

Abstract
Microfluidic systems show great promise for single-cell analysis, but as these technologies mature their utility must be validated by studies of biologically-relevant processes. An important biomedical application of these systems is characterization of tumor cell heterogeneity. In this work, we used a robust microfluidic platform to explore the heterogeneity of enzyme activity in single cells treated with a chemotherapeutic drug. Using chemical cytometry, we measured peptide degradation in the U937 acute myeloid leukemia (AML) cell line in the presence and absence of the aminopeptidase inhibitor Tosedostat (CHR-2797). Analysis of 99 untreated cells revealed rapid and consistent degradation of the peptide reporter within 20 min of loading. Results from drug-treated cells showed inhibited, but on-going degradation of the reporter. Because the device operates at an average sustained throughput of 37 ± 7 cells/h, we were able to sample cells over the course of this time-dependent degradation. In data from 498 individual drug-treated cells, we found a linear dependence of degradation rate on amount of substrate loaded superimposed upon substantial heterogeneity in peptide processing in response to inhibitor treatment. Importantly, these data demonstrated the potential of microfluidic systems to sample biologically-relevant analytes and time-dependent processes in large numbers of single cells.
Biological research is increasingly identifying important implications of cellular heterogeneity. For example, recent findings in cancer biology underscore the heterogeneity of individual tumor cells. This heterogeneity results in differential responses to chemotherapeutic drugs,1–3 including fractional killing of tumor cells,4 emergence of drug resistant subpopulations,5–6 and increased phenotypic heterogeneity.2 Characterization of cell-to-cell variability in response to chemotherapy will therefore inform both basic cancer biology research and personalized medicine for individual patients. Both applications require robust, high-throughput methods to evaluate cellular responses to drug treatment at the single-cell level.
Established single-cell analysis methods, such as flow and image cytometry are high throughput, analyzing up to 50,000 cell/s and ~1,000 cells/s, respectively;7 however, these methods most commonly use antibodies to label and detect proteins of interest. One drawback is that antibodies can only identify the presence of a protein of interest or its phosphorylated substrate but not the level of activity. Additionally, antibody-labeling suffers from nonspecific binding and reduces the generality of these methods because effective antibodies have not been developed for the active forms of many enzymes or the products of the reactions that they catalyze. As a result, single-cell methodologies based on antibody-labeling are poorly suited to direct measures of enzyme activity in drug-treated cells. Alternative methods use fluorescence resonance energy transfer (FRET) sensors or fluorogenic substrates to report on enzyme activity.8 While FRET and fluorogenic substrates provide spatial and temporal information, these substrates have several disadvantages. FRET sensors typically have limited dynamic range and require genetic engineering of the cells under study. Fluorogenic substrates generally lack specificity, and neither method corrects for variation in substrate concentration.8
In contrast, chemical cytometry addresses these concerns by providing a direct, quantitative readout of cell contents. In this technique, individual cells are lysed, and the released intracellular molecules are electrophoretically separated and detected by fluorescence or a similarly sensitive method. Chemical cytometry has been applied to small molecules, amino acids, native peptides and proteins, and fluorescent peptide substrates using both capillary- and microchip-based systems. Compared to more established techniques, however, the throughput of chemical cytometry systems has been limited. For example, state-of-the-art capillary systems have typically been applied to tens of cells per study,9–14 although work from this laboratory has assayed up to 28 cells in 16 min.15 Microfluidic systems with higher throughputs (10s of cells/min) have been demonstrated, but most of these devices have analyzed fluorescent dyes rather than biologically relevant molecules.16–18 Like capillary systems, most microfluidic devices applied to biologically relevant analytes have low throughput and/or analyze small numbers of cells.19–30 A few select studies have achieved high throughput analysis of biological analytes, such as amines, hemoglobin, and glutathione, in >50 single cells.31–33 However, to date, no comprehensive analysis has been performed on biological data from a large number of single cells analyzed by microfluidic chemical cytometry.
In this work, we use a robust microfluidic platform to explore the heterogeneity of peptide processing in acute myeloid leukemia, demonstrating drug-induced metabolic change in single cells using chemical cytometry. Although this device has been well-characterized,18, 34 it was previously applied only to analysis of fluorescent dye molecules.18 Here we demonstrate the applicability of this technology to a biologically relevant analyte in a study of peptidase-targeted chemotherapy. To assay this process, we investigate the degradation of a fragment of cyclin-dependent kinase 4 (cdk4) in acute myeloid leukemia (AML) cells.35 An aminopeptidase inhibitor, currently in clinical trials for AML treatment,36 slowed peptide processing sufficiently to observe this dynamic process over the time scale of the experiments (20–120 min) and revealed cellular heterogeneity in susceptibility to chemotherapeutic inhibition of peptide metabolism. To evaluate trends in these data sets, we performed a comprehensive analysis of the cell-to-cell variability in fragment peak profiles and degradation rates.
Experimental Section
Materials
The peptide reporter, YSYQMALTPVV(K-5-FAM)TL, was chosen based on the proteasomal products of cdk4 as a likely precursor peptide to the antigenic fragment ALTPVVVTL predicted by NetChop 3.0.37 One valine residue was changed to a FAM-tagged lysine to add a fluorescent label away from the potentially biologically relevant N- and C-termini, which bind to the major histocompatibility complex during antigen presentation.38 The labeled reporter was obtained from Anaspec and stored as a 2 mM stock in DMSO at −20 °C. Side-chain labeled lysine (K-FAM) was prepared by deprotecting Fmoc-K-FAM (Anaspec) using 30% piperidine in DMF, confirmed by LC-ESI-MS (Agilent, 1200 HPLC, 6110 quadrupole MS), and used as a standard. Small unilamellar vesicles were prepared by sonication at a concentration of 2 mg/mL phosphatidylcholine in 25 mM Tris, 150 mM NaCl, pH 8.4 as described previously.39 Extracellular buffer (ECB) contained 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), 135 mM NaCl, 5 mM KCl, 1 mM MgCl2, 1 mM CaCl2 and 10 mM glucose at pH 7.4. Silver-silver chloride wire electrodes were prepared by soaking silver wires overnight in aqueous 1 M iron (III) chloride and 0.1 M hydrochloric acid.
Device fabrication
Hybrid poly(dimethylsiloxane) (PDMS)-glass devices were fabricated using standard soft lithography. SU-8 masters were fabricated by spin-coating a 20 μm thick photoresist layer onto a silicon wafer. Masters were pre-baked for 5 min at 95 °C, exposed to 300 mJ UV light (Karl Suss MA6/BA6 mask aligner), and underwent a post-exposure bake of 5 min at 95 °C. The patterned wafer was then developed in propylene glycol methyl ether acetate for 2 min, rinsed with isopropyl alcohol, and hard-baked for 10 min at 90 °C and 70 min at 120 °C. A 10:1 mixture of PDMS prepolymer and curing agent (Sylgard 184, Corning) was degassed, spin-coated onto the SU-8 master at 300 rpm for 30 s, and cured for 15 min at 120 °C. Using an oxygen plasma (PDC-001, Harrick), PDMS channel structures were irreversibly bonded to glass coverslips (#1, Fisher Scientific) and siloxane tubing reservoirs (Masterflex) were bonded to the PDMS channel outlets. Immediately after bonding, the channels were filled with small unilamellar phosphatidylcholine vesicles, which spontaneously formed a supported bilayer membrane coating, as described previously.39 Assembled and coated devices were stored at 4 °C and used within two days.
Cell culture and treatment
The human acute myeloid leukemia cell line U937 was obtained from American Type Culture Collection (ATCC, Manassas, VA) and maintained at a density of 105 – 106 cells/mL in RPMI media supplemented with 10% fetal bovine serum, 50 units/mL penicillin and 50 μg/mL streptomycin at 37 °C and 5% CO2. For drug treatment experiments, cells were resuspended in fresh media containing 1 μM CHR-2797 (Tosedostat, SelleckChem) at a density of 5 × 105 cells/mL 24 h prior to pinocytic loading with the peptide reporter. Immediately prior to experimentation, all cells were loaded with carboxyfluorescein and the peptide reporter using Influx pinocytic loading reagent (Life Technologies). Briefly, 2–4 × 106 cells were pelleted, resuspended in 40 μL of hypertonic loading media containing 100 μM peptide and 25 μM carboxyfluorescein as an internal standard, and incubated for 10 min at 37 °C. Cells were then exposed to 10 mL of hypotonic media for 2 min to lyse pinosomes and release the peptide into the cytoplasm. The time at which the hypotonic media was applied was used as t = 0 min for all experiments. The cells were then washed once in complete media and twice in ECB. During optimization of pinocytic loading procedures, epifluorescence microscopy was used to confirming loading of the peptide and internal standard into the cells. For single-cell experiments, the cells were resuspended at a density of 2–3 × 106 cells/mL in ECB. For ensemble measurements, 107 cells were loaded, washed and pelleted as described above, resuspended in 20 μL ECB, and lysed by heat treatment at 90 °C for 5 min. The resulting lysate was centrifuged at 19000×g for 5 min at 4 °C, and the supernatant was collected and stored at −20 °C until analysis.
Ensemble Measurements
Simple cross chips 20 μm wide by 20 μm deep were fabricated as described above and flushed for 10 min with filtered ECB prior to use. Samples were prepared as described above and diluted in ECB and loaded into the sample reservoir. Silver-silver chloride wire electrodes were used to apply potentials from a high voltage sequencer (HVS448, LabSmith) for 500 ms gated injections and subsequent separation at a field strength of 210 V/cm. Laser-induced fluorescence detection was achieved 10 mm from the intersection using an epifluorescence detection scheme with a 60× objective, argon ion laser (488 nm), a 20 μm square pinhole formed using a Nikon PFX adaptor, a photomultiplier tube (PMT 2000, Advanced Research Instruments Corp.), current-to-voltage converter (PMT-5 amplifier, Advanced Research Instruments Corp.), 10 Hz hardware filter (Tuneable Active Filter 900, Frequency Devices, Inc.), and customized LabView software (National Instruments). Addition of 0.5 – 1 μL of carboxyfluorescein (cFL) or K-FAM standards to the sample was used to assist in peak identification.
Chemical cytometry
Microfluidic chemical cytometry was performed using a previously characterized device design (Figure 1).18, 34 Prior to use, chips were flushed for 10 min with 25 mM Tris, pH 8.3. Cells in ECB buffer were then loaded into the sample inlet, and pressure-driven flow rates were adjusted by changing the fluid level in the waste outlet reservoir. For these experiments, a 2-mm stir bar was placed in the cell reservoir and controlled using a cuvette stirrer (Starna) to prevent the cells from settling. Cells were detected by video and lysed approximately 50 μm upstream of the separation channel with the focused microbeam of a 532-nm pulsed microchip laser (FP-2 ILM, CRC) as described previously.40 Using platinum wire electrodes, a potential of 1500 V (CZE1000R, Spellman) was applied to the 35-mm long separation channel to draw charged cell contents, including the fluorescently labeled peptide fragments, into the channel and effect a separation. Laser-induced fluorescence detection was achieved using an argon ion laser, FITC filter, PMT (H7732-10, Hamamatsu), current-to-voltage converter, and 10 Hz hardware filter. Data were recorded at a rate of 350 Hz in a custom LabView program.
Figure 1.
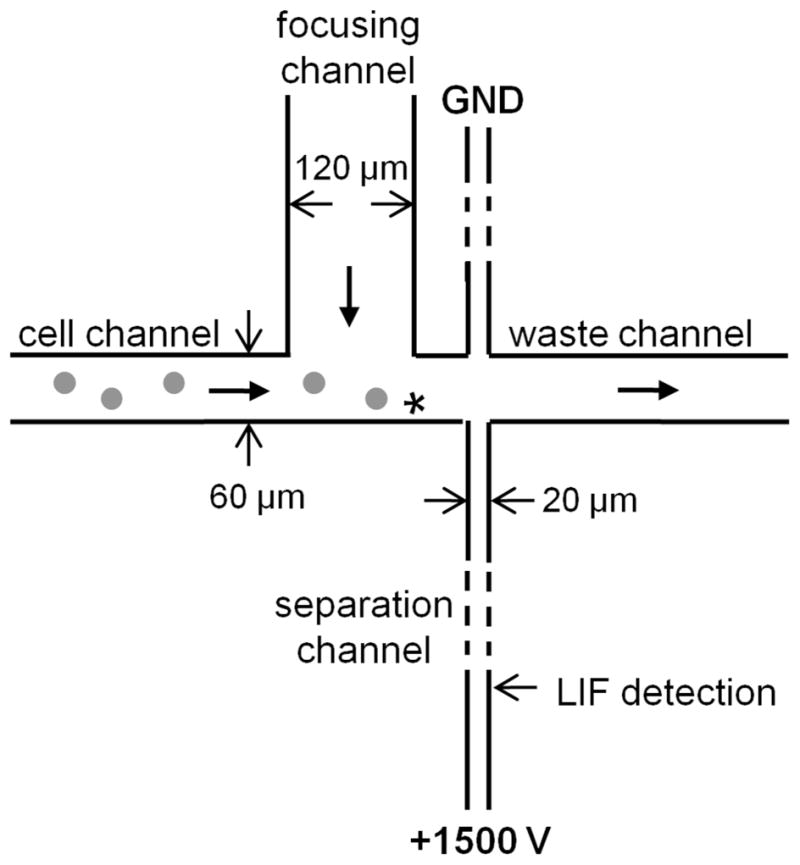
Schematic of the microfluidic device used. The asterisks indicates the focal point of the pulsed laser, i.e., the location of cell lysis.
Data analysis
Data analysis was performed in MATLAB using an in-house program for wavelet de-noising, baseline correction, and peak picking. Automated wavelet de-noising was performed using the symlet 8 wavelet with a soft threshold. The symlet 8 wavelet was selected based on empirical observations of its performance as well as previously reported data showing its effectiveness in processing electropherograms,41 and soft thresholding has been shown to produce optimal reconstructions when used in de-noising 1-dimensional signals.42 The edges of peaks were identified from the smoothed derivative of the data, and a median-value baseline subtraction was performed. Peaks were selected that met the following criteria, which were determined using a subset of the data as a training set: minimum width of 0.35 s, minimum height of twice the median baseline, and minimum area of 120× the minimum peak height. The automated data analysis was manually verified to confirm accurate peak identification.
Results and Discussion
In this work, a fluorescent peptide substrate was used to report peptidase activity in single cells. This combination of chemical cytometry and a fluorescent peptide substrate has previously been applied to studies of peptidases in single cells and allows researchers to identify the most susceptible sites for peptide breakdown and compare degradation patterns between intact cells and lysates.43–45 For these experiments, intact cells were loaded with the fluorescent peptide reporter using pinocytic loading, and the application of the hypotonic media was considered t = 0 for all experiments. At fixed time points after loading, ensemble samples were prepared by lysing a large number (107) of cells to yield an average degradation pattern for the population. Data from these samples were compared to measurements of peptide degradation in single cells analyzed using the device design shown in Figure 1. Briefly, cells were loaded on the device and hydrodynamically moved to the intersection. Flow from the focusing channel directed the cells toward the opposing edge of the cell channel for lysis by a laser pulse (asterisk, Figure 1) near the intersection with the separation channel. Released intracellular compounds, including fragments of the fluorescent reporter peptide, were electrophoretically drawn into the separation channel, separated, and detected.
In the absence of inhibitor, peptide degradation was extremely rapid. For both ensemble measurements (Figure 2a) and single cells (Figure 2b) degradation appeared to be complete by the earliest measured times, and a single peak was observed for 98% of cells (n = 99 cells). This was true across a wide range of peptide loading concentrations (Figure 2d). Addition of a K-FAM standard to the ensemble lysate sample confirmed that the single peak observed co-migrated with the labeled lysine residue, suggesting rapid degradation of this peptide to form the single side chain-labeled amino acid. To further validate this peak assignment, single cells were loaded with carboxyfluorescein and K-FAM for analysis (Figure 2c). In these cells, the normalized migration time of the K-FAM peak was similar that of the single peak in peptide-loaded cells. We suspect the slight difference in normalized migration time between Figure 2b and Figure 2c was due to chip-to-chip variation in channel length, surface properties, or other parameters. Although these data do not rule out the possibility that other fragments co-migrated with the single peak observed, degradation to individual amino acids is likely since many peptides are rapidly degraded in living cells.46 Cancer cells, in particular those derived from rapidly growing tumors like AML, over-express multiple types of peptidases and proteases, which can provide various survival advantages.47 For example, while aminopeptidase N (CD13) is expressed in some normal myeloid lineage cells, it is expressed in roughly 90% of AML cases and highly expressed in U937 cells.48 Rapidly growing cancers require a sufficient pool of amino acids for protein synthesis. Because aminopeptidases (including aminopeptidase N) generate free amino acids, their pharmacologic inhibition by agents such as Tosedostat can lead to amino acid deprivation and cell death.36, 49
Figure 2.

Electropherograms of peptide fragments from untreated samples, including (a) ensemble lysate formed from 107 cells and (b) single cells loaded with the internal standard (carboxyfluorescein, cFL) and the reporter peptide. Peaks in (a) were identified by addition of standards to the sample. (c) Electropherogram of the contents of a single cell loaded with cFL and K-FAM. Time since peptide loading was < 30 min for all traces. The time axis of each trace was normalized to the migration time of the internal standard for (b), and y-values of traces (b) and (c) were multiplied by 25 and 150, respectively. (d) Percent of total fragment peak area in peak 1 as a function of total fragment peak area for n = 99 untreated cells.
Interestingly, of the 99 untreated cells analyzed, 2 cells showed a second fragment peak migrating just after the major peak (Figure S-1). In both cases, this peak represented < 7% of the total fragment peak area (Figure 2d). Observation of the second fragment peak suggests heterogeneity in peptide processing among untreated cells, albeit on a time scale faster than was captured by our current methods.
To explore the heterogeneity of cellular response to an aminopeptidase inhibitor, we incubated cells with the drug candidate Tosedostat (CHR-2797) at a dose known to inhibit peptidase activity and to have anti-proliferative effects. Tosedostat is a membrane-permeable compound, which is converted to the active aminopeptidase inhibitor by esterases within cells.49 Previous studies have shown that application of 1 μM Tosedostat to cells for 24 h inhibits fragmentation of a fluorogenic substrate by approximately 50%,50 and prolonged exposure to this dose substantially inhibits proliferation of U937 cells.49 In this work, the inhibition of aminopeptidase activity resulted in slowed degradation of the reporter peptide and formation of multiple fragment peaks during the analysis time (Figure 3). In ensemble measurements and in most single cells, we observed three major peaks representing at least three peptide fragments. Capillary electrophoresis experiments with standard additions indicated that none of the three peaks co-migrated with the precursor peptide, YSYQMALTPVV(K-5-FAM)TL (Figure S-2), indicating that the three peaks were composed of fragments of the precursor peptide. Tosedostat inhibits three important aminopeptidases, leucine aminopeptidase (LAP), puromycin-sensitive aminopeptidase (PuSA), and aminopeptidase N, with IC50 values of 100, 150, and 220 nM, respectively;49 however, the drug has not been shown to inhibit endopeptidases and inhibits other aminopeptidases to a lesser extent. Consequently, the fragments observed are likely the products of uninhibited peptidase activity on the 14 amino acid precursor peptide.
Figure 3.

Electropherograms for (a) lysate from 107 cells and (b) single cells treated with Tosedostat and lysed at various time points after peptide loading. The first peak in each electropherogram was the internal standard, carboxyfluorescein, and the time axis of each trace was normalized to the migration time of the internal standard for the first trace.
Although inhibitor treatment slowed peptide processing, on-going degradation of the peptide reporter was evident both in ensemble and single-cell measurements. As noted previously, three major peaks were observed for the earliest time point (16 min). Over time, the first fragment peak, which co-migrated with K-FAM, increased in area while the other fragment peaks decreased in size (Figure 3). In ensemble measurements, the K-FAM-containing peak was the major feature observed after 2 h; in contrast, single cells showed greater heterogeneity in peptide fragment pattern as a function of time. While most single cell separations yielded the same peaks observed in bulk lysate (Figure 3), these peaks had different relative intensities from cell-to cell. Even cells lysed at similar time points post-peptide loading showed substantial heterogeneity in fragment peak patterns (Figure S-3). Additionally, some cells possessed peak patterns not observed in ensemble measurements. For example, 16% of single cells yielded electropherograms with two fragment peaks (Figure 4a), a pattern not observed at any time point in ensemble measurements (Figure 3). Based on comparisons of peak areas obtained from single cells to those of K-FAM standards, the peptide loaded into cells ranged from 4 × 10−19 to 5 × 10−16 mol per cell (see Supporting Info for details). Despite the wide variation in substrate loading from cell to cell, the number of fragment peaks observed in individual cells in this study was not correlated to the amount of peptide loaded (Figure 4a). Instead, the heterogeneity observed here likely reflects a combination of biological variation in normal peptide metabolism between cells, varying uptake and susceptibility to inhibitor, and biological noise.
Figure 4.

Distributions of cells with respect to total peak area (peptide loading), fragment peak number, and analysis time. (a) The distribution of total fragment peak area as a function of number of fragment peaks. (b) Histogram of the population distribution as a function of time elapsed after peptide loading. For both panels, n = 498 drug-treated cells.
Although chemical cytometry permits only one time point to be collected per cell, time-dependent processes can still be observed with single-cell resolution provided that a representative sample of the population can be analyzed for each relevant time window. Previous chemical cytometry studies of peptide degradation in single cells have analyzed tens of cells;12, 43–44 in contrast, a typical experiment in this work yielded useable data for 50 cells over a duration of 1.25 h, resulting in an average sustained throughput of 37 ± 7 cells/h. We conducted 11 such experiments with Tosedostat-treated cells, accumulating data for n = 498 cells distributed across multiple time points from 16 and 162 min after reporter loading. By pooling data from these experiments, we were able to sample at least 10 cells for every 15-min time window, at > 70 cells for each 15-min time window from 30–90 min (Figure 4b). Remarkably cells with each different peak pattern were present at almost all experimental times tested (30–165 min, Figure 4b). It is possible that in the presence of Tosedostat, some cells were able to fully metabolize the peptide while others could not. The ability to sample large numbers of cells across time allowed us to conduct a detailed analysis of the time-dependence of peptide degradation in drug-treated cells.
The majority of the cells (~60%) analyzed prior to 105 min showed three fragment peaks (Figure 4a), the first of which was identified as co-migrating with K-FAM on the basis of ensemble lysate measurements (Figure 3a). Because the resemblance to ensemble electropherograms facilitated peak identification in these samples, we conducted a more detailed analysis of the 287 single-cell electropherograms with 3 fragment peaks. The major trend apparent in these data was an increase in the relative proportion of fragment peak 1 over time since peptide loading (Figure 5a). Since K-FAM is the terminal degradation product for this peptide, it is likely the major component of Peak 1. As a result, it is not surprising that the area of peak 1 would increase for cells sampled later in the experiment. However, this general trend was overlaid by substantial heterogeneity. Importantly, cells analyzed late in experiments sometimes showed peak patterns similar to cells analyzed at early time points, suggesting that the time dependent variation observed represents real biological heterogeneity, not an artifact of device fouling.
Figure 5.

For cells with 3 fragments peaks, (a) a ternary plot of normalized peak areas for each of the three fragment peaks and (b) a plot of area under fragment peak 1 / time elapsed since peptide loading versus total fragment peak area, which should reflect degradation rate versus peptide concentration. Color mapping shows time elapsed between peptide loading and cell lysis (n = 287 cells).
We also evaluated the kinetics of peptide degradation in the cells that yielded three fragment peaks. As a proxy for degradation rate, we calculated the area under fragment peak 1 divided by the time elapsed since peptide loading. We then plotted this value as a function of total fragment peak area, which should be proportional to total cellular peptide concentration (Figure 5b). We observed a moderate correlation (R2 = 0.69), supporting observations from previous studies on smaller numbers of cells that peptide degradation rate depends on peptide concentration.12, 44 However, many cells deviated from the linear correlation between degradation rate and substrate concentration, demonstrating the heterogeneity of this process in inhibitor-treated cells. These data fit into a larger, and still developing, picture of cellular heterogeneity in response to drug treatment.1–2
Conclusions
Chemical cytometry provides complementary information to that obtained by flow and image cytometry. However, to be a viable alternative to these established single-cell analysis methods, chemical cytometry instrumentation must be robust and sufficiently high-throughput to sample representative populations. In this work, we have realized the potential of a microfluidic system to perform chemical cytometry measurements of a biological analyte in large numbers of cells. Importantly, the throughput of the system was sufficient to sample a time-dependent process. Because pinocytic loading requires multiple wash steps prior to on-chip analysis, the earliest time points assayed were ~20 min after addition of hypotonic media to release the peptide into the cells. While this process precluded more rapid characterization of peptide degradation in intact cells, we expect alternative loading methods, e.g. with a photo-releasable substrate or on-chip peptide-loading into cells, could provide a more complete picture of the variability of peptide metabolism in untreated cells.
Results obtained for drug-treated cells show substantial heterogeneity in peptide processing in cancer cells exposed to a chemotherapeutic aminopeptidase inhibitor. Because identification of the components of the fragment peaks was outside the scope of this work, these data represent the minimum amount of heterogeneity in processing of this peptide. Additional undetected heterogeneity may be present if, for example, the same peak pattern was produced from different fragment mixtures. Qualitative and quantitative analyses of the fragment peak patterns revealed interesting temporal and cell-to-cell variation. Single cell electropherograms revealed fragment patterns not observed in ensemble measurements, and kinetic data showed considerable variability in the extent of peptidase inhibition in individual cells. The collection of these data with a chip-based system demonstrates the on-going maturation of microfluidic instrumentation into a robust tool for single-cell analysis.
Supplementary Material
Acknowledgments
The authors thank Prof. David Lawrence for use of the LC-ESI-MS system. This work was supported by grants from the National Institutes of Health: Minority Opportunities in Research division of the National Institute of General Medical Sciences (NIGMS) grant #K12GM000678 (MLK), grants KL2TR000084 and K08HL113594 (PMA) and EB11763 (NLA).
Footnotes
The authors declare no competing financial interest.
Experimental details and electropherograms for additional single cells and ensemble measurements conducted on a commercial capillary electrophoresis system. This material available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Cohen AA, et al. Science. 2008;322:1511–1516. doi: 10.1126/science.1160165. [DOI] [PubMed] [Google Scholar]
- 2.Gascoigne KE, Taylor SS. Cancer Cell. 2008;14:111–122. doi: 10.1016/j.ccr.2008.07.002. [DOI] [PubMed] [Google Scholar]
- 3.Singh D, Ky CJ, Wichaidit C, Steininger R, III, Wu L, Altschuler S. Mol Syst Biol. 2010;6:369. doi: 10.1038/msb.2010.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Niepel M, Spencer S, Sorger P. Curr Opin Chem Biol. 2009;13:556–561. doi: 10.1016/j.cbpa.2009.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Greaves M, Maley C. Nature. 2012;481:306–313. doi: 10.1038/nature10762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ding L, et al. Nature. 2012;481:506–510. doi: 10.1038/nature10738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tárnok A, Pierzchalski A, Valet G. Curr Med Chem. 2010;17:1719–1729. doi: 10.2174/092986710791111251. [DOI] [PubMed] [Google Scholar]
- 8.Kovarik ML, Allbritton NL. Trends Biotechnol. 2011;29:222–230. doi: 10.1016/j.tibtech.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Han F, Lillard SJ. Anal Biochem. 2002;302:136–143. doi: 10.1006/abio.2001.5519. [DOI] [PubMed] [Google Scholar]
- 10.Xie W, Xu A, Yeung ES. Anal Chem. 2009;81:1280–1284. doi: 10.1021/ac802249m. [DOI] [PubMed] [Google Scholar]
- 11.Jiang D, Sims CE, Allbritton NL. Electrophoresis. 2010;31:2558–2565. doi: 10.1002/elps.201000054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brown RB, Hewel JA, Emili A, Audet J. Cytom A. 2010;77A:347–355. doi: 10.1002/cyto.a.20880. [DOI] [PubMed] [Google Scholar]
- 13.Nemes P, Knolhoff AM, Rubakhin SS, Sweedler JV. Anal Chem. 2011;83:6810–6817. doi: 10.1021/ac2015855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Essaka DC, Prendergast J, Keithley RB, Hindsgaul O, Palcic MM, Schnaar RL, Dovichi NJ. Neurochem Res. 2012;37:1308–1314. doi: 10.1007/s11064-012-0733-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jiang D, Sims C, Allbritton N. Electrophoresis. 2010;31:2558–2565. doi: 10.1002/elps.201000054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McClain MA, Culbertson CT, Jacobson SC, Allbritton NL, Sims CE, Ramsey JM. Anal Chem. 2003;75:5646–5655. doi: 10.1021/ac0346510. [DOI] [PubMed] [Google Scholar]
- 17.Wang HY, Lu C. Chem Comm. 2006:3528–3530. doi: 10.1039/b605722e. [DOI] [PubMed] [Google Scholar]
- 18.Phillips KS, Lai HH, Johnson E, Sims CE, Allbritton NL. Lab Chip. 2011;11:1333–1341. doi: 10.1039/c0lc00370k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu HK, Wheeler A, Zare RN. Proc Natl Acad Sci USA. 2004;101:12809–12813. doi: 10.1073/pnas.0405299101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gao J, Yin XF, Fang ZL. Lab Chip. 2004;4:47–52. doi: 10.1039/b310552k. [DOI] [PubMed] [Google Scholar]
- 21.Hellmich W, Pelargus C, Leffhalm K, Ros A, Anselmetti D. Electrophoresis. 2005;26:3689–3696. doi: 10.1002/elps.200500185. [DOI] [PubMed] [Google Scholar]
- 22.Xia F, Jin W, Yin XF, Fang Z. J Chromatogr, A. 2005;1063:227–233. doi: 10.1016/j.chroma.2004.12.022. [DOI] [PubMed] [Google Scholar]
- 23.Sun Y, Lu M, Yin XF, Gong XG. J Chromatogr, A. 2006;1135:109–114. doi: 10.1016/j.chroma.2006.09.020. [DOI] [PubMed] [Google Scholar]
- 24.Sun Y, Yin XF. J Chromatogr, A. 2006;1117:228–233. [Google Scholar]
- 25.Toriello NM, Douglas ES, Thaitrong N, Hsiao SC, Francis MB, Bertozzi CR, Mathies RA. Proc Natl Acad Sci USA. 2008;105:29173–20178. doi: 10.1073/pnas.0806355106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Greif D, Galla L, Ros A, Anselmetti D. J Chromatogr, A. 2008;1206:83–88. doi: 10.1016/j.chroma.2008.07.013. [DOI] [PubMed] [Google Scholar]
- 27.Zhao S, Li X, Liu YM. Anal Chem. 2009;81:3873–3878. doi: 10.1021/ac900391u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhao S, Huang Y, Liu YM. J Chromatogr, A. 2009;1216:6746–6751. doi: 10.1016/j.chroma.2009.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ye F, Huang Y, Xu Q, Shi M, Zhao S. Electrophoresis. 2010;31:1630–1636. doi: 10.1002/elps.200900665. [DOI] [PubMed] [Google Scholar]
- 30.Xu CX, Wang M, Yin XF. Analyst. 2011;136:3877–3883. doi: 10.1039/c1an15019g. [DOI] [PubMed] [Google Scholar]
- 31.Allen PB, Doepker BR, Chiu DT. Anal Chem. 2009;81:3784–3791. doi: 10.1021/ac900099y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mellors JS, Jorabchi K, Smith LM, Ramsey JM. Anal Chem. 2010;82:967–973. doi: 10.1021/ac902218y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xu CX, Yin XF. J Chromatogr, A. 2011;1218:726–732. doi: 10.1016/j.chroma.2010.11.049. [DOI] [PubMed] [Google Scholar]
- 34.Kovarik ML, Lai HH, Xiong JC, Allbritton NL. Electrophoresis. 2011;32:3180–3187. doi: 10.1002/elps.201100234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McGary C, Enyenihi AA, Hunsucker SA, Ropp PA, Rodriguez-Cruz T, Lizee G, Alatrash G, Glish GL, Armistead PM. Blood (ASH Annual Meeting Abstracts) 2011;118:4020. [Google Scholar]
- 36.Löwenberg B, et al. J Clin Oncol. 2010;28:4333–4338. doi: 10.1200/JCO.2009.27.6295. [DOI] [PubMed] [Google Scholar]
- 37.Nielsen M, Lundegaard C, Lund O, Kesmir C. Immunogenetics. 2005;57:33–41. doi: 10.1007/s00251-005-0781-7. [DOI] [PubMed] [Google Scholar]
- 38.Guo HC, Jardetzky TS, Garrett TP, Lane WS, Strominger JL, Wiley DC. Nature. 1992;360:364–366. doi: 10.1038/360364a0. [DOI] [PubMed] [Google Scholar]
- 39.Phillips KS, Kottegoda S, Kang KM, Sims CE, Allbritton NL. Anal Chem. 2008;80:9756–9762. doi: 10.1021/ac801850z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lai H, Quinto-Su P, Sims C, Bachman M, Li G, Venugopalan V, Allbritton N. J R Soc Interface. 2008;5:S113–S121. doi: 10.1098/rsif.2008.0177.focus. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Perrin C, Walczak B, Massart DL. Anal Chem. 2001;73:4903–4917. doi: 10.1021/ac010416a. [DOI] [PubMed] [Google Scholar]
- 42.Donoho DL. IEEE Trans Inf Theory. 1995;41:613–627. [Google Scholar]
- 43.Proctor A, Wang Q, Lawrence DS, Allbritton NL. Analyst. 2012;137:3028–3038. doi: 10.1039/c2an16162a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Proctor A, Wang Q, Lawrence DS, Allbritton NL. Anal Chem. 2012;84:7195–7202. doi: 10.1021/ac301489d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brown RB, Hewel JA, Emili A, Audet J. Cytom Part A. 2010;77A:347–355. doi: 10.1002/cyto.a.20880. [DOI] [PubMed] [Google Scholar]
- 46.Yewdell JW, Reits E, Neefjes J. Nat Rev Immunol. 2003;3:952–961. doi: 10.1038/nri1250. [DOI] [PubMed] [Google Scholar]
- 47.Dubowchik GM, Walker MA. Pharmacol Therapeut. 1999;83:67–123. doi: 10.1016/s0163-7258(99)00018-2. [DOI] [PubMed] [Google Scholar]
- 48.Knowles DM. In: Neoplastic Hematopathology. 2. Knowles DM, editor. Lippincott, Williams, and Wilkins; Philadelphia: 2001. pp. 156–157. [Google Scholar]
- 49.Krige D, et al. Cancer Res. 2008;68:6669–6679. doi: 10.1158/0008-5472.CAN-07-6627. [DOI] [PubMed] [Google Scholar]
- 50.Jenkins C, Hewamana S, Krige D, Pepper C, Burnett AK. Leukemia Res. 2011;35:677–681. doi: 10.1016/j.leukres.2010.10.030. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.