

Abstract
Persistent inflammation results in an increase in the magnitude and duration of high K+-evoked Ca2+ transients in putative nociceptive cutaneous dorsal root ganglion (DRG) neurons. The purpose of the present study was to determine whether recruitment of Ca2+-induced Ca2+ release (CICR) contributes to these inflammation-induced changes. Acutely dissociated, retrogradely labeled cutaneous DRG neurons from naïve and complete Freund’s adjuvant inflamed adult male Sprague Dawley rats were studied with ratiometric microfluorimetry. Ryanodine only attenuated the duration but not magnitude of the high K+-evoked Ca2+ transient in neurons from inflamed rats. However, there was no significant impact of inflammation on the potency or efficacy of ryanodine-induced block of the caffeine-evoked Ca2+ transient, or the impact of sarco-endoplasmic reticulum ATPase (SERCA) inhibition on the high K+-evoked Ca2+ transient. Furthermore, while there was no change in the magnitude, an inflammation-induced increase in the duration of the caffeine-evoked Ca2+ transient was only observed with a prolonged caffeine application. In contrast to the high K+-evoked Ca2+ transient, there was no evidence of direct mitrochondrial involvement or that of the Ca2+ extrusion mechanism, the Na+/Ca2+ exchanger, on the caffeine-evoked Ca2+ transient, and block of SERCA only increased the duration of this transient. These results indicate the presence of Ca2+ regulatory domains in cutaneous nociceptive DRG neurons within which cytosolic Ca2+ increased via influx and release are highly segregated. Furthermore, our results suggest that changes in neither CICR machinery nor the coupling between Ca2+ influx and CICR are primarily responsible for the inflammation-induced changes in the evoked Ca2+ transient.
Introduction
We previously demonstrated that persistent inflammation of peripheral tissue is associated with an increase in the magnitude and duration of the high K+-evoked Ca2+ transient in a subpopulation of putative nociceptive cutaneous dorsal root ganglion (DRG) neurons [1]. Identifying the underlying mechanism(s) of this change in Ca2+ signaling is important as an alteration in intracellular Ca2+ signaling may contribute to the pain and hypersensitivity of persistent inflammation both directly via the facilitation of transmitter release [2] and indirectly via changes in the regulation of proteins critical for the control of neuronal excitability [3] and/or changes in gene expression [4].
In sensory neurons, the high K+-evoked increase in the concentration of intracellular Ca2+ ([Ca2+]i) is initiated by a depolarization-induced activation of voltage-gated Ca2+ channels (VGCC) and further shaped by Ca2+ release from intracellular stores, sequestration into organelles, and extrusion from the cell [5–9]. Thus, there are a number of mechanisms that could contribute to the inflammation-induced change in Ca2+ signaling, several likely candidates of which we have previously ruled out. In particular, the inflammation-induced change in the high K+-evoked Ca2+ transient was neither the result of increased neuronal excitability nor an increase in the magnitude of the high K+-evoked depolarization [1]. We subsequently ruled out an increase in Ca2+ influx via VGCC as current density was selectively suppressed by inflammation in putative nociceptive cutaneous DRG neurons [10].
Another mechanism that may contribute to the inflammation-induced change in the regulation of [Ca2+]i in cutaneous neurons is an increase in the relative contribution of Ca2+-induced Ca2+ release (CICR) to the evoked transient. We [7] and others [11] previously demonstrated that CICR contributes to the magnitude of evoked Ca2+ transients in some DRG neurons from naïve animals. However this mechanism contributes little to the high K+-evoked Ca2+ transient in the subpopulation of afferents defined by a small cell body diameter (<30μM), IB4 binding and capsaicin sensitivity. This is also the subpopulation primarily impacted by inflammation [1]. Thus, any change in CICR machinery or the coupling between Ca2+ influx and CICR in these neurons could have a profound influence on the evoked transient. Therefore, in the present study we assessed the contribution of CICR to the inflammation-induced increase in the high K+-evoked Ca2+ transient.
Retrogradely labeled small diameter IB4 binding DRG neurons from naïve and inflamed rats were studied with ratiometric Ca2+ imaging in combination with a variety of pharmacological manipulations to assess the impact of inflammation on the mechanisms underlying CICR.
Methods
Adult male Sprague-Dawley rats (Harlan, 220–300g) were used for all experiments. Animals were housed two per cage in a temperature and humidity controlled animal facility on a 12:12 light:dark schedule with food and water freely available. All procedures were approved by the University of Pittsburgh Institutional Animal Care and Use Committee and performed in accordance with National Institutes of Health guidelines for the use of laboratory animals in research.
Fourteen to 17 days prior to tissue harvest, the retrograde tracer 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbo-cyanine perchlorate (DiI, Invitrogen, Carlsbad, CA) was injected into the glabrous skin of the hindpaw in order to label cutaneous afferents. The tracer was dissolved at 170 mg/mL in dimethylsufoxide (DMSO), diluted 1:10 in 0.9% sterile saline, and injected in 3–5 subcutaneous sites using a 30 g needle for a total volume of 10 μL per hindpaw under isoflurane (Abbott Laboratories, North Chicago, IL, USA) anesthesia. Three days prior to tissue harvest, rats were again anesthetized with isoflurane and inflammation was induced at the site of tracer injection in the left hindpaw with a 100 μL subcutaneous injection of complete Freud’s adjuvant (CFA, Sigma-Aldrich, St Louis MO), diluted 1:1 in 0.9 % sterile saline). Prior to tissue removal, animals were deeply anesthetized with an intraperitoneal injection of a cocktail containing ketamine (55 mg/kg), xylazine (5.5 mg/kg) and acepromazine (1.1 mg/kg) and the L4–L5 DRG were removed ipsilateral to labeling and CFA-induced inflammation. Ganglia were enzymatically treated, mechanically dissociated, and plated on laminin-(Invitrogen, Carlsbad, CA; 1mg/ml) and poly-L-ornithine-coated (Sigma-Aldrich; 1 mg/ml) glass cover slips as previously described [7]. All subsequent experiments were performed within 8 h of tissue harvest.
Calcium Imaging
Neurons were incubated with 2.5 μM Ca2+ indicator fura-2 AM ester with 0.025 % Pluronic F-127 for 20min at room temperature. Neurons were then labeled with FITC-conjugated IB4 (10 μg/ml) for 10 min at room temperature. Labeled neurons were placed in a recording chamber and continuously superfused via a gravity fed system with normal bath solution (mM: 130 NaCl, 3 KCl, 2.5 CaCl2, 0.6 MgCl2, 10 HEPES, 10 glucose, pH 7.4, osmolality 325 mOsm) or a ‘Ca2+-free’ bath solution (mM: 130 NaCl, 3 KCl, 10 MgCl2, 2 EGTA, 10 Hepes, 10 glucose, pH 7.4, osmolality 325 mOsm). Fluorescence data were acquired on a PC running Metafluor software (Molecular Devices, City State) via a CCD camera (Roper Scientific; model RTE/CCD 1300). The ratio (R) of fluorescence emission (510 nm) in response to 340/380nm excitation (controlled by a lambda 10–2 filter changer (Sutter Instrument, Novato, CA) was acquired at 1 Hz during drug application. [Ca2+]i was determined from fura-2 ratio following in situ calibration experiments according to the following equation:
Where Kd is the dissociation constant for fura-2 for Ca2+ at room temperature; Sf2/Sb2 is the fluorescence ratio of the emission intensity excited by 380 nm signal in the absence of Ca2+; Rmin and Rmax are the minimal and maximal fluorescence ratios respectively. Determination of these variables has been described in detail previously [7, 12, 13]. All drugs were applied through a computer-controlled perfusion fast-step system (switching time <20 ms; Warner Perfusion System, Hamden, CT, USA, Model SF-77B).
Chemicals and Reagents
Cyclopiazonic acid (CPA) (10μM, 100mM stock in DMSO) was purchased from Calbiochem (La Jolla, CA, USA), 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbo-cyanine perchlorate (DiI) was purchased from Invitrogen (Carlsbad, CA, USA). Fura-2 acetoxymethyl (AM) ester (2.5 μM) and Pluronic F-127 (0.025 %) were purchased from TEF Laboratories (Austin, TX, USA). Ryanodine (1–100 μM, 10 mM stock in DMSO) and H-89 (10μM, 100mM stock in DMSO) were purchased from R&D Systems (Minneapolis, MN, USA), FITC-conjugated IB4 (IB4, 10 ug/ml), caffeine (10 mM), carbonyl cyanide m-chlorophenyl hydrazone (CCCP) (10μM, 100mM stock in DMSO) were obtained from Sigma-Aldrich (St Louis, MO, USA).
Statistical Analysis
Data are expressed as mean ± s.e.m. Student’s t test was used for simple comparisons between groups. For experiments involving the application of test compounds, vehicle controls were always included. A two-way ANOVA was used for analysis of more than two groups with the Holm-Sidak test used for post-hoc analysis. The response to repeated caffeine application in the presence of different concentrations of ryanodine was assessed with a mixed design 3-way ANOVA. Statistical significance was assessed at p < 0.05.
Results
Data were collected from 315 cutaneous DRG neurons acutely dissociated from 54 male Sprague Dawley rats, of which 30 were control and 24 were subject to CFA-induced peripheral inflammation. Based on previous results indicating that the inflammation-induced increase in the high K+-evoked Ca2+ transient was primarily manifest in small (<30 μm) diameter, cutaneous, IB4 binding, capsaicin sensitive DRG neurons, this subpopulation was the focus of the present study and is subsequently referred to as a subpopulation of putative nociceptive cutaneous neurons. Consistent with our previous results [1], inflammation was associated with a significant increase in the magnitude (as measured by a change from baseline) and duration (as measured by the time to a 50, 75 and 90, but not 25% decrease from peak (T50, T75, T90 and T25)) of the high K+-evoked Ca2+ transient (Figure 1A & B and Table 1). These changes in the evoked Ca2+ transient were not associated with a change in resting [Ca2+]i, which was 139.09 ± 4.80 nM (n = 41) and 133.29 ± 6.33 nM (n = 39) in neurons from naïve and inflamed rats, respectively (p > 0.05).
Figure 1.

Inflammation increases the magnitude and duration of the high K+-evoked Ca2+ transient in putative nociceptive cutaneous DRG neurons. A) Typical high K+-evoked Ca2+ transients in neurons from naïve (gray trace) and inflamed (black trace) rats. B) Pooled data for the magnitude (change from baseline (Δ [Ca2+]i)) and decay (quantified as the time to 50% of peak, or T50) of the high K+ (30 mM for 4 sec) evoked Ca2+ transient in putative nociceptive cutaneous DRG neurons from naïve (n = 32) and inflamed (CFA, n = 29) rats. * is p < 0.05. ** is p < 0.01.
Table 1. Changes in the decay of the high K+ evoked Ca2+ transient in putative nociceptive cutaneous DRG neurons.
Neurons were challenged with high K+ (30 mM, 4s). T25, T50, T75 and T90 are the time to 25, 50, 75 and 90% decay of the peak increase evoked with high K+. Data were analyzed with a mixed design two-way ANOVA, which indicated that there were significant main effects associated with treatment (p < 0.01) and time (p < 0.01), as well as significant interaction between the two (p < 0.01). The Holm-Sidak test was used for post hoc comparisons. As our primary interest was the impact of treatment on the rate of decay, we have only provided the results of the comparisons within each time domain compared to naive.
| Treatment | n | T25 (s) | T50 (s) | T75 (s) | T90 (s) |
|---|---|---|---|---|---|
| Naïve | 41 | 4.66 ± 0.5 | 19.43 ± 2.4 | 50.36 ± 8.2 | 184.45 ± 21.0 |
| CFA | 36 | 3.08 ± 0.3** | 36.93 ± 5.2** | 98.19 ± 13.4** | 280.63 ± 26.3** |
| Naïve Post CPA | 13 | 15.37 ± 5.9** | 82.45 ± 17.0** | 167.7 ± 28.2** | 273.75 ± 34.8 |
| CFA Post CPA | 12 | 9.25 ± 3.7 | 142.56 ± 39.3** | 222.07 ± 47.4** | 304.6 ± 61.8 |
| Naïve Post CCCP | 11 | 15.00 ± 1.09** | 22.14 ± 1.55** | 42.42 ± 2.9** | 66.46 ± 4.85** |
| Naïve Post Na+ Free | 11 | 4.46 ± 0.6 | 31.29 ± 5.93 | 192.62 ± 33.77** | 331.33 ± 39.84** |
is p < 0.01.
Ryanodine attenuates the duration of the high K+-evoked Ca2+ transient in cutaneous neurons from inflamed rats
To assess the presence of an inflammation-induced change in the relative contribution of CICR to the high K+-evoked Ca2+ transient, high K+ (30 mM, 4 sec) was applied to cutaneous neurons from naïve and inflamed rats before and after the application of the ryanodine receptor (RyR) blocker, ryanodine (10 μM). To establish steady-state RyR block, caffeine (10 mM, 4 sec) was applied 4 times with a 2.5 min inter-stimulus interval (ISI) after the application of ryanodine and before the last challenge with high K+ (Figure 2A). A 4s application of 10mM caffeine was chosen based on the results of an analysis of the response to caffeine applied for durations ranging between 250 ms and 12 s: the magnitude of the caffeine-evoked Ca2+ transient saturated in all neurons tested (Δ[Ca2+]i = 453.48 ± 38.9 nM, n = 15) with a duration of application of 4s or greater. Of note, this change in magnitude was comparable to that evoked with high K+ (Figure 1). Interestingly, in control experiments in which caffeine was applied 4 times between high K+ applications in the absence of ryanodine, there was an increase in the T50 of decay of the second high K+-evoked transient (Figure 2A, B and C). The response to high K+ is highly reproducible with an ISI of at least 5 minutes, therefore, this increase in T50 of decay appears to be due to the actions of caffeine, possibly secondary to a shift in resting protein kinase A (PKA) activity as a result of caffeine-induced inhibition of phosphodiesterase [14]. To assess this possibility, we repeated the experiment with high K+ applied before and after repeated application of caffeine in neurons pre-incubated (20min) with the PKA inhibitor, H-89 (10 μM). Consistent with the suggested involvement of PKA in the caffeine-induced increase in the duration of the high K+-evoked Ca2+ transient, the presence of H-89 resulted in a significant attenuation of the caffeine-induced increase in the duration of the high K+-evoked transient from 164.59 ± 0.34 (n = 9) to 51.3 ± 0.34 (n = 7) %ΔT50 (p < 0.05). Regardless of the mechanism of the change in T50, because of its presence, the impact of ryanodine-induced RyR block was assessed relative to control neurons treated identically in the absence of ryanodine. Consistent with previous results indicating that the contribution of CICR to the high K+-evoked transient in putative nociceptive cutaneous DRG neurons is minimal [7], ryanodine (10 μM) had no detectable influence on the magnitude or decay of the high K+-evoked transient in neurons tested from naïve rats (Figure 2C). Comparable results were obtained with 100 μM ryanodine, where the high K+-evoked Ca2+ transient in the presence of ryanodine (T50 = 60.27 ± 9.43 s, n = 4) was not different (p > 0.05) from that observed in control neurons the absence of ryanodine (T50 = 46.69 ± 11.85 s, n = 9). In contrast, while ryanodine had no significant influence on the magnitude of the high K+-evoked Ca2+ transient in putative nociceptive cutaneous neurons from inflamed rats, the T50 of decay was significantly (p < 0.001) attenuated (Figure 2C). This observation is consistent with the recruitment of CICR as a mechanism contributing to at least part of the inflammation-induced change in the high K+-evoked transient.
Figure 2.

The impact of ryanodine on the inflammation-induced increase in the magnitude and time of decay of the high K+-evoked transient. A) Evoked Ca2+ transients in neurons from naïve (gray traces) and inflamed (black traces) rats challenged with high K+ (30 mM, 4 sec) before and after 4 applications of caffeine (10 mM, 4 sec). Neurons were challenged with caffeine in the presence (Ryanodine) or absence (Control) of ryanodine (10 μM). Note the increase in the duration of the high K+-evoked transient in control neurons following repeated caffeine administration. B) The change in the magnitude (ΔΔ [Ca2+]i (nM)) and decay (ΔΔ T50 (s)) of the high K+-evoked Ca2+ transient associated with the application of ryanodine (10 μM) was assessed as the difference between the high K+-evoked transients before and after the application of ryanodine. Control neurons (open bars, n = 12) treated with an identical protocol revealed a caffeine-induced increase in the T50 of decay, therefore, changes associated with ryanodine (black bars, n = 12) were assessed relative to control with a two way ANOVA. ** p < 0.01, * p < 0.05
Finally, recent evidence suggests that CICR via IP3 receptors contributes to the Ca2+ transient associated with transient receptor potential (TRP) channel activation in DRG neurons which appears to occur secondary to a Ca2+-dependent activation of phospholipase C [15, 16]. Because we have previously demonstrated that IP3 receptor activation contributes minimally to the high K+-evoked Ca2+ transient in putative nociceptive DRG neurons from naïve rats [7], we sought to rule out the possibility that recruitment of an IP3-dependent mechanism contributes to the inflammation-induced increase in the high K+-evoked Ca2+ transient. As we have previously demonstrated that 100 μM 2-APB was sufficient to attenuate an UTP-evoked Ca2+ transient in DRG neurons [7], the same concentration of this IP3 receptor blocker was used in the present experiment. We observed no significant (p > 0.05, n = 7) difference in either the magnitude (−5.74 ± 0.08 %) or duration (11.47 ± 0.11 %) of the high K+-evoked Ca2+ transient evoked in the presence or absence of 2-APB, respectively, in putative nociceptive cutaneous neurons from inflamed rats. These results argue against a role for IP3 receptor activation in the inflammation-induced changes in the high K+-evoked Ca2+ transient.
No detectable influence of inflammation on the magnitude or duration of the caffeine-evoked Ca2+ transient
Given that CICR reflects the activation of Ca2+ activated Ca2+ channels (i.e., RyRs) on the endoplasmic reticulum (ER) which enable the release of Ca2+ loaded into the ER via sarco-endoplasmic reticulum Ca2+ ATPase (SERCA), there are several mechanisms that could account for the recruitment of CICR as a contributing factor to the inflammation-induced increase in the time of decay of the high K+-evoked transient. These include: 1) an increase in releasable Ca2+ stored in the ER, 2) a shift in the expression of RyR subtypes from a receptor such as RyR1 with a low open channel probability to one such as RyR3 with a high open channel probability [17], 3) a decrease in the rate of SERCA uptake, 4) a shift in the coupling between VGCC Ca2+ influx and ER store release, and/or 5) a change in a SERCA/CICR-independent mechanism that enables the high K+-evoked transient to engage CICR to further amplify the evoked transient. To address the first possibility, we analyzed caffeine-evoked transients in putative nociceptive cutaneous neurons from naïve and inflamed rats.
Results from the first set of experiments suggested that there was no inflammation-induced change in the magnitude or decay of the caffeine-evoked transient. An additional set of neurons (n = 36 naïve, n = 29 CFA) was used to study the caffeine-evoked transients directly to avoid the potential confound associated with an initial challenge with high K+. Results with caffeine alone were consistent with our initial observations, indicating that there is no detectable influence of inflammation on either the magnitude or the decay of the caffeine-evoked transient (Figure 3A). To confirm that the caffeine-evoked transient was due to the release of Ca2+ from internal stores, caffeine was applied to neurons in the presence of Ca2+ free bath solution or following depletion of Ca2+ from the ER with the SERCA inhibitor, cyclopiazonic acid (CPA, 10 μM) (Figure 3B). In contrast to the results obtained in nodose ganglion neurons [18], there was no significant (p > 0.05) difference in the magnitude of the caffeine transient evoked in the presence or absence of extracellular Ca2+ (Figure 3B and C). Furthermore, the caffeine-evoked transient was completely blocked following depletion of ER stores with CPA (Figure 3B and C).
Figure 3.

Inflammation did not change in the magnitude or duration of the caffeine-evoked Ca2+ transient. A) The magnitude (Δ [Ca2+]i) and duration (T50) of caffeine (10 mM for 4 sec) evoked Ca2+ transients were determined in a manner identical to that used for the high K+-evoked transient. Pooled data from putative nociceptive cutaneous neurons from naïve (n = 36) and inflamed (n = 29) rats are plotted. B) Top: Example of caffeine-evoked Ca2+ transients in a putative nociceptive cutaneous neuron from a naïve rat in the presence (open square) and absence (closed square) of Ca2+ in the bath solution. Bottom: Example of caffeine-evoked Ca2+ transients in a putative nociceptive cutaneous neuron from a naïve rat before (open square) and after (closed square) the application of the SERCA inhibitor, CPA. C) Pooled data from neurons (n = 8) challenged with caffeine before and after the application Ca2+ free bath solution and from a second groups of neurons (n = 10) challenged before and after depletion of intracellular Ca2+ stores with CPA. D) Pooled concentration response data for putative cutaneous neurons from naïve (n = 13) and inflamed (n = 14) rats challenged with increasing concentrations of caffeine. Data from each neuron were fitted with a modified Hill equation to estimate the maximal size of the evoked transient (Emax) and this value was used to normalize data prior to pooling. Pooled data are also fitted with a modified Hill equation. ***p < 0.001
The concentration of caffeine used in these experiments is comparable to that used by other investigators [19–21]. However, to rule out the possibility that inflammation altered either the potency or efficacy of caffeine, concentration response data were collected from another group of neurons (n = 15 naïve, n = 14 CFA). Increasing concentrations of caffeine were applied to each neuron with an ISI of 5 minutes. Data for each neuron was fitted with a Hill equation to determine the concentration resulting in a response 50% of maximal (EC50) as well as the maximal response (Emax). Results of this analysis indicated that inflammation has no detectable influence on either the potency or efficacy of the caffeine-evoked transient, with EC50’s of 4.09 ± 0.39 and 5.29 ± 0.67 mM and Emax of 522.62 ± 58.93 and 500.84 ± 38.52 nM Ca2+ in neurons from naïve and inflamed rats, respectively (Figure 3D).
Inflammation does not affect the balance of RyR-mediated Ca2+ release and SERCA-mediated Ca2+ re-uptake
The rapid decay of the caffeine-evoked Ca2+ transient highlights the possibility that a shift in the relative balance of Ca2+ release to re-uptake contributes to the inflammation-induced increase in the high K+-evoked Ca2+ transient as it suggests that in putative nociceptive cutaneous neurons, release and re-uptake are tightly coupled. To determine whether inflammation is associated with a shift in the balance of these two processes, we assessed the extent of caffeine-induced depletion of intracellular stores in Ca2+ free bath solution. Results of this experiment indicated that even after 4 applications of 10 mM caffeine in Ca2+ free bath solution, there was no significant reduction in the caffeine-evoked transient (Figure 4A). Comparable results were obtained in neurons from naïve and inflamed rats (Figure 4B), arguing against an inflammation-induced shift in the balance of release and re-uptake, at least over a time frame relevant to the high K+-evoked Ca2+ transient.
Figure 4.
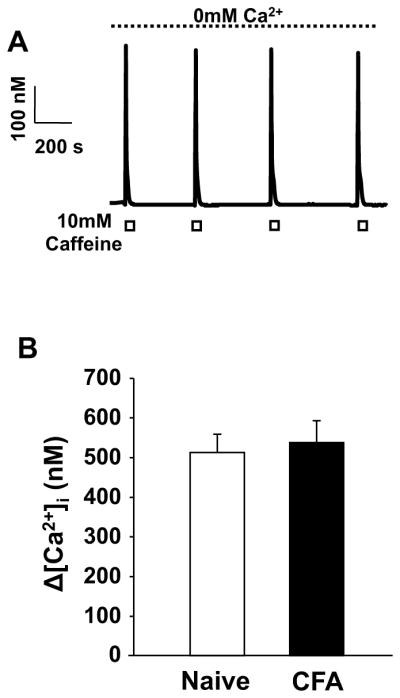
No significant reduction in the magnitude of caffeine-evoked transient in the absence of extracellular Ca2+. A) Caffeine-evoked Ca2+ transients in a putative nociceptive cutaneous neuron from a naïve rat. The neuron was continuously bathed in Ca2+ free bath solution. Caffeine was applied where indicated by the white boxes B) The magnitude of the caffeine-evoked Ca2+ transient in Ca2+ free bath solution was comparable in putative nociceptive cutaneous neurons from naïve (n = 37) and inflamed (n = 38) rats.
Heterogeneity among cutaneous neurons with respect to functional RyR subtypes does not contribute to the inflammation-induced changes in Ca2+ signaling
To further determine whether a shift in the balance of functional RyR subtypes contributes to the inflammation-induced increase in the high K+-evoked Ca2+ transient, we assessed the concentration dependence of ryanodine-induced block of the caffeine-evoked transient. Caffeine (10 mM) was applied before and then 4 times (ISI = 5 minutes) in the presence of 1, 10 or 100 μM ryanodine in Ca2+ free bath solution. Results of this experiment suggested the presence of at least 3 subpopulations of putative nociceptive cutaneous DRG neurons: those that were relatively resistant (resistant), those that were sensitive (sensitive), and those that were highly sensitive (highly sensitive) to ryanodine-induced block (Figure 5A and B). This difference between neurons was most readily apparent in the response to 10 μM ryanodine, depicted by plotting the magnitude of the transient evoked in response to the 4th application of caffeine in the presence of ryanodine normalized to the response prior to the application of ryanodine (Figure 5B); 10 μM ryanodine produced only ~20% block in resistant neurons, ~65% block in sensitive neurons and ~100% block of highly sensitive neurons. These subpopulations appeared to be less well defined in neurons from inflamed rats (Figure 5B). However there was no statistically significant difference in the average block produced by 10 μM ryanodine. Plotting the fractional block as a function of caffeine application and ryanodine concentration (Figure 5C and D), suggests the presence of an inflammation-induced increase in the sensitivity to ryanodine-induced block of the caffeine response. However, statistical analysis (mixed design 3 way ANOVA) revealed no significant interaction between inflammation and ryanodine concentration despite significant main effects associated with caffeine application and ryanodine concentration.
Figure 5.

Heterogeneity in the ryanodine receptor activity in neurons from both naïve and inflamed rats. A) Examples of caffeine-evoked Ca2+ transients in putative nociceptive cutaneous neurons before and after the application of 10 μM ryanodine in Ca2+ free bath. The neuron on the left was relatively resistant to ryanodine-induced block, while that on the right was highly sensitive. B) The percent of ryanodine-induced block of the caffeine-evoked Ca2+ transient was calculated from the response evoked by the 4th caffeine application relative to the response evoked prior to the application of ryanodine. Data from neurons from naïve (n = 15) and inflamed (n = 15) rats challenged with 10 μM ryanodine are plotted. C) Neurons (n = 31) from naïve rats were challenged with caffeine (10 mM) before and after the application of 1 of 3 concentrations of ryanodine (Ry). The response to each of 4 successive applications of caffeine was calculated as a percent of the response to caffeine prior to the application of caffeine. Pooled data over subsequent challenges are plotted relative to the concentration of ryanodine employed. D) Data from neurons (n = 33) from inflamed rats were collected and plotted as described in C. There was no significant interaction between inflammation and ryanodine concentration despite significant main effects associated with caffeine application and ryanodine concentration (Mixed 3 way ANOVA, p > 0.05).
To begin to assess the functional consequences of what appeared to be a differential distribution of RyRs among putative nociceptive cutaneous neurons, we analyzed the initial response to caffeine in subpopulations of neurons defined by their sensitivity to ryanodine-induced block. This analysis revealed that the magnitude of the Ca2+ transient in response to first caffeine application was significantly larger in the “highly sensitive” neurons (825.15 ± 113.05 nM) compared to those that were “sensitive” (501.33 ± 65.92 nM) or “resistant” (480.37 ± 67.38 nM) to the ryanodine-induced block. Nevertheless, while these results are consistent with a heterogeneous distribution of RyR subtypes among cutaneous nociceptive DRG neurons, there does not appear to be an inflammation-induced shift in this distribution.
No evidence of an inflammation-induced change in SERCA activity
While the results from the caffeine experiments suggest that the inflammation-induced increase in high K+-evoked Ca2+ transients is not due to a change in the relative level of SERCA activity, we directly tested this possibility by comparing the magnitude and decay of the high K+-evoked Ca2+ transient before and after the application 10 μM CPA in normal Ca2+-containing bath solution. To confirm complete block of SERCA, caffeine (10 mM) was applied after resting [Ca2+]i levels returned to baseline following CPA application (Figure 6A). Consistent with the suggestion that Ca2+ released from the ER does not engage the mechanisms underlying the inflammation-induced increase in the magnitude of the high K+-evoked Ca2+ transient, the CPA-induced Ca2+ transient was comparable in neurons from naïve and inflamed rats (p > 0.05, Figure 6B). There was no significant change in the magnitude of the high K+-evoked Ca2+ transient in neurons from either naïve or inflamed animals in the presence of CPA (Figure 6C). However, in both groups of neurons, the T50 of decay was significantly (p < 0.01) increased in the presence of CPA. Furthermore, the CPA-induced increase in T50 was significantly greater in neurons from inflamed rats than in neurons from naïve (Figure 6D). These results are consistent with our previous finding that in this subpopulation of neurons, SERCA plays a far more dominant role in the regulation of the duration of the Ca2+ transient than in the magnitude. They also argue against a role for an inflammation-induced change in SERCA activity as an underlying mechanism of the inflammation-induced increase in the high K+-evoked Ca2+ transient because an inflammation-induced decrease in SERCA activity would have attenuated the CPA-induced increase in T50 in neurons from inflamed rats.
Figure 6.

No inflammation-induced change in the influence of SERCA on the high K+-evoked transient. A) Ca2+ transients in neurons from a naïve (grey trace) and inflamed (black trace) rat evoked with high K+ before and after the application of CPA. Both neurons were challenged with caffeine to confirm depletion of Ca2+ from intracellular stores. B) The magnitude of the CPA-evoked Ca2+ transient was comparable in neurons from naïve (n = 11) and inflamed (n = 9) rats. C) CPA had no influence on the magnitude of the high K+-evoked Ca2+ transient, despite a significant main effect associated with inflammation (two-way ANOVA). Pooled data are responses obtained before (Veh) and after application of CPA. D) The decay of the high K+-evoked Ca2+ transient was calculated, pooled and plotted as in C. There were significant main effects associated with CPA and inflammation but no statistically significant interaction (two-way ANOVA). * p < 0.05; ** p < 0.01.
Selective influence of inflammation on the decay of the Ca2+ transient evoked with prolonged caffeine application
Having largely ruled out a change in the machinery underlying CICR as a mechanism contributing to the inflammation-induced increase in the high K+-evoked Ca2+ transient, we performed an experiment to begin to assess the presence of an inflammation-induced shift in the coupling between Ca2+ influx and release. The possibility of such a shift in coupling was suggested by the observation that the decay of the caffeine-evoked Ca2+ transient (average T50 = 9 sec) is much faster than that of the high K+-evoked Ca2+ transient (average T50 = 45 sec) despite the fact that the magnitude of the transient evoked with these two stimuli were comparable (p > 0.05). This observation suggests that very different Ca2+ regulatory mechanisms are engaged by Ca2+ influx and Ca2+ release in this subpopulation of sensory neurons. It also raises the possibility that increasing the duration of the caffeine application would prolong the Ca2+ transient sufficiently to enable Ca2+ access to the mechanisms altered by inflammation that contribute to the inflammation-induced increase in the high K+-evoked transient, where negative results would be consistent with a change in the association between influx and release. To test this possibility, caffeine was applied for 12 seconds to another group of neurons (n = 8 naïve, n = 10 CFA) (Figure 7A). Consistent with preliminary results indicating that the magnitude of the caffeine-evoked Ca2+ transient was saturated in response to caffeine applications of 4s or longer, there was not difference between a 4s and 12s application of caffeine with respect to the magnitude of the evoked transient in neurons from naïve and inflamed rats (Figure 7B). Interestingly, however, in contrast to the results obtained in neurons from naïve rats, where the T50 of decay to a 4 and 12 second application of caffeine were comparable (p > 0.05, Figure 7C), inflammation was associated with a significant (p < 0.05) increase in the T50 of decay in response to a 12 second caffeine application (Figure 7C).
Figure 7.
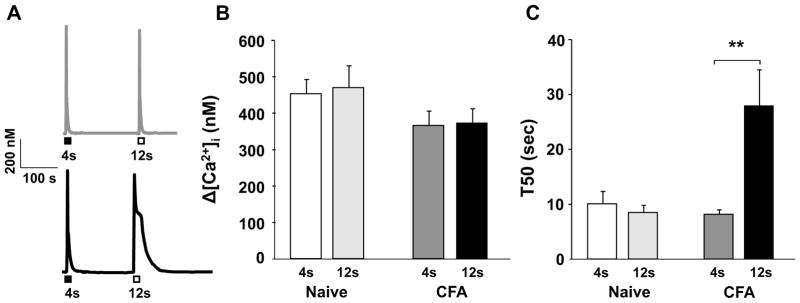
Ca2+ transients evoked in a neuron from naïve (grey trace) and inflamed (black trace) rats with a 4 second (closed box) or 12 second (open box) application of caffeine (10 mM, ISI = 5 min). Pooled magnitude (B) and duration (C) data from neurons from naïve (n = 8) and inflamed (n = 10) rats stimulated with caffeine as described in A are plotted. **p < 0.01
Different Ca2+ regulatory machinery are engaged with Ca2+ transients evoked with caffeine and high K+
The rapid decay of the caffeine-evoked Ca2+ transient not only suggests that in putative nociceptive cutaneous neurons ER Ca2+ release and re-uptake are tightly coupled, but that different Ca2+ regulatory machinery is engaged by Ca2+ influx via VGCC and release from the ER. The observation that repeated caffeine application results in an increase in the duration of the high K+-evoked transient (Figure 2), but no change in the duration of the caffeine-evoked transient (with a T50 of decay of 6.2 ± 0.9s after the first application, and 7.0 ± 1.2s after the fourth application, p > 0.05, n =10) is also consistent with the suggestion that the Ca2+ transients evoked by these stimuli engage distinct Ca2+ regulatory machinery. To further explore these suggestions, we performed three additional experiments: First, caffeine or high K+ were applied in the presence of 10μM CCCP, a mitochondrial proton pump inhibitor; Second, caffeine was applied in the presence of SERCA inhibitor CPA; and third caffeine or high K+ were applied in the presence of Na+ free bath to inhibit the Na+/Ca2+ exchanger (NCX).
For the first experiment, CCCP was co-applied with caffeine or high K+ to minimize the potential impact of a decrease in ATP on Ca2+ regulatory machinery. However, because CCCP was associated with a transient increase in intracellular Ca2+ which influenced the magnitude of the evoked transient relative to baseline, CCCP was first applied alone and the magnitude of the CCCP-evoked transient was subtracted from the caffeine or high K+-evoked transient prior to calculating the percent change from the transient evoked in the absence of CCCP. Results of this first experiment indicated that, while the complex decay of the high K+-evoked Ca2+ transient is due in part to mitochondrial buffering, mitochondria do not appear to influence either the magnitude or decay of the caffeine evoked Ca2+ transient. That is, both the magnitude (32.85 ± 0.8%, p < 0.05, n = 7) and decay (p < 0.01, Table 1) of the high K+-evoked Ca2+ transient were significantly increased in the presence of CCCP. Furthermore, not only was the decay of the high K+-evoked transient significantly increased, most dramatically during the initial phase of decay (i.e., at T25, Table 1), but the shape of the transient was dramatically altered, with no evidence of a “shoulder” just above the T50 of decay (Figure 8A). In contrast, changes in neither the magnitude (−8.2± 0.04%) nor duration (25.13 ± 0.02%) of the caffeine-evoked transient in the presence of CCCP, were significant (p > 0.05, n = 6, Figure 8A).
Figure 8.

High K+- and caffeine-evoked Ca2+ transients engage distinct Ca2+ regulatory machinery. Putative nociceptive cutaneous DRG neurons from naive rats were challenged with either high K+ (30 mM, 4s, Left Traces) or caffeine (10 mM, 4s, Right Traces), before and after the application of CCCP (10 μM, A), CPA (10 μM, B), or Na+ Free bath (C). Pooled magnitude (peak change in concentration of intracellular Ca2+ ([Ca2+]i) from baseline) and duration (time of decay) data obtained before and after the application of CCCP, CPA and Na+ Free bath are plotted. For the high K+ experiments, the number of neurons studied were 7, 11, and 12, with CCCP, CPA and Na+ free bath, respectively, while for the caffeine experiments, the number of neurons studied were 6, 6 and 6 for CCP, CPA and Na+ free bath, respectively. Data were analyzed with a mixed design two-way ANOVA: differences between before and after the application of CCCP, CPA or Na+ Free bath, as well as between high K+ and caffeine are indicated. * is p < 0.05, and ** is p < 0.01.
For the second experiment, caffeine was also co-applied with CPA to prevent depletion of the ER that occurs with the pre-treatment protocol used for the high K+ experiment (Figure 6). Interestingly, the impact of CPA on the caffeine-evoked Ca2+ transient was similar to that on the high K+-evoked Ca2+ transient (Figure 6), where there was no influence on the magnitude (−4.59 ± 0.09% of control, p > 0.05, n = 6) but a significant increase in the duration (210.42 ± 0.57% of control, p < 0.05) of the caffeine-evoked Ca2+ transient (Figure 8B, Table 1). These results suggest that the magnitude of the caffeine-evoked Ca2+ transient, at least in response to a saturating concentration and duration of caffeine, is largely determined by the concentration of Ca2+ in the ER and the density and distribution of RyRs, while the duration of the transient is largely determined by SERCA.
Finally, the reproducible response to caffeine even in a Ca2+ free bath (Figure 4) suggests that extrusion mechanisms contribute minimally to both the magnitude and duration of the caffeine-evoked transient. In contrast, we have previously demonstrated the Ca2+ extrusion via NCX influences the duration of the high K+-evoked Ca2+ transient in putative nociceptive DRG neurons [7]. Therefore, to confirm that extrusion, at least via NCX, contributes minimally to the regulation of the caffeine-evoked transient in cutaneous nociceptive neurons, we assessed the response to caffeine before and after block of NCX with zero Na+ bath (where NaCl was replaced with choline-Cl). The results of this experiment indicated that block of NCX had no significant (p > 0.05, n = 6) influence on either the magnitude (−6.07 ± 0.04% of control) or duration (−6.4 ± 0.06% of control) of the caffeine-evoked Ca2+ transient (Figure 8C, Table 1). In contrast, and consistent with our previous results in unlabeled DRG neurons, inhibition of NCX had no significant influence on the magnitude of the high K+-evoked transient in cutaneous nociceptive neurons, but was associated with a significant (p < 0.01, n = 12) increase in the duration (490.54 ± 0.89%) of the high K+-evoked transient (Figure 8C).
Discussion
In the present set of experiments, we sought to determine whether a change in mechanisms underlying CICR and/or the association between influx and release of Ca2+ could contribute to the inflammation-induced increase in the high K+-evoked Ca2+ transient. Towards that end, we were able to reproduce our previous results indicating inflammation is associated with an increase in both the magnitude and duration of the high K+-evoked Ca2+ transient. While there was no evidence for an inflammation-induced recruitment of IP3 receptor mediated release, the inflammation-induced increase in duration, but not magnitude of the high K+-evoked Ca2+ transient was at least partially blocked by the RyR blocker, ryanodine. Interestingly, however, there was no influence of inflammation on either the magnitude or duration of transients evoked with a brief (4 second) application of caffeine, or the potency or efficacy of the caffeine-induced release of Ca2+ from the ER. There was also no influence of inflammation on the response to repeated caffeine application in Ca2+ free bath solution. Furthermore, there was no detectable influence of inflammation on the potency or efficacy of the ryanodine-induced block of the caffeine-evoked Ca2+ transients. There was also no evidence that inflammation was associated with a decrease in SERCA activity. Finally, inflammation was associated with a selective increase in the duration of the Ca2+ transient in response to prolonged (12 second) caffeine application.
These observations have several interesting implications. Most relevant to the purpose of the present study, these data suggest that mechanism(s) other than a change in CICR or the coupling between Ca2+ influx and CICR underlie the inflammation-induced changes in the high K+-evoked Ca2+ transient. This was most readily demonstrated by a lack of evidence for the involvement of CICR in the regulation of the magnitude of the high K+-evoked Ca2+ transient, in the face of a clear inflammation-induced increase in this parameter. Furthermore, evidence that mechanisms underlying CICR are comparable in neurons from naïve and inflamed animals leaves only a change in coupling between influx and release as a possible mechanism contributing to the inflammation-induced increase in the duration of the high K+-evoked Ca2+ transient. However, while we have not conclusively ruled out a shift in the coupling, which enabled the Ca2+ influx via VGCC to engage CICR, the results from the prolonged caffeine application experiment demonstrate that an inflammation-induced change in another Ca2+ regulatory mechanism can now be engaged via Ca2+ release from the ER. Given our present results with a zero Na+ bath, our previous results [7] as well as those of others [22, 23] suggesting that the plasma membrane Ca2+-ATPase (PMCA) and NCX play a greater role in regulating the duration rather than the magnitude of the depolarization-induced Ca2+ transient, an inflammation-induced decrease in the rate of Ca2+ extrusion could account for increased duration of the Ca2+ transient evoked by both depolarization and prolonged release from the ER.
There are distinct Ca2+ regulatory machinery engaged by Ca2+ entering the cytosol via voltage-gated Ca2+ channels versus release from the ER, as suggested by several lines of evidence. These include 1) differences in kinetics of the high K+ and caffeine-evoked Ca2+ transients despite the comparable magnitude of the transients evoked with these two stimuli, 2) the impact of inflammation on the duration (and magnitude) of the high K+ but not caffeine-evoked transient, 3) the impact of an increase in PKA activity on the duration of the high K+ but not the caffeine-evoked transient, 4) the contribution of mitochondria to the regulation of both the magnitude and decay of the high K+-evoked transient, but neither the magnitude nor decay of the caffeine-evoked transient, and 5) the impact of NCX block on the duration of the high K+ but not the caffeine-evoked transient. Of note, while the absence of an inflexion on the falling phase of the caffeine-evoked transient, a signature of mitochondria-mediated buffering [24], in combination with the absence of a detectable influence of CCCP on the caffeine-evoked transient in putative nociceptive cutaneous DRG neurons argue strongly against a direct role for mitochondria in the regulation of the caffeine-evoked transient, these observations are in contrast to previous results from unlabeled sensory neurons indicating mitochondria are involved in buffering the Ca2+ released from the ER [25]. Nevertheless, we are not the first to report that mitochondria are differentially engaged by Ca2+ influx and release, as comparable results have been previously reported by others in mouse DRG neurons [26]. We suggest that heterogeneity among DRG neurons is the most likely explanation for the apparent difference between out results and those in these previous studies.
The term microdomain is generally applied to spatially isolated elementary events or larger Ca2+ transients that results from the combination of smaller events [27]. While we have not provided direct evidence for the spatial segregation of the transients associated with Ca2+ release and influx in putative nociceptive cutaneous neurons, we suggest that the functional segregation of the regulatory machinery engaged by these two sources of cytoplasmic Ca2+ imply the presence of microdomains. Most convincing was the observation that caffeine-evoked transients were stable in the presence of Ca2+ free bath solution and unaffected by NCX block suggesting that the majority of Ca2+ released during caffeine application is pumped back into the ER by SERCA. The result appears to be a tightly regulated microdomain through which the magnitude and decay of the caffeine-evoked Ca2+ transient are determined by the amount of Ca2+ in the ER, the density and distribution of ryanodine receptors, and SERCA activity. Strikingly, even a 12 second application of caffeine to this subpopulation of neurons from naïve rats appeared to be insufficient to engage mitochondrial Ca2+ buffering. An additional interesting note is that while Ca2+ influx via VGCC was insufficient to activate RyRs in neurons from naïve rats, SERCA still contributes to the regulation of the duration of the high K+-evoked Ca2+ transient as illustrated by the results obtained with CPA. This observation suggests that it is SERCA that largely defines the barrier separating Ca2+ influx from release. Given evidence for the dynamic regulation of SERCA activity in DRG neurons [28], a SERCA-mediated barrier would provide a sensitive mechanism for the modulation of the coupling between Ca2+ influx and release in these neurons.
The observation that the inflammation-induced changes in high K+-evoked transient are manifest despite a decrease in VGCC current density [10] argues that regulation of the magnitude and duration of the Ca2+ transient is largely independent on the magnitude of the initial Ca2+ influx. Furthermore, the observation that inflammation is associated with an increase in both the magnitude and duration of the high K+-evoked Ca2+ transient, in the face of evidence that there are mechanisms such as SERCA and NCX that influence the duration but not the magnitude of the high K+-evoked Ca2+ transient raises the possibility that there are two Ca2+ regulatory processes that are altered in the presence of inflammation. Interestingly, despite recent evidence suggesting that Ca2+ influx via TRP channels may drive CICR secondary to the activation of IP3 receptor [15], such a mechanism does not appear to contribute to the high K+-evoked Ca2+ transient. While this may be a unique feature of the subpopulation of neurons studied here, this difference raises the possibility that Ca2+ influx via TRP channels engages Ca2+ regulatory processes distinct from those engaged by caffeine or high K+. While future experiments would be needed to address this possibility, the further definition of the microdomain engaged following Ca2+ influx through VGCC serves to limit the number of mechanisms that may ultimately be responsible for the inflammation-induced changes in the high K+-evoked Ca2+ transient.
While not the focus of the present study, results obtained with CCCP suggest mitochondria are not a potential mechanisms contributing to the inflammation-induced change in the high K+-evoked Ca2+ transient. That is, because CCCP results in a significant increase in the duration of the high K+-evoked Ca2+ transient in cutaneous neurons from naïve rats, albeit with altered decay kinetics, one would predict that if a decrease in mitochondrial buffering of the high K+-evoked transient contributed to the changes observed in the presence of inflammation, the impact of CCCP on the high K+-evoked Ca2+ transient in neurons from inflamed rats should be attenuated. Preliminary results with a pre-application protocol similar to that employed previously [7] suggest that this is not the case, as CCCP (10 μM) was associated with an increase in the duration of the high K+-evoked transient (T50 = 113.2 ± 14.2 s, n = 9) that was, if anything even larger than that observed in neurons from naïve rats (T50 = 72.9 ± 11 s, n = 11). However, given the complex way in which mitochondria contribute to the regulation of [Ca2+]i, this issue may need to be addressed more systematically.
The suggestion that distinct mechanisms underlie the inflammation-induced increase in the magnitude and duration of the high K+-evoked Ca2+ transient raises the possibility that these mechanisms are separable within the neuron. The functional implications of this possibility will depend on where the changes are manifest. For example, activation of a sustained increase in [Ca2+]i in central or peripheral afferent terminals would facilitate transmitter release, thus augmenting neurogenic inflammation in the periphery or increasing the transmission of nociceptive information at the central terminal. A comparable increase in the duration of the transient at a spike initiation zone or along an axon where Ca2+-dependent K+ channels may be localized would influence spike pattern or the relative refractory period. In the cell body, the dynamics of the Ca2+ transient may lead to very different effects on gene expression [4]. Conversely, an increase in the magnitude of the Ca2+ transient independent of a change in duration may lead to differential activation of regulatory proteins or second messenger pathways based on Ca2+ affinity, alternative gene transcription and translation, and/or excitotoxicity [4, 24, 29].
In contrast to previous results indicating that 10 μM ryanodine is sufficient to completely block caffeine-evoked Ca2+ transients in sensory neurons [11, 30, 31], we only observed a ~60% block of the caffeine-evoked transient at this concentration in cutaneous DRG neurons. We have previously observed that differences in how the neurons were processed and the amount of time in culture prior to study are factors that to influence the properties of Ca2+ transients in DRG neurons (data not shown). Nevertheless, we suggest that the most likely explanation for the difference between our results and previous studies is due to heterogeneity among DRG neurons. We have previously reported that there are significant differences between subpopulations of DRG neurons with respect to the relative impact of CICR to the high K+-evoked Ca2+ transient [7]. Furthermore, the results of the present study indicate that even within a small subpopulation of cutaneous DRG neurons, there is heterogeneity with respect to the impact of ryanodine on the caffeine-evoked Ca2+ transient. Indeed, consistent with these previous results, we observed some neurons in which 10 μM ryanodine was sufficient to completely block the caffeine-evoked transient, arguing against a problem with how the compound was prepared or the neurons were challenged. However, this was clearly not the case in all neurons. We also suggest that a differential expression of RyR subtypes within the subpopulation of interest is likely to account for this heterogeneity in the actions of ryanodine. That is, while RyR 3 is similar to RyR 2 in its threshold of Ca2+-induced activation (1μM), the two receptors differ in open channel probability (Po) in response to comparable levels of Ca2+: RyR 3 has a Po of ~1 while that of RyR 2 is ~0.6 and RyR 1 is ~0.15 at ~100μM Ca2+. Furthermore, RyR 3 is able to maintain its open state and requires higher Ca2+ concentrations for inactivation compared to RyR1 or RyR 2 [32–34]. Ryanodine is an open channel blocker, therefore, the difference in Po would suggest that RyR 1 is the dominant receptor in neurons “resistant” to ryanodine and RyR 3 is the dominant receptor in neurons “highly sensitive” to ryanodine. This would also account for the larger caffeine-evoked transient in “highly sensitive” neurons. Importantly, a differential distribution of RyR subtypes both within and between neurons would result in very different responses to stimuli capable of engaging CICR.
In summary, while our results argue against a change in CICR or the coupling between CICR and Ca2+ influx via VGCC as the mechanism underlying the inflammation-induced increase in the high K+-evoked Ca2+ transient, we further characterized the regulation of [Ca2+]i in putative nociceptive cutaneous neurons, revealing several aspects of the Ca2+ regulation that may have a profound impact on the function of this subpopulation of neurons. This includes evidence that mechanisms underlying the inflammation-induced increase in magnitude and duration of the high K+-evoked transient are distinct, that SERCA serves as a barrier isolating Ca2+ regulatory domains accessed by release from the ER and influx via VGCC, and that there is a differential distribution of RyR subtypes among subpopulations of these neurons. Future experiments will be needed to assess the functional consequences of these unique features.
Acknowledgments
This work was supported by Grants from the National Institutes of Health (NS04499 and DE018252), as well as intramural support from the Department of Anesthesiology at the University of Pittsburgh. The authors would like to thank Drs. William de Groat, Gerald Gebhart, Steve Meriney, Derek Molliver for their constructive feedback during the preparation of this manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lu SG, Gold MS. Inflammation-induced increase in evoked calcium transients in subpopulations of rat dorsal root ganglion neurons. Neuroscience. 2008;153:279–288. doi: 10.1016/j.neuroscience.2008.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Richardson JD, Vasko MR. Cellular mechanisms of neurogenic inflammation. J Pharmacol Exp Ther. 2002;302:839–845. doi: 10.1124/jpet.102.032797. [DOI] [PubMed] [Google Scholar]
- 3.Zhang XL, Mok LP, Lee KY, Charbonnet M, Gold MS. Inflammation-induced changes in BKCa currents in cutaneous dorsal root ganglion neurons from the adult rat. Mol Pain. 2012;8:37. doi: 10.1186/1744-8069-8-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fields RD, Lee PR, Cohen JE. Temporal integration of intracellular Ca2+ signaling networks in regulating gene expression by action potentials. Cell Calcium. 2005;37:433–442. doi: 10.1016/j.ceca.2005.01.011. [DOI] [PubMed] [Google Scholar]
- 5.Gemes G, Bangaru ML, Wu HE, Tang Q, Weihrauch D, Koopmeiners AS, Cruikshank JM, Kwok WM, Hogan QH. Store-operated Ca2+ entry in sensory neurons: functional role and the effect of painful nerve injury. J Neurosci. 2011;31:3536–3549. doi: 10.1523/JNEUROSCI.5053-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jackson JG, Thayer SA. Mitochondrial modulation of Ca2+-induced Ca2+-release in rat sensory neurons. J Neurophysiol. 2006;96:1093–1104. doi: 10.1152/jn.00283.2006. [DOI] [PubMed] [Google Scholar]
- 7.Lu SG, Zhang X, Gold MS. Intracellular calcium regulation among subpopulations of rat dorsal root ganglion neurons. J Physiol. 2006;577:169–190. doi: 10.1113/jphysiol.2006.116418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Usachev YM, Thayer SA. All-or-none Ca2+ release from intracellular stores triggered by Ca2+ influx through voltage-gated Ca2+ channels in rat sensory neurons. J Neurosci. 1997;17:7404–7414. doi: 10.1523/JNEUROSCI.17-19-07404.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Usachev YM, Thayer SA. Ca2+ influx in resting rat sensory neurones that regulates and is regulated by ryanodine-sensitive Ca2+ stores. J Physiol. 1999;519(Pt 1):115–130. doi: 10.1111/j.1469-7793.1999.0115o.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lu SG, Zhang XL, Luo ZD, Gold MS. Persistent inflammation alters the density and distribution of voltage-activated calcium channels in subpopulations of rat cutaneous DRG neurons. Pain. 2010;151:633–643. doi: 10.1016/j.pain.2010.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Usachev Y, Shmigol A, Pronchuk N, Kostyuk P, Verkhratsky A. Caffeine-induced calcium release from internal stores in cultured rat sensory neurons. Neuroscience. 1993;57:845–859. doi: 10.1016/0306-4522(93)90029-f. [DOI] [PubMed] [Google Scholar]
- 12.Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- 13.Kao CH, Wang SJ, Chen CY, Yeh SH. Detection of esophageal carcinoma using Tc-99m MIBI SPECT imaging. Clin Nucl Med. 1994;19:1069–1074. doi: 10.1097/00003072-199419120-00007. [DOI] [PubMed] [Google Scholar]
- 14.Leijten PA, van Breemen C. The effects of caffeine on the noradrenaline-sensitive calcium store in rabbit aorta. J Physiol. 1984;357:327–339. doi: 10.1113/jphysiol.1984.sp015502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rohacs T, Thyagarajan B, Lukacs V. Phospholipase C mediated modulation of TRPV1 channels. Mol Neurobiol. 2008;37:153–163. doi: 10.1007/s12035-008-8027-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rohacs T. Teaching resources. TRP channels. Sci STKE. 2005;2005:tr14. doi: 10.1126/stke.2822005tr14. [DOI] [PubMed] [Google Scholar]
- 17.Meissner G. Ryanodine receptor/Ca2+ release channels and their regulation by endogenous effectors. Annu Rev Physiol. 1994;56:485–508. doi: 10.1146/annurev.ph.56.030194.002413. [DOI] [PubMed] [Google Scholar]
- 18.Hoesch RE, Weinreich D, Kao JP. A novel Ca(2+) influx pathway in mammalian primary sensory neurons is activated by caffeine. J Neurophysiol. 2001;86:190–196. doi: 10.1152/jn.2001.86.1.190. [DOI] [PubMed] [Google Scholar]
- 19.Cheng LZ, Lu N, Zhang YQ, Zhao ZQ. Ryanodine receptors contribute to the induction of nociceptive input-evoked long-term potentiation in the rat spinal cord slice. Mol Pain. 2010;6:1. doi: 10.1186/1744-8069-6-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eun SY, Jung SJ, Park YK, Kwak J, Kim SJ, Kim J. Effects of capsaicin on Ca(2+) release from the intracellular Ca(2+) stores in the dorsal root ganglion cells of adult rats. Biochem Biophys Res Commun. 2001;285:1114–1120. doi: 10.1006/bbrc.2001.5272. [DOI] [PubMed] [Google Scholar]
- 21.Lokuta AJ, Komai H, McDowell TS, Valdivia HH. Functional properties of ryanodine receptors from rat dorsal root ganglia. FEBS Lett. 2002;511:90–96. doi: 10.1016/s0014-5793(01)03312-9. [DOI] [PubMed] [Google Scholar]
- 22.Pottorf WJ, Thayer SA. Transient rise in intracellular calcium produces a long-lasting increase in plasma membrane calcium pump activity in rat sensory neurons. J Neurochem. 2002;83:1002–1008. doi: 10.1046/j.1471-4159.2002.01221.x. [DOI] [PubMed] [Google Scholar]
- 23.Werth JL, Usachev YM, Thayer SA. Modulation of calcium efflux from cultured rat dorsal root ganglion neurons. J Neurosci. 1996;16:1008–1015. doi: 10.1523/JNEUROSCI.16-03-01008.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Berridge MJ, Bootman MD, Roderick HL. Calcium signalling: dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol. 2003;4:517–529. doi: 10.1038/nrm1155. [DOI] [PubMed] [Google Scholar]
- 25.Friel DD, Tsien RW. An FCCP-sensitive Ca2+ store in bullfrog sympathetic neurons and its participation in stimulus-evoked changes in [Ca2+]i. J Neurosci. 1994;14:4007–4024. doi: 10.1523/JNEUROSCI.14-07-04007.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Svichar N, Kostyuk P, Verkhratsky A. Mitochondria buffer Ca2+ entry but not intracellular Ca2+ release in mouse DRG neurones. Neuroreport. 1997;8:3929–3932. doi: 10.1097/00001756-199712220-00017. [DOI] [PubMed] [Google Scholar]
- 27.Berridge MJ. Calcium microdomains: organization and function. Cell Calcium. 2006;40:405–412. doi: 10.1016/j.ceca.2006.09.002. [DOI] [PubMed] [Google Scholar]
- 28.Usachev YM, Marsh AJ, Johanns TM, Lemke MM, Thayer SA. Activation of protein kinase C in sensory neurons accelerates Ca2+ uptake into the endoplasmic reticulum. J Neurosci. 2006;26:311–318. doi: 10.1523/JNEUROSCI.2920-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Carafoli E, Santella L, Branca D, Brini M. Generation, control, and processing of cellular calcium signals. Crit Rev Biochem Mol Biol. 2001;36:107–260. doi: 10.1080/20014091074183. [DOI] [PubMed] [Google Scholar]
- 30.Shmigol A, Verkhratsky A, Isenberg G. Calcium-induced calcium release in rat sensory neurons. J Physiol. 1995;489(Pt 3):627–636. doi: 10.1113/jphysiol.1995.sp021078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Solovyova N, Fernyhough P, Glazner G, Verkhratsky A. Xestospongin C empties the ER calcium store but does not inhibit InsP3-induced Ca2+ release in cultured dorsal root ganglia neurones. Cell Calcium. 2002;32:49–52. doi: 10.1016/s0143-4160(02)00094-5. [DOI] [PubMed] [Google Scholar]
- 32.Chen SR, Li X, Ebisawa K, Zhang L. Functional characterization of the recombinant type 3 Ca2+ release channel (ryanodine receptor) expressed in HEK293 cells. J Biol Chem. 1997;272:24234–24246. doi: 10.1074/jbc.272.39.24234. [DOI] [PubMed] [Google Scholar]
- 33.Jeyakumar LH, Copello JA, O’Malley AM, Wu GM, Grassucci R, Wagenknecht T, Fleischer S. Purification and characterization of ryanodine receptor 3 from mammalian tissue. J Biol Chem. 1998;273:16011–16020. doi: 10.1074/jbc.273.26.16011. [DOI] [PubMed] [Google Scholar]
- 34.Meissner G. Regulation of mammalian ryanodine receptors. Front Biosci. 2002;7:d2072–2080. doi: 10.2741/A899. [DOI] [PubMed] [Google Scholar]