genomes. Although they constitute the major component of centromeres and telomeres, microsatellite repeats are not uncommon in the promoters and transcribed segments of genes [1]. Microsatellite repeats are typically less stable than other genomic sequences, lengthening (expanding) or shortening (contracting) the repeat tract at relatively high frequencies. In some cases, expansions of repeats are associated with human diseases. For example, several neurodegenerative diseases are caused by the germline expansion of trinucleotide repeat (TNR) tracts to lengths that have pathological consequences in the next generation [2–6]. Expansion-biased TNR instability is also found in somatic tissues including brain, the major tissue affected in these individuals [7]. The mechanisms of TNR instability are not well defined, and may differ in germline and somatic cells, in different somatic tissues, and in the different genomic contexts surrounding disease genes. For neurons, which no longer divide, the ongoing instability of their repeats cannot depend on DNA replication, a process initially thought to be the major source for TNR instability [3,8–10].
Recently, it was shown both in human cells and in a fly model that transcription can stimulate CAG repeat instability [7,11–13]. These studies, together with previous findings that transcription destabilizes TNRs in bacteria [14,15], strongly suggest that transcription can trigger TNR instability, providing a potential mechanism for the ongoing repeat instability in neurons. Transcription-induced TNR instability arises through a mechanism that involves the DNA repair proteins associated with nucleotide excision repair (NER) and mismatch repair (MMR) [11,12,16–18].
The primary role of MMR is to remove mismatched bases and small insertions and deletions (IDs) introduced during DNA replication [19,20]. In mammalian cells, MMR is initiated by mismatch recognition via one of two complexes: MutSα (MSH2/MSH6) and MutSβ (MSH2/MSH3). Although MutSα prefers to bind mismatched bases and MutSβ prefers IDs, they have somewhat overlapping specificities [21,22]. MMR is critical for the maintenance of genomic integrity in humans. MMR deficiency lowers the fidelity of DNA replication and gives rise to human diseases [23,24]. Beyond this primary role, MMR proteins monitor mismatches that arise in heteroduplexes during DNA recombination, remove certain types of damaged bases, and act as accessory factors in other repair pathways such as NER [22].
MMR plays a key role in microsatellite instability. For example, the elevated instability of CA dinucleotide repeats that is common in hereditary nonpolyposis colon cancer (HNPCC), is commonly caused by mutation of MSH2, MSH6, MLH1, or PMS2, which are core MMR genes [25,26]. These mutations prevent MMR from repairing the ID mismatches that arise by slippage of the primer strand relative to the template during DNA replication. In E. coli [27–29] and yeast [30,31], deficiency of MMR apparently increases TNR instability in the same way, causing mostly the one- or two-unit expansions and contractions typical of slippage during replication. In sharp contrast, MMR deficiencies in mouse models and human cells stabilize TNRs. In transgenic mouse models of Huntington’s disease (HD) and myotonic dystrophy (DM1), deficiency of either MSH2 or MSH3 dramatically decreased expansions of the repeat tract in the germline and in somatic tissues [32–36], while MSH6 deficiency caused only slight changes in repeat instability [34]. Deficiency of PMS2, a downstream factor involved in MMR, decreased expansions and increased contractions in some somatic tissues [37]. In human cells, siRNA knockdown of MSH2 or MSH3, but not of MSH6, reduced transcription-induced CAG repeat instability, as measured by contractions of the repeat tract [12], providing the first evidence linking MMR and transcription-induced repeat instability. These studies in mouse models and human cells reinforce the idea that components of MMR normally act to destabilize TNRs, but they do not define a mechanism.
In this study, we investigated the roles of MLH1 and PMS2 in transcription-induced CAG repeat instability. MLH1 and PMS2 form a complex, MutLα, that acts downstream of mismatch recognition. We used siRNA knockdowns of these proteins to assess their effects on repeat stability in a well-characterized genetic assay that can selectively detect CAG contractions that arise during transcription [12,18]. We show that knockdown of either MLH1 or PMS2 stimulates CAG repeat contraction. In addition, we examined the effects of double knockdown of MLH1 in combination with other proteins—MSH2, XPA, BRCA1, and DNMT1—that we previously indentified as modulators of CAG repeat instability in human cells [12,18,38]. Our results show that mismatch recognition proteins and downstream processing factors oppositely affect CAG repeat contractions. We discuss potential mechanism that might underlie these opposite effects of mismatch recognition and processing.
2. Materials and Methods
2.1. Cell culture and HPRT+ selection
FLAH25 cells, whose construction was described previously [12,18], were derived from HT1080 cells (a human fibrosarcoma cell line) that carried a nonreverting mutation in the endogenous HPRT gene. FLAH25 cells, which are HPRT−, contain an HPRT minigene that is inactivated by insertion of a CAG95 tract in its intron. Transcription of the HPRT minigene, which is controlled by the pTRE-CMVmin promoter, was induced by addition of doxycycline (2 μg/ml) to the medium. FLAH25 cells were incubated at 37°C with 5% CO2 in DMEM/F-12 medium supplemented with 10% dialyzed serum and 1% MEM nonessential amino acids. HPRT+ cells were selected by plating 500,000 cells on 100 mm dishes in HAT medium [0.1 mM hypoxanthine, 0.4 μM aminopterin, and 16 μM thymine (Sigma)] plus doxycycline for two weeks, with addition of fresh doxycycline after the first week of selection. Contraction frequencies, which were calculated as the number of HPRT+ colonies divided by the number of viable cells, are the average of at least 6 independent experiments.
2.2. Real-time RT-PCR
About two million cells were harvested for preparation of total RNA using RNeasy mini kits (Qiagen). For measurements of HPRT expression, doxycycline was added one day before RNA was extracted. For real-time RT-PCR, 50 ng of total RNA per reaction was assayed using the SYBR Green RT-PCR kit (Qiagen). Results were normalized to the concentration of β-actin RNA, which was also determined by real-time RT-PCR. HPRT expression was measured relative to the RNA level in untreated cells, which was arbitrarily defined as 1. For expression of target genes in siRNA knockdown experiments, the RNA levels in cells treated with vimentin siRNA, which does not affect RNA levels, was defined as 1 and used as the reference. Conditions for real-time RT-PCR were 50°C for 30 min, 95°C for 15 min, followed by 45 cycles of 94°C for 15 sec, 50°C for 30 sec, and 72°C for 30 sec. The relative levels of mRNA were calculated by comparing the number of cycles (generally between 15 and 25 cycles) at which the PCR products became detectable above the basal threshold. Primers used for real-time RT-PCR are shown in Table 1.
Table 1.
RT-PCR primers and siRNA used in this study.
| Real-Time RT-PCR Primers | |||
|---|---|---|---|
| Gene | Forward | Reverse | siRNAs (Reference) |
| β-Actin | AGAGAGGCATCCTCACCCTG | CATGAGGTAGTCAGTCAGGT | |
| BRCA1 | AATGCCAGTCAGGCACAG | CAGCTCTGGGAAAGTATCGC | 1.AGGAGCUUUCAUCAUUCACCC [73] |
| GACCATTCTGCTCCGTTTG | TACTGAGCCACAGATCCTACAAGAG | ||
| DNMT1 | 1.GAGUGUGUGAGGGAGAAA [18] | ||
| HPRT | CGGCTACAAGGACGACTCTAG | TTGATGTAATCCAGCAGGTCAGC | |
| MLH1 | CACTACTAGTAAACTGCAGTCC | CTTGATTGCCAGCACATGG | 1. AUCAGGCAGGUUAGCAAGCUG |
| AAGAGAGGACCTACTTCCAG | ATGCAACATCTCCCGGAG | 2. CCCAGAAUGUGGAUGUUAAUGUGCA | |
| MLH3 | GCTTGTGAAGCTGATGTGAC | TGCATGAGTGAGAGAGCTTC | 1. GCCACACAGGAUAAUUCCUUUAAU |
| GGAGCTACTCCAGACCAC | ATGAGGACAGAGCTTCAATAAGG | 2. GGACAAUCCAGUAUUUGCCCGUUAU | |
| MSH2 | CAGATGATTCCAACTTTGGACAG | CCAGAGACTGAGAGCCAG | 1. UCUGCAGAGUGUUGUGCUU [12] |
| ATGTAGAACCAATGCAGACACTC | CAACACAAGCATGCCTGG | ||
| PMS2 | GACTGATGTTTGATCACAATGGG | GTAAGACCTGGACCATTTTGG | 1. ACUGAUUUCCUUGCCAACUAGUAAA |
| CACTTCCGTGGATTCTGAGG | TCGTCAGTTTCAGGCGC | 2. GGCCAACCAUGAGACACAUCGCCAA | |
| Vimentin | 1. GAAUGGUACAAAUCCAAGU [12] | ||
| XPA | GCGGCGGCTTTAGAGCAAC | CATAGCCATGCCTCCAGTAGCC | 1. GCUACUGGAGGCAUGGCUA [12] |
| GAGGTCTCTTGAAGTTTGGGGTAG | TCTCCCTTTTCCACACGCTG | ||
2.3. Treatments with siRNA
The siRNAs used in this study are shown in Table 1. For siRNA treatments, 100,000 FLAH25 cells were plated in a 100-mm plate on day −4. On day −3, siRNAs (Invitrogen) at a total concentration of 200 nM were transfected into cells using oligofectamine (Invitrogen). Each specific siRNA was tested at 100 nM in the presence of 100-nM vimentin-siRNA or 100 nM of a second specific siRNA. Treatments with 200-nM vimentin-siRNA, which does not affect repeat contraction, served as controls. On day 0, the cells were again transfected with siRNA, and cultures were then grown in the presence of doxycycline. Induction of HPRT transcription by doxycycline and knockdown of target gene expression by siRNAs were evaluated by real time RT-PCR on day 1 (Table 2). Selection for HPRT+ colonies was begun on day 3 by growth in HAT medium supplemented with doxycycline. At the same time, 200 cells were plated in nonselective medium and were allowed to form colonies as a measure of the effects of treatment on cell viability (Table 2).
Table 2.
SiRNA knockdowns and their effects on cells.
| siRNA | Cell number x10−6 (%)a | Proliferation rate (%)b | Plating efficiencyc (%) | HPRT mRNAd (fold) | Target mRNAe (%) |
|---|---|---|---|---|---|
| Vimentin | 4.5 (100) | 0.92 (100) | 100 | 23 | |
| BRCA1-1 | 1.2 (27) | 0.60 (66) | 73 | 20 | 36 |
| DNMT1-1 | 4.2 (93) | 0.90 (98) | 62 | 22 | 34 |
| MLH1-1 | 4.3 (95) | 0.90 (99) | 76 | 25 | 17 |
| MLH1-2 | 3.6 (79) | 0.87 (95) | 89 | 20 | 29 |
| MLH3-1 | 4.5 (100) | 0.92 (100) | 98 | 21 | 24 |
| MLH3-2 | 3.7 (82) | 0.88 (96) | 95 | 26 | 26 |
| MSH2-1 | 4.3 (95) | 0.90 (99) | 78 | 22 | 38 |
| PMS2-1 | 4.4 (98) | 0.91 (99) | 98 | 17 | 19 |
| PMS2-2 | 4.5 (100) | 0.92 (100) | 96 | 19 | 15 |
| XPA-1 | 4.1 (91) | 0.89 (98) | 93 | 27 | 21 |
Cell number refers to the total number of cells per plate after 6 days of siRNA treatment. Each plate began with 105 cells. Cell number with vimentin siRNA treatment was used as the reference and defined as 100%.
Proliferation rate is the number of cell doublings per day required to increase the cell population from 100,000 cells per plate to the number present at the end of the 6-day treatment. As an example, for vimentin siRNA treatment cell number increased 45-fold over 6 days, which equals 5.5 population doublings per 6 days, or 0.92 doublings per day. The proliferation rate during treatment with vimentin siRNA was used as the reference and defined as 100%.
Plating efficiency was measured at the time cells were replated for selection for HPRT+ cells. It is the number of colonies per 100 cells that formed in nonselective medium. The absolute plating efficiency for vimentin siRNA-treated cells was 47%, which was used as the reference and defined as 100%.
Induced HPRT mRNA levels in cells treated with specific siRNAs in the presence of doxycycline are expressed relative to HPRT mRNA levels in cells treated with vimentin siRNA in the absence of induction by doxycycline, which was used as the reference and defined as 1.0.
Target mRNA is the amount of target gene mRNA present in cells treated with the specific siRNA relative to that present in cells treated with vimentin siRNA.
2.4. Characterization of repeat length in HPRT+ clones
Genomic DNA was prepared from about one million cells of each HPRT+ clone using a kit (GE Healthcare). The CAG repeat tract in each DNA preparation was amplified by PCR. The forward primer for PCR was HPRT1-1 (CTTCTCAAGCACTGGCCT) and the reverse primer was HPRT1-2 (TAGTGGACTGGTTAAATCAGTTATG). The PCR products were separated by electrophoresis on 1.5% agarose gels, and track lengths were measured relative to a ladder of CAG tracks (from CAG15 to CAG36) whose lengths had been determined by DNA sequencing. This method is accurate to ±2 CAG repeats, as shown previously [12].
3. Results
3.1. Knockdown of MLH1 or PMS2 enhances CAG contractions
We previously developed a sensitive genetic assay to detect CAG contraction in human cells, based on the selectable marker HPRT (Fig. 1). Briefly, a long CAG95 tract inserted into the intron of the HPRT minigene produces a nonfunctional protein by inclusion of the repeat tract into the final spliced product. When big deletions (more than 55 repeats) occur, however, the shortened CAG repeat does not efficiently support aberrant splicing, rendering cells HPRT+ and allowing them to survive in HAT selection. Thus, large CAG contractions are readily detected by the formation of HPRT+ colonies during selection [12,18,39]. Using this novel assay in which transcription through the repeat tract is driven by a doxycycline-inducible promoter, we have shown that transcription-induced TNR instability in human cells requires several components of both the MMR and NER pathways, which appear to be part of the same mechanism, since various pairwise knockdowns yielded the same effect as single knockdowns [12,18].
Fig. 1.
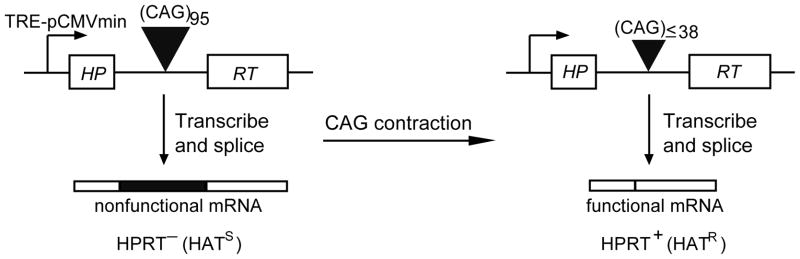
Selection assay for transcription-induced contraction of CAG repeats. When long CAG tracts are present in the intron of the HPRT minigene, they are incorporated into the mRNA, rendering it nonfunctional and giving the cells an HPRT− (HAT-sensitive) phenotype. When contraction of the repeat generates a tract with fewer than 39 repeats, it is not efficiently incorporated into the mRNA, allowing sufficient normal protein to be made to give an HPRT+ (HAT-resistant) phenotype [39]. The HPRT minigene is controlled by the TRE-pCMVmini promoter, which can be turned on by addition of doxycycline. When the reverse tetracycline transcription activator (rtTA) binds doxycycline, it is converted to an active form that binds to the promoter.
To probe more deeply into the role of MMR in TNR instability during transcription, we knocked down key downstream components and assayed the effects on repeat contraction. We chose to test three components: MLH1, PMS2, and MLH3. MLH1 and PMS2 form a complex, MutLα, which plays a major role in completing MMR after DNA mismatches have been recognized and bound by MutS complexes. MLH3 forms a similar complex with MLH1 (MutLγ), but functions primarily in meiotic recombination, although it may provide some backup for MutLα in MMR [21]. To knock down these proteins, we transfected FLAH25 cells with siRNAs targeting their respective mRNAs. For each protein we tested two different siRNAs to minimize the possibility that a detected consequence might be due to an off-target effect. The knockdown efficiency was determined using real-time RT-PCR to measure mRNA levels; all siRNAs knocked down their target mRNAs by 62% to 85% (Table 2).
Treatment with the two siRNAs against MLH1 increased the frequency of transcription-induced CAG contraction by 2.1-fold (p<0.0001) and 2.0-fold (p<0.0001) (Fig. 2). Similarly, two siRNA knockdowns of PMS2 led to increases in contraction of 1.9-fold (p<0.001) and 2.5-fold (p<0.001) (Fig. 2). None of these siRNAs changed the frequency of CAG contraction in the absence of doxycycline, when transcription through the repeat was turned off (data not shown). In contrast, knockdown of MLH3 did not affect the frequency of contractions (p>0.2) (Fig. 2). Because transcription through the repeat drives CAG contraction in this system, it was critical to determine whether the siRNA knockdowns altered the levels of transcription of the HPRT minigene. Using real-time RT-PCR, we showed that the HPRT mRNA level in cells treated with control or target-specific siRNAs did not substantially change (Table 2), arguing against the possibility that these siRNAs affected TNR instability indirectly through effects on HPRT transcription. Several HPRT+ clones that arose after siRNA knockdown of MLH1 or PMS2 were picked for characterization of the repeat tract. More than 95% of the clones arose by CAG contraction (Fig. 3), as we previously observed for siRNA knockdowns of other MMR components [12,18]. Even though the types of event are the same, stimulation of contraction by siRNAs against the processing components, MLH1 and PMS2, contrasts sharply with reduction of contractions by siRNAs against the recognition components, MSH2 and MSH3 [12].
Fig. 2.
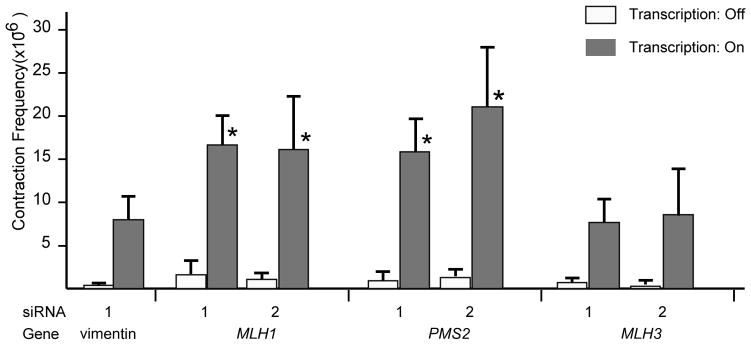
Effects of siRNA-mediated knockdowns on transcription-induced CAG repeat contraction. Contraction frequencies were calculated as the number of HPRT+ colonies divided by the number of viable cells, averaged over at least five independent experiments. Values obtained in the absence of doxycycline (transcription turned off) are shown in unfilled boxes. Values obtained in the presence of doxycycline are shown in black boxes. Transcription-induced frequencies that are significantly different from the vimentin siRNA control are indicated with an asterisk; specific p values are indicated in the text.
Fig. 3.

PCR analysis of the lengths of CAG repeat tracts. The CAG repeat tracts in HPRT+ colonies were amplified by PCR and the products were separated by electrophoresis. The number of CAG repeats in PCR products from independent HPRT+ colonies (lanes 9–28) was determined by reference to a ladder of length markers (lanes 1–8). Lanes 1 to 8 contain tracts of 15, 18, 20, 24, 27, 30, 33, and 36 CAG repeats, respectively.
3.2. Interactions of MLH1 with other components of transcription-induced repeat instability
We previously identified nine proteins whose knockdown reduces the frequency of CAG contractions induced by transcription [12,18]. These proteins include recognition elements for MMR (MSH2, MSH3), components of transcription-coupled NER (CSB, XPA, ERCC1, XPG), and proteins that may deal with stalled RNA polymerase II molecules (BRCA1, BARD1, and TFIIS). Pairwise knockdowns of these components did not further reduce contraction frequencies relative to single knockdowns, suggesting that all these proteins may function in the same pathway for repeat instability [18]. We selected one member of each group—MSH2, XPA, and BRCA1—to test in combination with MLH1. In all cases, pairwise knockdowns yielded frequencies of contraction that were intermediate between the reductions caused by knockdown of MSH2, XPA, or BRCA1 alone and the stimulation caused by knockdown of MLH1 (Fig. 4). In each instance, the pairwise knockdown was significantly lower than MLH1 knockdown alone, and significantly higher than the single knockdown of MSH2, XPA, or BRCA1 (Table 3). Intermediate values for these pairwise knockdowns suggest that MutLα in some way interacts with the transcription-induced pathway for CAG repeat instability, as we consider in the Discussion.
Fig. 4.

Transcription-induced CAG repeat contraction after single or double siRNA-mediated knockdowns. In all cases the concentration of total siRNA was 200 nM. For single knockdowns, 100 nM of siRNA was used and the total concentration of siRNA was adjusted to 200 nM with vimentin siRNA. For double knockdowns, 100 nM of each siRNA was used. Contraction frequencies are the average results of at least five experiments.
Table 3.
P values for comparisons of double knockdowns with single knockdowns.
| siRNA double knockdowns | versus siRNA single knockdown | p values by Student’s t test |
|---|---|---|
| MLH1-1 + MSH2-1 | MSH2-1 | <0.01 |
| MLH1-1 | <0.001 | |
| MLH1-1 + XPA-1 | XPA-1 | <0.001 |
| MLH1-1 | <0.01 | |
| MLH1-1 + BRCA1-1 | BRCA1-1 | <0.05 |
| MLH1-1 | <0.001 | |
| DNMT1-1 + MSH2-1 | MSH2-1 | <0.001 |
| DNMT1-1 | <0.01 | |
| DNMT1-1 + XPA-1 | XPA-1 | <0.05 |
| DNMT1-1 | <0.01 | |
| DNMT1-1 + MLH1-1 | MLH1-1 | <0.0001 |
| DNMT1-1 | <0.001 |
3.3. Simultaneous knockdown of MLH1 and DNMT1 additively increases repeat instability
We previously found that knockdown of DNMT1—the major maintenance DNA methyltransferase in human cells—stimulates CAG contraction about 2 fold in human cells and that DNMT1 haploinsufficiency increases germline instability in a SCA1 mouse model [38]. Stimulation of CAG contraction was dependent on transcription through the repeat; in the absence of doxycycline, when transcription through the repeat was turned off, siRNA knockdown of DNMT1 did not change the frequency of CAG contraction (data not shown). The function of DNMT1 in repeat instability is undefined and may be complicated, given the role of DNMT1 in various complexes involved in chromatin structure [40–42], DNA replication [43,44], and DNA damage response [45–47]. DNMT1 also appears to participate directly or indirectly in the MMR pathway since DNMT1-deficient mouse cells are defective in MMR [48] and display increased microsatellite instability [49].
To test the interactions of DNMT1 with MMR and NER, we knocked down DNMT1 in combination with MSH2, MLH1, and XPA. As shown in Figure 4, pairwise knockdowns of DNMT1 with MSH2 or XPA gave contraction frequencies that were intermediate between the reductions caused by the single knockdowns. In each case, the pairwise knockdown was significantly lower than DNMT1 knockdown alone, and significantly higher than the single knockdown of MSH2 or XPA (Table 3). By contrast, the pairwise knockdown of DNMT1 and MLH1 increased contraction frequency by 4.8 fold, which is close to the sum of the single knockdown frequencies and significantly higher than either single knockdown (Table 3). These results suggest that deficiency of MLH1 and DNMT1 interact with the transcription-induced pathway of TRN instability in distinct ways.
4. Discussion
In previous studies we identified a transcription-induced pathway for repeat instability in human cells that involves elements of MMR and TC-NER [12,18]. Knockdown of any one of nine different proteins—MSH2, MSH3, CSB, XPA, ERCC1, XPG, BRCA1, BARD1, and TFIIS—reduced the frequency of CAG contractions induced by transcription [12,18]. Because pairwise knockdowns produced no greater effect than knockdowns of individual components, we proposed a working model in which all nine proteins functioned in the same pathway [18]. In this speculative model, MutSβ (MSH2/MSH3) binds to transcription-induced CTG hairpins in the template strand and blocks progress of RNA polymerase II (RNAPII) molecules. Stalled RNA polymerase is a classic signal for engaging TC-NER (CSB, XPA, ERCC1, and XPG) [50]. The necessity for dealing with a stalled RNAPII may account for the involvement of TFIIS, which aids in backing RNAPII away from a lesion to allow access to repair proteins [51,52], and for BRCA1 and BARD1, which form a complex that functions as an E3 ligase, one of whose targets is RNAPII [53,54]. Involvement of an E3 ligase is consistent with the requirement for proteasome activity in the transcription-induced pathway, which was demonstrated using the proteasome inhibitor, MG132 [18].
In this working model, there is no obvious role for the downstream processing components of MMR. Thus, we were surprised to find that knockdown of MLH1 or PMS2 stimulates CAG repeat contractions in our system in human cells. MLH1 and PMS2 form the MutLα complex, which serves as a molecular matchmaker during replication to link the mismatch recognition elements, MutSα or MutSβ, to proteins that define the newly synthesized strand, allowing misincorporated nucleotides to be removed by exonuclease action [21]. Stimulation of repeat contractions by knockdown of MutLα components raises two related questions. How does MutLα influence the transcription-induced pathway for repeat instability? And why do MutLα and MutSβ affect the pathway in opposite directions?
Below we briefly consider four ways in which MutLα might influence the transcription-induced pathway of repeat instability. The central idea in each is that MutSβ binds to and stabilizes the hairpins, while MutLα acts to divert intermediates from the transcription-induced pathway to side pathways whose products we do not detect in our selection assay (Fig. 5). According to this idea, knockdown of MutLα permits an increased flow along the transcription-induced pathway, whose products—contractions—we can detect. First, MutLα could trigger completion of normal MMR. Because MutSβ binds to the hairpin instead of the main duplex [55], it is not exactly clear what the product of such repair might be. If the product were a small change to the repeat length, it would be undetectable in our assay, yet it would eliminate the transcription block caused by MutSβ. Knocking down MutLα would then increase the number of transcription blocks, hence increase the number of detectable contractions. Second, MutLα could stimulate removal of MutSβ from the hairpin. MutSα, which is better studied, binds to a mismatch to form a sliding clamp that can move along the duplex [56]. Because MutSβ binds to the hairpin, it might with some frequency slide off the end of the hairpin. If MutLα stimulated this removal, its knockdown would increase the residence time for MutSβ and thereby increase repeat contractions. Third, the binding of MutLα to hairpin-bound MutSβ could trigger an apoptotic pathway, leading to cell death. MutSα and MutLα have been shown to activate ATR kinase directly in response to O6-methyl-G, without a requirement for the usual intermediate of single-stranded DNA, which is thought to be the major signal for activating the ATR pathway [57]. If MutSβ and MutLα activated ATR when bound to CAG or CTG hairpins, they might trigger apoptosis. In that case, knockdown of MutLα would increase the survival of cells that contain hairpins, thereby allowing more contractions to occur and be detected. Fourth, the high density of mismatches in the hairpin (one out of every three nucleotides) could initiate a process akin to heteroduplex rejection, in which MMR proteins engage a helicase to unwind the heteroduplex formed during recombination between homeologous sequences [58]. Heteroduplex rejection is best defined in yeast, where it depends strongly on MutSα and to a lesser extent on MLH1 [59]. If MutSβ and MutLα acted in an analogous way on CAG or CTG hairpins (that is, by engaging a helicase to unwind the hairpin), then knockdown of MutLα would allow more hairpins to survive, thereby increasing the number of contractions. In support of this idea, yeast carrying mutant forms of helicase Srs2, which normally unwinds repeat hairpins [60], display increased frequencies of repeat instability [61].These potential interactions between MutSβ and MutLα, which are not mutually exclusive, will likely require biochemical studies to sort them out.
Fig. 5.

Possible interactions between MLH1, DNMT1, and the transcription-induced pathway for repeat instability. Transcription through a repeat tract is envisioned to generate a slipped-strand duplex structure with CTG hairpins on one strand and CAG loops on the other. Shown here is a single CTG hairpin, bound by MutSβ, serving as a block to progression of a subsequent RNAPII molecule. This intermediate is processed by TC-NER to produce large contractions, which can be detected in our assay system. MutLα may act to divert intermediates to products that we cannot detect. DNMT1 could act to inhibit formation of the intermediate (by inhibiting transcription), promote an alternative pathway for processing the intermediate (like MutLα), or remove products from detection (for example, by silencing).
The link between DNMT1 and the pathway for transcription-induced CAG repeat contraction is unclear; however, two observations indicate that some connection exists. First, the stimulation of CAG repeat contraction by knockdown of DNMT1 depends on transcription through the repeat. When the HPRT promoter is turned off, CAG contractions remain at background levels even when DNMT1 has been knocked down. Second, double knockdowns of DNMT1 plus MSH2 or DNMT1 plus XPA yield frequencies of contractions that are intermediate between the single knockdowns. If the instability due to DNMT1 knockdown were completely independent of transcription-induced instability, one would expect the contraction frequency for double knockdowns to equal the sum of the single-knockdown frequencies. One straightforward possibility is that demethylation induced by DNMT1 knockdown enhances transcription through the CAG repeat tract, thereby increasing formation of hairpins and stimulating transcription-induced repeat instability (Fig. 5). In its simplest form this possibility is ruled out by measurements that show no change in HPRT transcription after DNMT1 knockdown (Table 2). It should be noted, however, that a substantial increase in HPRT transcription in a subpopulation of cells might escape our detection, yet be sufficient to elevate the overall frequency of contractions.
Several observations in the literature suggest a link between DNMT1 and MMR. Mutations in the DNMT1 gene were picked up in a screen for genes defective in MMR [48] and ES cells deficient for DNMT1 display increased microsatellite instability, a hallmark of MMR deficiency [48,49,62]. Although no direct interaction between MMR components and DNMT1 has yet been identified [63], both DNMT1 and MutSβ bind to PCNA [44,64,65] and this common platform for binding could serve as the basis for a functional interaction. Other studies have demonstrated that DNMT1 is recruited to sites of DNA damage [66] and that it methylates about half the products of double-strand break repair and silences their expression [67]. If a similar DNMT1-directed silencing mechanism operated on the products of the transcription-induced pathway for CAG contraction, it would render some products undetectable in our system (Figure 5). Discovering the basis for our observations will require additional investigation.
A striking result in these studies is that double knockdown of DNMT1 and MLH1 produced additive effects on repeat contraction. The possibilities illustrated in Figure 5 can account for this result in a general way. For example, if DNMT1 knockdown increased transcription to generate more intermediates, and if MLH1 knockdown reduced the removal of intermediates, then the double knockdown would be expected to increase the frequency of contractions additively. Similarly, if MLH1 knockdown reduced the removal of intermediates and if DNMT1 knockdown reduced the removal of selectable products, then the double knockdown would also additively increase contractions. Which of the possibilities implied in Figure 5 forms the specific basis for additivity is not clear.
In previous studies and this one, we have tested seven MMR genes and shown that they have diverse effects on the stability of CAG repeat tracts in human cells [12,18,38]. Knockdown of MSH2 or MSH3 decreases the frequency of repeat contractions, suggesting that MutSβ normally operates to destabilize repeats. Knockdown of MLH1, PMS2, or DNMT1 increases repeat contractions, suggesting that MutLα and DNMT1 normally act to stabilize CAG repeats. Finally, knockdown of MSH6 and MLH3 had no effect on repeat stability, suggesting that MutSα and MutLγ are not involved in generating CAG contractions. These diverse effects on CAG repeats stand in contrast to the uniformly destabilizing effects of deficiencies in MSH2, MSH3, MLH1, and PMS2 on mono- and dinucleotide repeats that has been observed in human and mouse cells [25,26,65]. These differences are consistent with the distinct mechanisms of instability proposed for the two processes: transcription-induced DNA repair for CAG repeats and DNA replication-induced slippage events for mono- and dinucleotide repeats.
The goal of our studies in human cells is to gain insight into processes that lead to TNR expansions in humans. Yet our genetic assay selectively detects large contractions (>55 repeat units), which represent only a narrow segment of the spectrum of TNR instability, one that is at the opposite end from the changes that are most important for human disease. This consideration raises the natural question: Are observations made in this selection system relevant to CAG repeat stability in vivo? Or framed slightly differently: Do genes that alter CAG stability in this human-cell assay also alter repeat stability in vivo? In this report and previous ones, we have analyzed the effects of 18 siRNA-mediated knockdowns of genes involved in DNA metabolism: 12 significantly altered CAG repeat stability and 6 had no effect [12,18,38]. Ten of these same genes have been tested in mouse or fly models. Among those that altered repeat stability in human cells, mutations in MSH2 [32,33,68], MSH3 [34], PMS2 [37], DNMT1 [38], and XPA (Hubert, Lin, and Wilson, unpublished data) also altered repeat stability in mouse models, and mutations in Mus201, the fly homolog of XPG, altered repeat stability in a fly model [11]. Among those that showed little effect on repeat stability in human cells, mutations in XPC [69], FEN1 [70,71], and MSH6 [34] had little effect on repeat stability in mouse models. In only one instance was there a discordance between systems: mutation of OGG1, which encodes a glycosylase involved in base excision repair (BER), decreased somatic CAG repeat instability in a mouse model [72], whereas siRNA knockdowns of OGG1 (or of APEX1, which controls the next step in the BER pathway) did not alter repeat stability in human cells [18]. Overall, the general agreement between our assay in human cells and assays in mouse models suggests that it will be a productive strategy to screen genes in human cells and then test positive candidates in mouse models, where germline and somatic instability can be assayed.
Acknowledgments
We thank members of the Wilson lab for helpful discussions. This work was supported by a grant from the NIH (GM38219) to JHW.
References
- 1.Bacolla A, Larson JE, Collins JR, Li J, Milosavljevic A, Stenson PD, Cooper DN, Wells RD. Abundance and length of simple repeats in vertebrate genomes are determined by their structural properties. Genome Res. 2008;18:1545–1553. doi: 10.1101/gr.078303.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gatchel JR, Zoghbi HY. Diseases of unstable repeat expansion: mechanisms and common principles. Nat Rev Genet. 2005;6:743–755. doi: 10.1038/nrg1691. [DOI] [PubMed] [Google Scholar]
- 3.Pearson CE, Edamura KN, Cleary JD. Repeat instability: mechanisms of dynamic mutations. Nat Rev Genet. 2005;6:729–742. doi: 10.1038/nrg1689. [DOI] [PubMed] [Google Scholar]
- 4.Orr HT, Zoghbi HY. Trinucleotide repeat disorders. Annu Rev Neurosci. 2007;30:575–621. doi: 10.1146/annurev.neuro.29.051605.113042. [DOI] [PubMed] [Google Scholar]
- 5.Usdin K, Grabczyk E. DNA repeat expansions and human disease. Cell Mol Life Sci. 2000;57:914–931. doi: 10.1007/PL00000734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Riley BE, Orr HT. Polyglutamine neurodegenerative diseases and regulation of transcription: assembling the puzzle. Genes Dev. 2006;20:2183–2192. doi: 10.1101/gad.1436506. [DOI] [PubMed] [Google Scholar]
- 7.Lin Y, Dion V, Wilson JH. Transcription and triplet repeat instability. In: Wells R, Ashizawa T, editors. Genetic Instability and Neurological Diseases. Elsevier; Amsterdam: 2006. pp. 691–704. [Google Scholar]
- 8.Kovtun IV, McMurray CT. Features of trinucleotide repeat instability in vivo. Cell Res. 2008;18:198–213. doi: 10.1038/cr.2008.5. [DOI] [PubMed] [Google Scholar]
- 9.Mirkin SM. Expandable DNA repeats and human disease. Nature. 2007;447:932–940. doi: 10.1038/nature05977. [DOI] [PubMed] [Google Scholar]
- 10.Wells RD, Dere R, Hebert ML, Napierala M, Son LS. Advances in mechanisms of genetic instability related to hereditary neurological diseases. Nucleic Acids Res. 2005;33:3785–3798. doi: 10.1093/nar/gki697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jung J, Bonini N. CREB-binding protein modulates repeat instability in a Drosophila model for polyQ disease. Science. 2007;315:1857–1859. doi: 10.1126/science.1139517. [DOI] [PubMed] [Google Scholar]
- 12.Lin Y, Dion V, Wilson JH. Transcription promotes contraction of CAG repeat tracts in human cells. Nat Struct Mol Biol. 2006;13:179–180. doi: 10.1038/nsmb1042. [DOI] [PubMed] [Google Scholar]
- 13.Lin Y, Hubert L, Jr, Wilson JH. Transcription destabilizes triplet repeats. Mol Carcinog. 2009;48:350–361. doi: 10.1002/mc.20488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bowater RP, Jaworski A, Larson JE, Parniewski P, Wells RD. Transcription increases the deletion frequency of long CTG.CAG triplet repeats from plasmids in Escherichia coli. Nucleic Acids Res. 1997;25:2861–2868. doi: 10.1093/nar/25.14.2861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schumacher S, Pinet I, Bichara M. Modulation of transcription reveals a new mechanism of triplet repeat instability in Escherichia coli. J Mol Biol. 2001;307:39–49. doi: 10.1006/jmbi.2000.4489. [DOI] [PubMed] [Google Scholar]
- 16.Parniewski P, Bacolla A, Jaworski A, Wells RD. Nucleotide excision repair affects the stability of long transcribed (CTG*CAG) tracts in an orientation-dependent manner in Escherichia coli. Nucleic Acids Res. 1999;27:616–623. doi: 10.1093/nar/27.2.616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Oussatcheva EA, Hashem VI, Zou Y, Sinden RR, Potaman VN. Involvement of the nucleotide excision repair protein UvrA in instability of CAG*CTG repeat sequences in Escherichia coli. J Biol Chem. 2001;276:30878–30884. doi: 10.1074/jbc.M104697200. [DOI] [PubMed] [Google Scholar]
- 18.Lin Y, Wilson JH. Transcription-induced CAG repeat contraction in human cells is mediated in part by transcription-coupled nucleotide excision repair. Mol Cell Biol. 2007;27:6209–6217. doi: 10.1128/MCB.00739-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kunkel TA, Erie DA. DNA mismatch repair. Annu Rev Biochem. 2005;74:681–710. doi: 10.1146/annurev.biochem.74.082803.133243. [DOI] [PubMed] [Google Scholar]
- 20.Modrich P. Mechanisms in eukaryotic mismatch repair. J Biol Chem. 2006;281:30305–30309. doi: 10.1074/jbc.R600022200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jiricny J. The multifaceted mismatch-repair system. Nat Rev Mol Cell Biol. 2006;7:335–346. doi: 10.1038/nrm1907. [DOI] [PubMed] [Google Scholar]
- 22.Iyer RR, Pluciennik A, Burdett V, Modrich PL. DNA mismatch repair: functions and mechanisms. Chem Rev. 2006;106:302–323. doi: 10.1021/cr0404794. [DOI] [PubMed] [Google Scholar]
- 23.Slean MM, Panigrahi GB, Ranum LP, Pearson CE. Mutagenic roles of DNA “repair” proteins in antibody diversity and disease-associated trinucleotide repeat instability. DNA Repair (Amst) 2008;7:1135–1154. doi: 10.1016/j.dnarep.2008.03.014. [DOI] [PubMed] [Google Scholar]
- 24.Wang G, Vasquez KM. Non-B DNA structure-induced genetic instability. Mutat Res. 2006;598:103–119. doi: 10.1016/j.mrfmmm.2006.01.019. [DOI] [PubMed] [Google Scholar]
- 25.Peltomaki P. Deficient DNA mismatch repair: a common etiologic factor for colon cancer. Hum Mol Genet. 2001;10:735–740. doi: 10.1093/hmg/10.7.735. [DOI] [PubMed] [Google Scholar]
- 26.Jacob S, Praz F. DNA mismatch repair defects: role in colorectal carcinogenesis. Biochimie. 2002;84:27–47. doi: 10.1016/s0300-9084(01)01362-1. [DOI] [PubMed] [Google Scholar]
- 27.Jaworski A, Rosche WA, Gellibolian R, Kang S, Shimizu M, Bowater RP, Sinden RR, Wells RD. Mismatch repair in Escherichia coli enhances instability of (CTG)n triplet repeats from human hereditary diseases. Proc Natl Acad Sci U S A. 1995;92:11019–11023. doi: 10.1073/pnas.92.24.11019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schmidt KH, Abbott CM, Leach DR. Two opposing effects of mismatch repair on CTG repeat instability in Escherichia coli. Mol Microbiol. 2000;35:463–471. doi: 10.1046/j.1365-2958.2000.01727.x. [DOI] [PubMed] [Google Scholar]
- 29.Hashem VI, Rosche WA, Sinden RR. Genetic assays for measuring rates of (CAG).(CTG) repeat instability in Escherichia coli. Mutat Res. 2002;502:25–37. doi: 10.1016/s0027-5107(02)00026-x. [DOI] [PubMed] [Google Scholar]
- 30.Miret JJ, Pessoa-Brandao L, Lahue RS. Instability of CAG and CTG trinucleotide repeats in Saccharomyces cerevisiae. Mol Cell Biol. 1997;17:3382–3387. doi: 10.1128/mcb.17.6.3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schweitzer JK, Livingston DM. Destabilization of CAG trinucleotide repeat tracts by mismatch repair mutations in yeast. Hum Mol Genet. 1997;6:349–355. doi: 10.1093/hmg/6.3.349. [DOI] [PubMed] [Google Scholar]
- 32.Kovtun IV, McMurray CT. Trinucleotide expansion in haploid germ cells by gap repair. Nat Genet. 2001;27:407–411. doi: 10.1038/86906. [DOI] [PubMed] [Google Scholar]
- 33.Savouret C, Garcia-Cordier C, Megret J, te Riele H, Junien C, Gourdon G. MSH2-dependent germinal CTG repeat expansions are produced continuously in spermatogonia from DM1 transgenic mice. Mol Cell Biol. 2004;24:629–637. doi: 10.1128/MCB.24.2.629-637.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.van den Broek WJ, Nelen MR, Wansink DG, Coerwinkel MM, te Riele H, Groenen PJ, Wieringa B. Somatic expansion behaviour of the (CTG)n repeat in myotonic dystrophy knock-in mice is differentially affected by Msh3 and Msh6 mismatch-repair proteins. Hum Mol Genet. 2002;11:191–198. doi: 10.1093/hmg/11.2.191. [DOI] [PubMed] [Google Scholar]
- 35.Foiry L, Dong L, Savouret C, Hubert L, Riele HT, Junien C, Gourdon G. Msh3 is a limiting factor in the formation of intergenerational CTG expansions in DM1 transgenic mice. Hum Genet. 2006 doi: 10.1007/s00439-006-0164-7. [DOI] [PubMed] [Google Scholar]
- 36.Manley K, Shirley TL, Flaherty L, Messer A. Msh2 deficiency prevents in vivo somatic instability of the CAG repeat in Huntington disease transgenic mice. Nat Genet. 1999;23:471–473. doi: 10.1038/70598. [DOI] [PubMed] [Google Scholar]
- 37.Gomes-Pereira M, Fortune MT, Ingram L, McAbney JP, Monckton DG. Pms2 is a genetic enhancer of trinucleotide CAG.CTG repeat somatic mosaicism: implications for the mechanism of triplet repeat expansion. Hum Mol Genet. 2004;13:1815–1825. doi: 10.1093/hmg/ddh186. [DOI] [PubMed] [Google Scholar]
- 38.Dion V, Lin Y, Hubert L, Jr, Waterland RA, Wilson JH. Dnmt1 deficiency promotes CAG repeat expansion in the mouse germline. Hum Mol Genet. 2008;17:1306–1317. doi: 10.1093/hmg/ddn019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gorbunova V, Seluanov A, Dion V, Sandor Z, Meservy JL, Wilson JH. Selectable system for monitoring the instability of CTG/CAG triplet repeats in mammalian cells. Mol Cell Biol. 2003;23:4485–4493. doi: 10.1128/MCB.23.13.4485-4493.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rountree MR, Bachman KE, Baylin SB. DNMT1 binds HDAC2 and a new co-repressor, DMAP1, to form a complex at replication foci. Nat Genet. 2000;25:269–277. doi: 10.1038/77023. [DOI] [PubMed] [Google Scholar]
- 41.Robertson KD, Ait-Si-Ali S, Yokochi T, Wade PA, Jones PL, Wolffe AP. DNMT1 forms a complex with Rb, E2F1 and HDAC1 and represses transcription from E2F-responsive promoters. Nat Genet. 2000;25:338–342. doi: 10.1038/77124. [DOI] [PubMed] [Google Scholar]
- 42.Esteve PO, Chin HG, Smallwood A, Feehery GR, Gangisetty O, Karpf AR, Carey MF, Pradhan S. Direct interaction between DNMT1 and G9a coordinates DNA and histone methylation during replication. Genes Dev. 2006;20:3089–3103. doi: 10.1101/gad.1463706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schermelleh L, Haemmer A, Spada F, Rosing N, Meilinger D, Rothbauer U, Cardoso MC, Leonhardt H. Dynamics of Dnmt1 interaction with the replication machinery and its role in postreplicative maintenance of DNA methylation. Nucleic Acids Res. 2007;35:4301–4312. doi: 10.1093/nar/gkm432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chuang LS, Ian HI, Koh TW, Ng HH, Xu G, Li BF. Human, DNA-(cytosine-5) methyltransferase-PCNA complex as a target for p21WAF1. Science. 1997;277:1996–2000. doi: 10.1126/science.277.5334.1996. [DOI] [PubMed] [Google Scholar]
- 45.Reale A, Matteis GD, Galleazzi G, Zampieri M, Caiafa P. Modulation of DNMT1 activity by ADP-ribose polymers. Oncogene. 2005;24:13–19. doi: 10.1038/sj.onc.1208005. [DOI] [PubMed] [Google Scholar]
- 46.Trinh BN, Long TI, Nickel AE, Shibata D, Laird PW. DNA methyltransferase deficiency modifies cancer susceptibility in mice lacking DNA mismatch repair. Mol Cell Biol. 2002;22:2906–2917. doi: 10.1128/MCB.22.9.2906-2917.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen T, Hevi S, Gay F, Tsujimoto N, He T, Zhang B, Ueda Y, Li E. Complete inactivation of DNMT1 leads to mitotic catastrophe in human cancer cells. Nat Genet. 2007;39:391–396. doi: 10.1038/ng1982. [DOI] [PubMed] [Google Scholar]
- 48.Guo G, Wang W, Bradley A. Mismatch repair genes identified using genetic screens in Blm-deficient embryonic stem cells. Nature. 2004;429:891–895. doi: 10.1038/nature02653. [DOI] [PubMed] [Google Scholar]
- 49.Wang KY, James CK. Shen DNA methyltransferase Dnmt1 and mismatch repair. Oncogene. 2004;23:7898–7902. doi: 10.1038/sj.onc.1208111. [DOI] [PubMed] [Google Scholar]
- 50.Tornaletti S, Patrick SM, Turchi JJ, Hanawalt PC. Behavior of T7 RNA polymerase and mammalian RNA polymerase II at site-specific cisplatin adducts in the template DNA. J Biol Chem. 2003;278:35791–35797. doi: 10.1074/jbc.M305394200. [DOI] [PubMed] [Google Scholar]
- 51.Donahue BA, Yin S, Taylor JS, Reines D, Hanawalt PC. Transcript cleavage by RNA polymerase II arrested by a cyclobutane pyrimidine dimer in the DNA template. Proc Natl Acad Sci U S A. 1994;91:8502–8506. doi: 10.1073/pnas.91.18.8502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Izban MG, Luse DS. The RNA polymerase II ternary complex cleaves the nascent transcript in a 3′----5′ direction in the presence of elongation factor SII. Genes Dev. 1992;6:1342–1356. doi: 10.1101/gad.6.7.1342. [DOI] [PubMed] [Google Scholar]
- 53.Kleiman FE, Wu-Baer F, Fonseca D, Kaneko S, Baer R, Manley JL. BRCA1/BARD1 inhibition of mRNA 3′ processing involves targeted degradation of RNA polymerase II. Genes Dev. 2005;19:1227–1237. doi: 10.1101/gad.1309505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kim HS, Li H, Cevher M, Parmelee A, Fonseca D, Kleiman FE, Lee SB. DNA damage-induced BARD1 phosphorylation is critical for the inhibition of messenger RNA processing by BRCA1/BARD1 complex. Cancer Res. 2006;66:4561–4565. doi: 10.1158/0008-5472.CAN-05-3629. [DOI] [PubMed] [Google Scholar]
- 55.Owen BA, Yang Z, Lai M, Gajek M, Badger JD, 2nd, Hayes JJ, Edelmann W, Kucherlapati R, Wilson TM, McMurray CT. (CAG)(n)-hairpin, DNA binds to Msh2-Msh3 and changes properties of mismatch recognition. Nat Struct Mol Biol. 2005;12:663–670. doi: 10.1038/nsmb965. [DOI] [PubMed] [Google Scholar]
- 56.Mendillo ML, Mazur DJ, Kolodner RD. Analysis of the interaction between the Saccharomyces cerevisiae MSH2-MSH6 and MLH1-PMS1 complexes with DNA using a reversible DNA end-blocking system. J Biol Chem. 2005;280:22245–22257. doi: 10.1074/jbc.M407545200. [DOI] [PubMed] [Google Scholar]
- 57.Yoshioka K, Yoshioka Y, Hsieh P. ATR kinase activation mediated by MutSalpha and MutLalpha in response to cytotoxic O6-methylguanine adducts. Mol Cell. 2006;22:501–510. doi: 10.1016/j.molcel.2006.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Waldman AS. Ensuring the fidelity of recombination in mammalian chromosomes. Bioessays. 2008;30:1163–1171. doi: 10.1002/bies.20845. [DOI] [PubMed] [Google Scholar]
- 59.Sugawara N, Goldfarb T, Studamire B, Alani E, Haber JE. Heteroduplex rejection during single-strand annealing requires Sgs1 helicase and mismatch repair proteins Msh2 and Msh6 but not Pms1. Proc Natl Acad Sci U S A. 2004;101:9315–9320. doi: 10.1073/pnas.0305749101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dhar A, Lahue RS. Rapid unwinding of triplet repeat hairpins by Srs2 helicase of Saccharomyces cerevisiae. Nucleic Acids Res. 2008 doi: 10.1093/nar/gkn225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bhattacharyya S, Lahue RS. Saccharomyces cerevisiae Srs2 DNA helicase selectively blocks expansions of trinucleotide repeats. Mol Cell Biol. 2004;24:7324–7330. doi: 10.1128/MCB.24.17.7324-7330.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kim M, Trinh BN, Long TI, Oghamian S, Laird PW. Dnmt1 deficiency leads to enhanced microsatellite instability in mouse embryonic stem cells. Nucleic Acids Res. 2004;32:5742–5749. doi: 10.1093/nar/gkh912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cannavo E, Gerrits B, Marra G, Schlapbach R, Jiricny J. Characterization of the interactome of the human MutL homologues MLH1, PMS1, and PMS2. J Biol Chem. 2007;282:2976–2986. doi: 10.1074/jbc.M609989200. [DOI] [PubMed] [Google Scholar]
- 64.Umar A, Buermeyer AB, Simon JA, Thomas DC, Clark AB, Liskay RM, Kunkel TA. Requirement for PCNA in DNA mismatch repair at a step preceding DNA resynthesis. Cell. 1996;87:65–73. doi: 10.1016/s0092-8674(00)81323-9. [DOI] [PubMed] [Google Scholar]
- 65.Hsieh P, Yamane K. DNA mismatch repair: molecular mechanism, cancer, and ageing. Mech Ageing Dev. 2008;129:391–407. doi: 10.1016/j.mad.2008.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mortusewicz O, Schermelleh L, Walter J, Cardoso MC, Leonhardt H. Recruitment of DNA methyltransferase I to DNA repair sites. Proc Natl Acad Sci U S A. 2005;102:8905–8909. doi: 10.1073/pnas.0501034102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cuozzo C, Porcellini A, Angrisano T, Morano A, Lee B, Di Pardo A, Messina S, Iuliano R, Fusco A, Santillo MR, Muller MT, Chiariotti L, Gottesman ME, Avvedimento EV. DNA damage, homology-directed repair, and DNA methylation. PLoS Genet. 2007;3:e110. doi: 10.1371/journal.pgen.0030110. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 68.Savouret C, Brisson E, Essers J, Kanaar R, Pastink A, te Riele H, Junien C, Gourdon G. CTG repeat instability and size variation timing in DNA repair-deficient mice. Embo J. 2003;22:2264–2273. doi: 10.1093/emboj/cdg202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dragileva E, Hendricks A, Teed A, Gillis T, Lopez ET, Friedberg EC, Kucherlapati R, Edelmann W, Lunetta KL, Macdonald ME, Wheeler VC. Intergenerational and striatal CAG repeat instability in Huntington’s disease knock-in mice involve different DNA repair genes. Neurobiol Dis. 2009;33:37–47. doi: 10.1016/j.nbd.2008.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.van den Broek WJ, Nelen MR, van der Heijden GW, Wansink DG, Wieringa B. Fen1 does not control somatic hypermutability of the (CTG)(n)*(CAG)(n) repeat in a knock-in mouse model for DM1. FEBS Lett. 2006;580:5208–5214. doi: 10.1016/j.febslet.2006.08.059. [DOI] [PubMed] [Google Scholar]
- 71.Spiro C, McMurray CT. Nuclease-deficient FEN-1 blocks Rad51/BRCA1-mediated repair and causes trinucleotide repeat instability. Mol Cell Biol. 2003;23:6063–6074. doi: 10.1128/MCB.23.17.6063-6074.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kovtun IV, Liu Y, Bjoras M, Klungland A, Wilson SH, McMurray CT. OGG1 initiates age-dependent CAG trinucleotide expansion in somatic cells. Nature. 2007;447:447–452. doi: 10.1038/nature05778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Quinn JE, Kennedy RD, Mullan PB, Gilmore PM, Carty M, Johnston PG, Harkin DP. BRCA1 functions as a differential modulator of chemotherapy-induced apoptosis. Cancer Res. 2003;63:6221–6228. [PubMed] [Google Scholar]