

Abstract
Nutritional epidemiology aims to identify dietary and lifestyle causes for human diseases. Causality inference in nutritional epidemiology is largely based on evidence from studies of observational design, and may be distorted by unmeasured or residual confounding and reverse causation. Mendelian randomization is a recently developed methodology that combines genetic and classical epidemiological analysis to infer causality for environmental exposures, based on the principle of Mendel’s law of independent assortment. Mendelian randomization uses genetic variants as proxiesforenvironmentalexposuresofinterest.AssociationsderivedfromMendelian randomization analysis are less likely to be affected by confounding and reverse causation. During the past 5 years, a body of studies examined the causal effects of diet/lifestyle factors and biomarkers on a variety of diseases. The Mendelian randomization approach also holds considerable promise in the study of intrauterine influences on offspring health outcomes. However, the application of Mendelian randomization in nutritional epidemiology has some limitations.
Keywords: Mendelian randomization, nutritional epidemiology, review
INTRODUCTION
The basic aim of nutritional epidemiology is to elucidate the roles of dietary and lifestyle factors in determining the risk of human diseases.1 Over the past couple of decades, nutritional epidemiology has had notable success in identifying many modifiable exposures that increase or decrease disease risk.2–4 The vast majority of nutritional epidemiological studies have been based on observational design. Even though randomized controlled trials (RCTs) can provide more rigorous evidence for causality, long-term RCTs with disease outcomes are not feasible due to ethical issues and lack of compliance. The interpretation of positive associations in observational studies, however, is not straightforward. This is especially true for dietary and lifestyle factors that occur in clusters or are highly correlated, e.g., people with healthy diets often have other healthy habits.5 Although several analytic strategies can control for confounding,6 unmeasured and residual confounding often remain a concern. Another issue of concern is reverse causality in which preclinical or disease status influences dietary exposure rather than vice versa. This is particularly a problem in studies obtaining exposure information retrospectively. Evidence from genetic studies can be used to control for these biases.
Mendelian randomization is a recent development in genetic epidemiology and is based on the random assortment of alleles/genotypes transferred from parent to offspring at the time of gamete formation.7,8 The independent distribution of alleles/genotypes means that a study relating health outcome to genetic variation would not be affected by confounders that often distort the interpretation of findings. In addition, because the random assignment to genotype takes place at conception, the association between a genetic variant and a disease is free of reverse causation. Thus, a comparison of groups of individuals defined by a genetic variant is analogous to an RCT (Figure 1). In addition, Mendelian randomization studies may have particular relevance in assessing the effects of long-term (lifetime) exposures, such as dietary intake, lifestyle, and excessive adiposity, whereas RCTs can examine only short-term effects. Interest in using Mendelian randomization in causality inference has been growing rapidly over the past 5 years. This article reviews the history of the concept of Mendelian randomization and analytic approaches, particularly discusses the application of this method in nutritional epidemiological studies, and summarizes the potential limitations and challenges.
Figure 1. Parallel relationship between Mendelian randomization and randomized controlled trial (RCT).
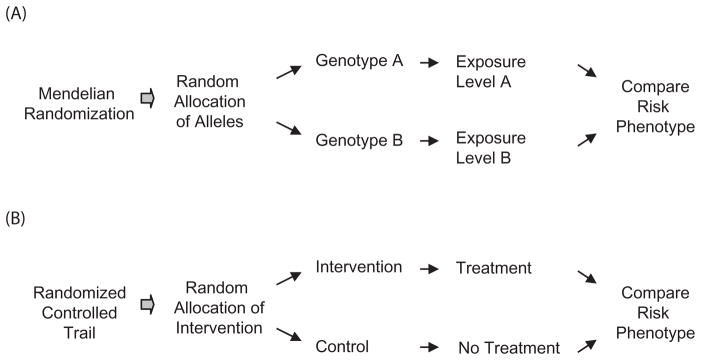
Genotypes A and B refer to the genotypes of selected polymorphisms with established relation with the exposure of interest.
MENDELIAN RANDOMIZATION, THE CONCEPT AND ANALYSIS
The principle underlying Mendelian randomization originates from the “The Law of Independent Assortment” also known as Mendel’s second law,9 which states that inheritance of one trait is independent of other traits. The idea of using Mendelian randomization in controlling biased associations was first put forth by Katan,10 who suggested that the evidence for APOE (apolipoprotein E gene) genetic variants and cholesterol association might help explain the causal effects of very low cholesterol on cancer. APOE has a key role in the clearance of cholesterol from plasma11 and APOE genotypes (E-2, E3, and E4) are related to different average levels of serum cholesterol. If low circulating cholesterol levels were causally related to cancer, individuals with the genotype associated with lower cholesterol would have higher cancer risk. Otherwise, there would be a null association between APOE genotype and cancer. Several excellent review articles have outlined the potential application of Mendelian randomization in studying the causality for environmental exposures.7,12,13 Briefly, a Mendelian randomization study uses genetic variants as robust proxies for modifiable exposures to make causal inferences about the outcomes of the modifiable exposures.14
Mendelian randomization is particularly intended to address two major biases affecting observational associations: confounding and reverse causation. As mentioned above, genetic variants should not be related to the confounding factors that are inherited independent of the disease outcome of interest, and the onset of disease will not change the genotype. Therefore, the genotype–disease relation is essentially free of confounding and reverse causation. A genetic association study mimics an RCT (Figure 1).7,13 A standard Mendelian randomization analysis requires four different relations to be estimated: 1) exposure of interest (e.g. diet, lifestyle, obesity, and biomarker) and outcome (continuous phenotype or disease risk); 2) a selected genotype and exposure; 3) the selected genotype and outcome; and 4) the predicted relationship between genotype and outcome (derived from the first two estimates), which is then compared with the third estimate (Figure 2). In practice, the association data among genotype, exposure, and outcome can be obtained from the same study sample15–17 or from different study samples.18,19 If the exposure–outcome estimate is biased by confounding or reverse causation, the predicted genotype–outcome association will be different from the observed association. In contrast, if the exposure–outcome estimate is true (causal effect), the observed and predicted genotype–outcome associations will be similar.
Figure 2. Estimates of relations among selected genotype, exposure, and outcome in a standard Mendelian randomization study.
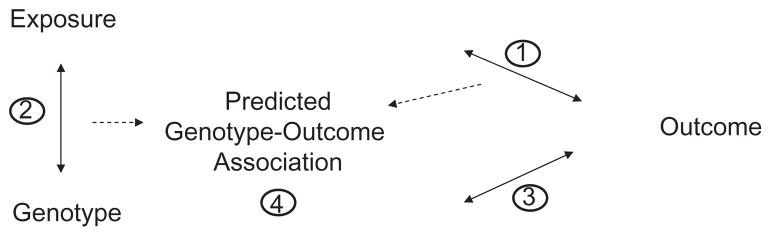
Relations include the associations between (1) exposure and outcome; (2) genotype and exposure; (3) genotype and outcome; and (4) the predicted genotype–outcome association based on estimates (1) and (2). The predicted estimate (4) is then compared with observed estimate (3).
Recently, the instrumental variable (IV) method was introduced as a formal test of Mendelian randomization using the genetic markers as IVs.8,14 This method calculates the predicted effect of exposure on outcome, given the associations between genotype and exposure and between genotype and outcome. The IV approach is widely used in econometrics to deal with “endogeneity,” a broad term covering confounding, reverse causality, and regression dilution bias. An IV should be: 1) not correlated with the confounders; 2) related to the exposure of interest; and 3) related to the outcome only through the exposure of interest.20 In the context of Mendelian randomization analysis, the genetic variant, which is not associated with confounding variables, can act as an IV for the environment exposure. The two-stage least squares (2SLS) method is most widely used to fit the IV models.14 In brief, in the first stage of the analysis, IV is used to generate predicted values for the independent exposure (endogenous) variable. The predicted value is then used in a second-stage regression to explain the variation in outcome. The F-statistics from the first-stage regressions can be used to evaluate the strength of the IV in estimating the exposure.21 A value greater than 10 is generally assumed to be a sign of sufficient strength. Because genotype is associated with the exposure but not the confounders, the predicted exposure–outcome association is not confounded and is likely the estimate for the causal effect. Other methods such as a structural equation model have also been used in Mendelian randomization analysis.22 In the case of non-linear variables, the structural equation approach allows a simpler interpretation of the regression coefficients when compared with an IV approach.
MENDELIAN RANDOMIZATION ANALYSIS ON DIET, LIFESTYLE, AND OBESITY
In nutritional epidemiology studies, the principal exposures of interest are related to dietary consumption (e.g., foods, alcohol, and other beverages), lifestyle (e.g., physical activity and smoking), and obesity. Many potentially disease-modifying factors, however, are interrelated and also closely correlated with socioeconomic status. Moreover, information on dietary and lifestyle factors is often collected retrospectively. Even in prospective studies, the baseline subclinical status may affect reporting of the exposures. Being aware of these potential biases, several studies have examined the causal effects of dietary and lifestyle factors, such as alcohol consumption, dairy and cruciferous vegetable intake, and obesity, on various disorders, using the Mendelian randomization approach (Table 1).
Table 1.
Selected Mendelian randomization studies on dietary/lifestyle factors and biomarkers.
| Author, year | Study design, subjects | Exposure variables | Gene, variants | Outcomes | Major findings |
|---|---|---|---|---|---|
| Minelli, (2004)18 | Meta-analysis | Homocysteine | MTHFR C677T (rs1801133) | CHD | Estimated OR for CHD per unit (1.0 μmol/liter) change in homocysteine level was 1.07 (1.02–1.14) |
| Lewis, (2005)25 | Meta-analysis | Alcohol | ALDH2, glutamic acid to lysine variant at residue 487 | Esophageal cancer | Expected relative risk (RR = 2.54; *1*1 vs *2*2 genotypes) was comparable to the meta-analysis on the unconfounded genetic association (RR = 2.77) |
| Davey Smith, (2005)47 | 3,529 women, 60–79 y | CRP | CRP, rs1800947 | Blood pressure, hypertension | In IV analysis, the estimated causal effect for a doubling of CRP was 0.08 (−6.52–6.69) mmHg for systolic blood pressure and OR for hypertension was 1.03 (0.61–1.73) |
| Casas, (2005)43 | Meta-analysis | Homocysteine | MTHFR C677T (rs1801133) | Stroke | The genetic associations (OR = 1.26, 1.14–1.40) for TT vs. CC homozygotes were similar to the expected OR of 1.20 (P = 0.29) |
| Timpson, (2005)15 | 3,218 women, 60–79 y | CRP | CRP haplotype; rs1800947, rs1130864, and rs1205 | Metabolic syndrome | Linear regression and IV analysis gave conflicting results for the associations of CRP with BMI (P = 0.0002), IR (P = 0.0139), triglycerides (P = 0.0313), and HDL-C (P = 0.0688) |
| Keavney, (2006)42 | 3,460 (from 8,145) men and women, 30–64 y; and meta-analysis | Fibrinogen | FGB (FIBB), –148C/T (rs# not available) | MI | Fibrinogen corresponding to the genetic effect of 0.14 g/l difference per allele was not associated with MI risk |
| Casas, (2006)49 | 4,659 (association with CRP) and 6,201 (association with MI) men | CRP | CRP, rs1130864 | Coronary event | The observed OR for non-fatal MI corresponding to the genetic difference in CRP (0.68 mg/l) was 1.01 (0.74–1.38), lower than expected ORs (>1.20) based on observed data or meta-analysis |
| Sacerdote, (2007)29 | 634 healthy men and women | Dairy and cruciferous vegetables | LCT rs4988235; and TAS2R38 rs713598 and rs1726866 | Cancer | LCT variant was associated with consumption of dairy products (ice cream, P = 0.004); the haplotypes of TAS2R38 were related to cruciferous vegetable intake (P = 0.02) |
| Kivimaki, (2007)16 | 1,609 men and women, 24–39 y | CRP | CRP, rs2794521, rs3091244, rs1800947, rs1130864, rs1205 | CIMT | Associations from standard regression analysis and IV analysis were conflicting |
| Qi, (2007)41 | 1,348 (from 1730) women | IL-6 | IL6R, rs8192284 | Type 2 diabetes | IV analysis estimate of diabetes risk 1.59 (0.45–5.66) per unit log(IL-6) was similar to the estimate from logistic regression analysis, 1.78 (1.49–2.10) |
| Frayling, (2007)40 | 1,671 men and women, 65–80 y | IL-18 | IL18, rs5744256 | Physical function | IL-18 was significantly associated with Short Physical Performance Battery Score in both logistic regression analysis (P = 0.00016) and IV analysis (P = 0.019) |
| Chen, (2008)19 | Meta-analysis | Alcohol | ALDH2, glutamic acid to lysine variant at residue 487 | Blood pressure and hypertension | IV meta-analysis estimates of the effect of alcohol intake on diastolic blood pressure was 0.16 (0.11–0.21) mmHg per g/d; on systolic blood pressure it was 0.24 (0.16–0.32) mmHg per g/d |
| Kivimaki, (2008)17 | 2,230 men and women; 24–39 y | Body mass index | FTO, rs9939609 | CIMT and atherosclerotic risk factors | Standard regression analysis and IV analysis showed consistency in the association with adult CIMT, systolic blood pressure, and glucose and insulin resistance |
| Brunner, (2008)52 | 4,674 (from 5,274) men and women | CRP | CRP, rs11308641, rs1205, and rs3093077 | HbA1c and HOMA-IR; type 2 diabetes | Association between CRP levels and HbA1c and HOMA-IR obtained from standard regression and IV estimates were different. CRP haplotypes were not associated with diabetes risk in three samples |
| Kivimaki, (2008)51 | 4,435 (from 5,274) men and women | CRP | CRP, rs11308641, rs1205, and rs3093077 | CIMT | No inferred association between CRP and CIMT in IV analysis |
| Linsel-Nitschke, (2008)22 | 1,324 CAD cases and 6,255 controls | LDL-C | LDLR, rs2228671 | CAD | Adjustment for LDL-C levels in Mendelian randomization model abolished the significant association between rs2228671 with CAD |
Abbreviations: ALDH2, aldehyde dehydrogenase 2 family gene; CAD, coronary artery disease; CHD, coronary heart disease; CIMT, carotid intima-media thickness; CRP, C-reactive protein; CRP, CRP gene; FGB, fibrinogen beta chain gene; FTO, fat mass and obesity associated gene; GSTM1, glutathione S-transferase M1 gene; GSTT1, glutathione S-transferase theta 1 gene; HbA1c, hemoglobin A1c; HDL-C, high-density lipoprotein cholesterol; HOMA-IR, homeostasis model assessment insulin resistance; IL-6, interleukin-6; IL6R, IL-6 receptor gene; IL-18, interleukin-18; IL18, IL-18 gene; IV, instrumental variable; LCT, lactase gene; LDL-C, low-density lipoprotein cholesterol; LDLR, low-density lipoprotein receptor gene; MI, myocardial infarction; MTHFR, 5,10-methylenetetrahydrofolate reductase gene; OR, odds ratio; TAS2R38, taste receptor, type 2, member 38 gene.
Alcohol consumption has been related to the risk of esophageal cancer,23 partly through the carcinogenic effects of alcohol’s principal metabolite acetaldehyde.24 However, alcohol intake tends to be correlated with other lifestyle risk factors such as smoking and diet. Lewis et al. examined the causal relation between alcohol and esophageal cancer risk using genetic data on the ALDH2 (aldehyde dehydrogenase 2 family) gene,25 which encodes the major enzyme eliminating acetaldehyde. A single point mutation in ALDH2 results in the 2*2 allele (glutamic acid to lysine substitution at residue 487) and inactivation of the enzyme. This in turn leads to an accumulation of acetaldehyde after alcohol intake. Individuals who are homozygous for the 2*2 allele have peak blood alcohol levels that are 18 times higher and heterozygotes 5 times higher than *1*1 homozygotes.26 Drinking alcohol is likely to cause unpleasant symptoms such as nausea, drowsiness, and headache in the carriers. Therefore, *2*2 homozygotes drink significantly less alcohol than the wild-type carriers, and heterozygotes fall somewhere in the middle. The authors performed a meta-analysis including 905 cases of esophageal cancer and 2,078 controls from seven studies carried out in Japan, Taiwan, and Thailand. The relative risk (RR) of esophageal cancer for *1*1 homozygotes versus *2*2 homozygotes was 2.77 [95% confidence interval (CI), 1.25–6.12]. The authors predicted RR for genotype *1*1 based on the following data: 1) A meta-analysis that found light, moderate, and heavy drinkers had 1.8-, 2.38-, and 4.36-fold higher risk of cancer, respectively, than nondrinkers.27 2) One study reported that 9.4%, 28.2%, 39.6%, and 22.9% of *1*1 individuals were non-, light, moderate, and heavy drinkers, respectively; whereas all 2*2 individuals were virtually nondrinkers.28 The overall RR for *1*1 homozygotes was then derived with a formula RR = RRi × Pi, where i denotes the drinking category (non-, light, moderate, or heavy), RRi is the relative risk in the ith drinking category estimated by a meta-analysis, and Pi is the assumed proportion of ith drinking category among controls. The predicted RR was 2.54, comparable to the meta-analysis on the non-confounded genetic association (RR = 2.77). The data support a causal effect of drinking on esophageal cancer risk.
Recently, Chen et al.19 performed a meta-analysis on the association between ALDH2 genotype and blood pressure (N = 7,685 from five studies) and hypertension (N = 4,219 from three studies). The majority of participants were Japanese. In males, the overall odds ratios (ORs) were 2.42 (95% CI, 1.66–3.55) and 1.72 (95% CI, 1.17–2.52) comparing the *1*1, *1*2 with *2*2 genotypes. Systolic blood pressure (SBP) was 7.44 (5.39–9.49) and 4.24 (2.18–6.31) mmHg greater among individuals with *1*1 and *1*2 genotypes than those with *2*2 genotype. There were not significant associations in women due to their generally low alcohol consumption. Based on the data from three studies reporting genotype means of both alcohol consumption and blood pressure, the authors further conducted a Mendelian randomization analysis on the causal relation between alcohol intake and blood pressure, using an IV approach and assuming the effect is linear. The first-stage F-statistics of 2SLS analysis (ranging from 41 to 1,962) indicated that genotype was a strong instrument for estimating alcohol consumption. Under the assumption that there was not a correlation between alcohol and blood pressure within each genotype, the summary IV estimate of the effect of alcohol intake on diastolic blood pressure (DBP) was 0.16 (0.11–0.21) mmHg per g/d and the effect on SBP was 0.24 (0.16–0.32) mmHg per g/d. The results did not change in the sensitivity analysis assuming a correlation of 0.2 between alcohol and blood pressure within each genotype. These findings support a causal effect of alcohol intake on high blood pressure and the risk of hypertension.
Some studies highlighted the potential use of some dietary factors in Mendelian randomization analysis. In 634 healthy subjects from the Italian EPIC cohort, Sacerdote et al. assessed whether LCT (lactase) and TAS2R38 (taste receptor, type 2, member 38) gene variants could be used as proxies for dairy and cruciferous vegetable intakes in Mendelian randomization analysis.29 The LCT gene determines lactase persistence. People homozygous for allele-C of LCT SNP rs4988235 C>T have almost undetectable levels of intestinal lactase production compared with people with TC and TT genotypes.30,31 The LCT variant was associated with consumption of dairy products such as ice cream (P = 0.004) and less so for milk intake. The TAS2R38 gene is a member of the bitter taste receptor family. Two SNPs in this gene, rs713598 (Pro49Ala) and rs1726866 (Ala262Val), form two haplotypes that refer to taster and non-taster status for a bitter chemical phenylthiocarbamide.32 Bitter-tasting compounds in cruciferous vegetables resemble phenylthiocarbamide. The haplotypes of TAS2R38 gene variants were significantly related to cruciferous vegetable intake (P = 0.02). The data suggest that LCT and TAS2R38 genetic variants can be used as proxies for intakes of dairy and cruciferous vegetable in Mendelian randomization studies. However, a considerable geographic difference (northern to southern Europe) in the frequency of the LCT variant was noted.33 Therefore, population stratification may bias the associations with this genetic marker.
In a recent study, Kivimaki et al.17 examined the causal effect of lifetime adiposity on carotid intima-media thickness (CIMT, a preclinical marker for atherosclerosis risk) and atherosclerotic risk factors in 2,230 individuals from the Cardiovascular Risk in Young Finns study, using a variant in the FTO gene (fat mass and obesity associated). The life-course phenotype measures were derived from repeated assessment. FTO polymorphism rs9939609, which has been reproducibly associated with high adiposity,34 was used as an IV for lifetime body mass index (BMI). SNP rs9939609 was not associated with potential confounding factors, such as age, sex, socioeconomic circumstances, education, or risk behavior. The standard regression analysis suggests positive associations of lifetime BMI with adult CIMT, lifetime systolic blood pressure, and adult glucose and insulin resistance. The F-statistics from the first-stage regression were <10 (range, 7.5–9.2), indicating weakness of the instrument. In the IV regression analysis, all the associations of BMI with CIMT and other phenotypes were in the same direction as in the standard regression analysis but were less precisely estimated (P-values range between 0.02 and 0.15). To improve the power, the authors imputed the missing data and performed a simulation analysis, in which the F-statistic from the first-stage regression was 31.6 and all of the associations of lifetime BMI with CIMT and atherosclerotic risk factors became robust and of greater magnitude (P-values range between 0.0001 and 0.02). The authors concluded that the mutually supportive results from Mendelian randomization and standard regression models supported a causal effect of lifetime BMI on atherosclerosis risk.
MENDELIAN RANDOMIZATION ANALYSIS ON BIOMARKERS
In nutritional epidemiology, biomarkers offer important opportunities to advance research with more objective quantification of nutritional exposures and intermediate metabolic changes. Numerous epidemiological studies have documented associations of various biomarkers such as lipids and inflammatory markers with disease risk.35–37 As with dietary intake, the associations between biomarkers and disease risk may also be biased by confounding and reverse causation. For instance, many biomarkers can be affected by environmental factors such as smoking, physical activity, and dietary intake.38,39 In addition, the expected random distribution of biomarkers in a population would be skewed in individuals of predisease status (e.g., insulin resistance or early-stage atherosclerosis). Even worse, in cross-sectional studies, biomarkers are usually measured after the incidence of disease. Mendelian randomization can provide an opportunity to assess the causality of biomarkers for disease. A good deal of attention has been given to inflammatory marker C-reactive protein (CRP) and some other biomarkers such as fibrinogen, homocysteine, interleukin-6 (IL-6), and interleukin-18 (IL-18)37,40–43 (Table 1).
C-Reactive protein
CRP is a non-specific marker of systemic inflammation. Raised concentrations of circulating CRP are associated with increased risk of various diseases including cardiovascular disease, hypertension, type 2 diabetes, and metabolic syndrome.37,44,45 These associations, however, may be not causal. Obesity, smoking, and physical inactivity can increase CRP levels and also influence the disease endpoints,46 thereby confounding the observations. Davey Smith et al.47 investigated whether CRP causally influenced blood pressure in 3,529 women from the British Women’s Heart and Health Study. A CRP SNP rs1800947, which was related to low CRP levels,48 was used as a proxy for CRP level. In IV analysis, the estimated causal effects for a doubling of CRP were 0.08 (−6.52 to 6.69) mmHg for systolic blood pressure and an OR of 1.03 (95% CI, 0.61–1.73) for hypertension, indicating no causal relation between CRP and these outcomes. In another study, Casas et al.49 examined the causal effects of CRP on nonfatal myocardial infarction (MI), using the genetic data on CRP +1444C>T polymorphism (rs1130864). The weighted mean difference (WMD) in CRP concentration between individuals homozygous for the T allele and carriers of the C allele was 0.68 mg/L (0.31–1.10; in 4,659 European men). The predicted ORs for non-fatal MI corresponding to this difference was 1.25 (95% CI, 1.09–1.43) based on the estimated CRP-MI association from a meta-analysis.50 The observed OR for non-fatal MI was 1.01 (95% CI, 0.74–1.38) comparing TT subjects with C allele carriers, lower than the predicted OR. The results suggest that the previously reported associations between CRP and coronary events may be not causal.
Kivimaki et al.16 examined the causality between CRP and CIMT by determining haplotypes from genetic variants in the CRP gene (rs2794521, rs3091244, rs1800947, rs1130864, and rs1205) in 1,609 young Finns (768 men and 841 women). Higher CRP levels were associated with greater CIMT in the standard regression analysis. However, the analysis with CRP haplotypes as IV showed inverse associations, suggesting CRP might not causally affect CIMT and CHD risk. The same group recently performed a similar IV analysis in 4,941 men and women from the Whitehall II Study, and the results supported their earlier conclusion.51 Timpson et al. examined associations between serum CRP concentration and metabolic syndrome phenotypes in 3,218 women from the British Women’s Heart and Health Study.15 Linear regression analysis and Mendelian randomization IV analysis (CRP haplotypes from rs2794521, rs1800947, rs1130864, and rs1205 as instrument) generated conflicting associations of CRP levels with BMI (P = 0.0002), insulin resistance (P = 0.0139), triglycerides (P = 0.0313), and HDL cholesterol (P = 0.0688). The data do not support causal effects of CRP on the metabolic syndrome.
In a recent study,52 the causal effects of CRP on diabetes were examined using a Mendelian randomization approach, in which CRP haplotypes (from rs3093077, rs1800947, and rs1205) were used as IVs for CRP level. Serum CRP was measured twice, once at mean age 49 y (baseline; N = 4,674) and once at 61 y (N = 5,274). Homeostasis model assessment of insulin resistance (HOMA-IR) and hemoglobin A1c (HbA1c), which were measured at mean age 61 y, were used as markers for diabetes risk. F-statistics from the first-stage regression were greater than 10 for both previous and contemporaneous CRP levels. Results of the Durbin-Wu-Hausman test for difference between the standard linear regression and IV estimates approached significance and were highly significant for contemporaneous serum CRP with HOMA-IR as outcome. In addition, analyses of the association between CRP haplotype and diabetes risk were consistently null in three study samples. The results indicate that the systemic CRP levels may not be causally responsible for development of insulin resistance, hyperglycemia, or diabetes.
Other biomarkers
Blood concentration of fibrinogen has been associated with coronary disease in many epidemiological studies,53 but it is uncertain whether this association is causal or reflects residual confounding by other risk factors such as smoking, obesity, and plasma lipid profile. In 1,923 female and 1,537 male control subjects from the International Studies of Infarct Survival (ISIS) study, Keavney et al.42 estimated a fibrinogen-raising effect of 0.14 g/L per allele of FGB (or FIBB, fibrinogen beta chain) –148C/T polymorphism (P < 0.0001). Such a difference in fibrinogen was not significantly related to MI risk in a multivariable analysis (OR = 1.03; 95% CI, 1.00–1.05), which was similar to the estimates of the risk per higher-fibrinogen allele in ISIS alone (OR = 1.06; 95% CI, 0.96–1.16) and in a meta-analysis (OR = 1.00; 95% CI, 0.95–1.04; ISIS and 19 other studies). The results indicate that long-term differences in fibrinogen concentrations are not a causal determinant for MI.
Casas et al.43 examined the influence of plasma homocysteine level on the risk of stroke using genetic data on the MTHFR (5,10-methylenetetrahydrofolate reductase) C677T polymorphism, which leads to a high homocysteine level.54 Based on a meta-analysis of 15,635 individuals, the weighted mean difference in homocysteine concentration between TT and CC homozygotes was 1.93 μmol/L (1.38–2.47). The expected OR for stroke corresponding to this difference based on previous observational studies was 1.20 (95% CI, 1.10–1.31). In the genetic meta-analysis (n = 13,928) the OR for stroke was 1.26 (95% CI, 1.14–1.40) for TT versus CC homozygotes, similar to the expected OR (P = 0.29). The authors concluded that plasma homocysteine might be causally related to stroke. In another study, Minelli et al.18 performed a meta-analysis on the associations of MTHFR C677T with circulating homocysteine levels and CHD risk. The estimates obtained from studies reporting both homocysteine change and CHD were similar to those reporting either outcome. By pooling all data, it was found that the genotype (TT vs. CC)-associated OR for CHD was 1.21 (95% CI, 1.06–1.40) and the mean difference in homocysteine was 2.71 (2.02–3.41) μM/L. Based on these data, it was estimated that a per-unit (1.0-μM/L) change in homocysteine level was associated with unconfounded 1.07 (1.02–1.14)-fold increased risk of CHD.
The missense variant Asp358Ala (rs8192284) in the interleukin-6 receptor gene (IL6R) was significantly related to circulating IL-6 levels.55 Using this SNP as IV for IL-6 concentration, we have tested the causal relation between IL-6 and diabetes risk.41 The risk estimate from Mendelian randomization IV analysis (OR = 1.59; 95% CI, 0.45–5.66) was slightly lower than that obtained from logistic regression (OR = 1.78, 95% CI, 1.49–2.10). The data tend to support a causal relation for IL-6 and diabetes risk. However, the wide confidence interval for the IV estimate suggests a larger sample size is needed for further confirmation. In another study, Frayling et al.40 used IL18 SNP rs5744256 as IV and demonstrated that IL-18 concentrations may causally affect physical function in an older population (65–80 y). Recently, Linsel-Nitschke et al.22 explored the causal relation between LDL-C and coronary artery disease (CAD). SNP rs2228671 in the LDLR gene was consistently related to a decrease in LDL-C levels by 0.19 mmol/L and 18% (11–24%) decreased risk of CAD (per T allele) in multiple European samples. The Mendelian randomization analysis was performed in 1,324 CAD cases and 6,255 controls using a structural equation model. The analysis revealed a highly significant association between the rs2228671 genotype and LDL-C as well as between LDL-C and CAD risk. The association between rs2228671 and the risk of CAD was abolished with adjustment for LDL-C levels. The findings indicate a causal link among LDLR variant, change in LDL-C levels, and CAD risk.
INTRAUTERINE INFLUENCE ON OFFSPRING HEALTH OUTCOMES
A potentially important application of Mendelian randomization is to investigate the intrauterine influence on offspring health outcomes.56 Exposure of the fetus to the intrauterine milieu can have profound effects on health in adulthood. A body of data shows that low birth weight and fetal overgrowth carry an elevated risk of developing metabolic diseases, such as obesity, insulin resistance, dyslipidemia, hypertension, CHD, and stroke, during adulthood.57,58 However, there has been long-term controversy about whether intrauterine development is involved in causal pathways underlying these observations. Confounding is a particular problem in studying intrauterine origins of diseases, because the information on confounders may not be collected adequately, especially when there is a long temporal gap between the exposure and outcome. Various approaches have been proposed to infer causal effects of intrauterine exposures on offspring health, as described in detail elsewhere.56 Here we discuss the potential application of the Mendelian randomization approach, in which maternal genotype is taken to be a proxy for the factor that influences the intrauterine environment experienced by her offspring.
The mechanisms by which intrauterine exposures lead to later metabolic disorders are complex and multi-factorial. There has been a major focus on the effects of maternal overnutrition. According to the developmental overnutrition hypothesis, greater maternal adiposity (lifetime and especially during pregnancy) is associated with a greater risk of metabolic abnormalities such as insulin resistance and glucose intolerance, which likely result in permanent changes in appetite control, neuroendocrine functioning, or energy metabolism in the developing fetus and lead to greater adiposity and risk of obesity in later life. In a recent study, Lawlor et al.14 performed a Mendelian randomization analysis, using maternal FTO SNP rs9939609 (T>A) genotype as IV, to test the causal effect of maternal adiposity on offspring obesity risk. The participants were from a longitudinal, population-based birth cohort study (the Avon Longitudinal Study of Parents and Children; ALSPAC) that recruited 14,541 pregnant women. Because intrauterine effects are markedly different for singletons and multiple births, only singleton births were used. Measurements of offspring at 9 y (N = 7,221) and 11 y (N = 6,710) were analyzed. Of these eligible participants, 3,263 had all of the relevant data for the Mendelian randomization analysis.
Neither maternal nor offspring FTO genotypes were associated with any of the potential confounding factors of the association of maternal BMI with offspring fat or lean mass, including social class, mother/father university degree, mother/father smoking during pregnancy, and parity. Each allele-A of the maternal FTO genotype was associated with a linear increase in maternal BMI of 0.09 (0.04–0.14) standard deviation (SD). Each allele-A of the offspring’s genotype was associated with 0.13 (0.09–0.18) SD of total fat mass and 0.03 SD (0.00–0.06) of lean mass. In the Mendelian randomization IV analysis, the first stage F-statistic without adjustment for offspring FTO was 12.9, and 10.1 with adjustment for offspring FTO genotype, indicating that maternal FTO genotype is a strong proxy for maternal obesity. There was not an association of maternal BMI with offspring fat mass in the IV analysis. The data do not support a causal effect of greater maternal BMI during pregnancy on offspring adiposity at age 9–11 y. However, the estimates from Mendelian randomization analysis had wide 95% confidence intervals, highlighting the importance of larger sample sizes in future studies. In addition, maternal FTO is an unconfounded proxy for maternal adiposity across her lifespan, including the periods of pregnancy and breastfeeding. Thus, the findings cannot specifically address the developmental overnutrition hypothesis.
POTENTIAL LIMITATIONS OF MENDELIAN RANDOMIZATION
While the Mendelian randomization method shows considerable promise in integrating genetic findings into nutritional epidemiology research, its application has several potential limitations. Mendelian randomization analysis is based on a number of underlying assumptions that are hard to verify in practice. When some key assumptions are violated, any inference of the causal effect would be biased. First of all, the association of the genetic markers used in Mendelian randomization studies with the exposures of interest should be reliably demonstrated, i.e., reproducible in multiple independent samples and functionally related to the exposures. However, the vast majority of the genetic associations are hard to replicate, and the functional testing for the related variants are largely lacking.
A principal assumption of Mendelian randomization analysis is that the effect of a genetic variant on an outcome acts only via the intermediate exposure (e.g., lifestyle factor or biomarker). However, a genetic variant may cause multiple biological alterations, i.e., pleiotropy. If the additional alterations also independently affect outcomes of interest, this may lead to spurious associations. For instance, an APOE gene variant may affect various lipids (total cholesterol, LDL cholesterol, and apo B) and also cause other metabolic abnormalities59,60; all can influence the same outcome such as cardiovascular disease. Therefore, APOE genotype would not be a valid instrument for a specific lipid marker in Mendelian randomization analysis. In addition, biological compensation (canalization), the buffering of the genetic effects during development, can also lead to forged causal inference. Developmental and physiological adaptation may occur during the lifelong exposure to the gene-related changes through permanent alterations in tissue structure and function that offset the genetic influence.
In addition, genetic confounding may also result in violation of Mendelian randomization assumptions, primarily through linkage disequilibrium (LD) between genetic variants and population stratification. A genetic marker under investigation may be in LD with another polymorphism (in the same gene or a different gene) that has not been detected but also independently affects the disease risk. In this case, the link between the investigated marker and disease is not merely through the intermediate exposure tested. As such, the genetic marker cannot be an instrument in Mendelian randomization study. Unknown population substructures (population stratification) may cause confounding of genetic association if subgroups have diverse genetic architecture and different distributions of the disease.61 Population stratification can seriously affect the Mendelian randomization estimates when gene-exposure and gene-outcome associations are derived from different populations. Another possible source for genetic confounding is transmission ratio distortion (TRD), i.e., the distribution of alleles at a particular locus in the surviving offspring differs from that expected on the basis of Mendelian proportions. TRD may result from selective survival between conception and entry into the study if the genetic variant of interest causes early mortality and may be widespread in the human genome.62 Such distortion may result in violation of the assumption of random assortment of genes. In addition, some genes may be silent or functionally active depending on whether a particular variant was maternally or paternally inherited (imprinting).63 The genes of parent-of-origin effect are not appropriate for Mendelian randomization.
This limitation of Mendelian randomization is further compounded by random measurement errors, moderate genetic effects, and selection bias. In Mendelian randomization analysis, the statistical uncertainty in both genotype–exposure and genotype–outcome association is taken into account. Measurement errors in genotype, exposure, and outcome can lead to spurious associations. Therefore, reducing errors in these measurements would significantly improve the causal inference. Recent advances in genetic studies indicate that individual genetic variants are likely to explain only a very small proportion of the variation in phenotypic traits. Thus, a very large sample size is required to estimate reliable genetic associations. While meta-analysis can help to improve statistical power, this approach may be subject to the problems of publication and reporting bias.18 In addition, because studies are usually based on a subset of the total population in which specific exposures or outcomes (e.g., biomarkers) are measured, they are likely to suffer from the usual limitations of selection bias, which is characterized by considerably unequal coverage of cases and controls that would lead to biased associations with disease status. When multiple studies are involved, heterogeneity among various populations may also distort assessments of the magnitude of the associations. This is particularly the case when studies using different methods in measuring dietary intakes and biomarkers are inappropriately combined.
CONCLUSION
Unmeasured and residual confounding remains a major challenge in traditional nutritional epidemiologic studies. The prospects for nutritional epidemiology in the post-genome era depend on understanding how to use genetic associations to test hypotheses about causal pathways, in addition to modeling the joint (interactive) effects of genotype and environment. Mendelian randomization analysis offers a potential research framework to infer causality and may provide unique insights into etiological mechanisms and inform preventative strategies. Some pioneering studies have examined the causal relation of dietary and lifestyle factors such as alcohol consumption, intakes of dairy and cruciferous vegetables, and obesity, as well as biochemical markers such as CRP, fibrinogen, IL-6, and IL-18 with various health outcomes using the Mendelian randomization approach. The data suggest that some associations found in the observational studies might be biased by confounding or reverse causation. Mendelian randomization approach may also be useful in studying intrauterine influence on offspring health.
As discussed above, there are still many challenges in applying this novel approach to nutritional epidemiology research. We would only ever countenance the use of genetic variants in Mendelian randomization studies if they have clearly established robust associations with the risk factor of interest, and if the study is carefully designed to reduce genetic confounding. The discovery of new genetic associations and subsequent functional assessment could tell us which genotype–exposure relation may be pursued in Mendelian randomization studies focusing on diet and lifestyle factors, e.g., FTO and MC4R genes and obesity,34,64 and nicotinic acetylcholine receptor subunit (CHRNA5, CHRNA3 and CHRNB4) genes and smoking.65 The genes relevant to most dietary factors, physical activity, and other lifestyle factors, however, have yet to be established. Because Mendelian randomization analysis combines possible errors in measurements of multiple parameters (genotype, exposure, and outcome), and the associations are generally modest (especially the genetic association), a crucial requirement for a Mendelian randomization study is that the design have adequate statistical power to evaluate the likelihood and magnitude of any causal association. Large-scale collaboration is a promising way to overcome this challenge.
Acknowledgments
Funding. Dr. Qi’s research is supported by NIH grant HL071981, American Heart Association Scientist Development Award, and the Boston Obesity Nutrition Research Center (DK46200).
References
- 1.Willett W. Nutritional Epidemiology. New York Oxford: Oxford University Press; 1998. [Google Scholar]
- 2.Willett WC, Stampfer MJ, Manson JE, et al. Intake of trans fatty acids and risk of coronary heart disease among women. Lancet. 1993;341:581–585. doi: 10.1016/0140-6736(93)90350-p. [DOI] [PubMed] [Google Scholar]
- 3.Li G, Zhang P, Wang J, et al. The long-term effect of lifestyle interventions to prevent diabetes in the China Da Qing Diabetes Prevention Study: a 20-year follow-up study. Lancet. 2008;371:1783–1789. doi: 10.1016/S0140-6736(08)60766-7. [DOI] [PubMed] [Google Scholar]
- 4.Lindstrom J, Ilanne-Parikka P, Peltonen M, et al. Sustained reduction in the incidence of type 2 diabetes by lifestyle intervention: follow-up of the Finnish Diabetes Prevention Study. Lancet. 2006;368:1673–1679. doi: 10.1016/S0140-6736(06)69701-8. [DOI] [PubMed] [Google Scholar]
- 5.Smith GD, Lawlor DA, Harbord R, Timpson N, Day I, Ebrahim S. Clustered environments and randomized genes: a fundamental distinction between conventional and genetic epidemiology. PLoS Med. 2007;4:e352. doi: 10.1371/journal.pmed.0040352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Phillips A. Commentary: balancing quantity and quality when designing epidemiological studies. Int J Epidemiol. 2003;32:58–59. doi: 10.1093/ije/dyg043. [DOI] [PubMed] [Google Scholar]
- 7.Davey Smith G, Ebrahim S. What can Mendelian randomisation tell us about modifiable behavioural and environmental exposures? BMJ. 2005;330:1076–1079. doi: 10.1136/bmj.330.7499.1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Didelez V, Sheehan N. Mendelian randomization as an instrumental variable approach to causal inference. Stat Methods Med Res. 2007;16:309–330. doi: 10.1177/0962280206077743. [DOI] [PubMed] [Google Scholar]
- 9.Morgan TH. Physical Basis of Heredity. Philadelphia: JB Lipincott Company; 1919. [Google Scholar]
- 10.Katan MB. Apolipoprotein E isoforms, serum cholesterol, and cancer. Lancet. 1986;1:507–508. doi: 10.1016/s0140-6736(86)92972-7. [DOI] [PubMed] [Google Scholar]
- 11.Mahley RW. Apolipoprotein E: cholesterol transport protein with expanding role in cell biology. Science. 1988;240:622–630. doi: 10.1126/science.3283935. [DOI] [PubMed] [Google Scholar]
- 12.Smith GD, Ebrahim S. Mendelian randomization: prospects, potentials, and limitations. Int J Epidemiol. 2004;33:30–42. doi: 10.1093/ije/dyh132. [DOI] [PubMed] [Google Scholar]
- 13.Nitsch D, Molokhia M, Smeeth L, DeStavola BL, Whittaker JC, Leon DA. Limits to causal inference based on Mendelian randomization: a comparison with randomized controlled trials. Am J Epidemiol. 2006;163:397–403. doi: 10.1093/aje/kwj062. [DOI] [PubMed] [Google Scholar]
- 14.Lawlor DA, Harbord RM, Sterne JA, Timpson N, Davey Smith G. Mendelian randomization: using genes as instruments for making causal inferences in epidemiology. Stat Med. 2008;27:1133–1163. doi: 10.1002/sim.3034. [DOI] [PubMed] [Google Scholar]
- 15.Timpson NJ, Lawlor DA, Harbord RM, et al. C-reactive protein and its role in metabolic syndrome: Mendelian randomisation study. Lancet. 2005;366:1954–1959. doi: 10.1016/S0140-6736(05)67786-0. [DOI] [PubMed] [Google Scholar]
- 16.Kivimaki M, Lawlor DA, Eklund C, et al. Mendelian randomization suggests no causal association between C-reactive protein and carotid intima-media thickness in the young Finns study. Arterioscler Thromb Vasc Biol. 2007;27:978–979. doi: 10.1161/01.ATV.0000258869.48076.14. [DOI] [PubMed] [Google Scholar]
- 17.Kivimaki M, Davey Smith G, Timpson NJ, et al. Lifetime body mass index and later atherosclerosis risk in young adults: examining causal links using Mendelian randomization in the Cardiovascular Risk in Young Finns study. Eur Heart J. 2008;25:2552–2560. doi: 10.1093/eurheartj/ehn252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Minelli C, Thompson JR, Tobin MD, Abrams KR. An integrated approach to the meta-analysis of genetic association studies using Mendelian randomization. Am J Epidemiol. 2004;160:445–452. doi: 10.1093/aje/kwh228. [DOI] [PubMed] [Google Scholar]
- 19.Chen L, Davey Smith G, Harbord RM, Lewis SJ. Alcohol intake and blood pressure: a systematic review implementing a Mendelian randomization approach. PLoS Med. 2008;5:e52. doi: 10.1371/journal.pmed.0050052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Greenland S. An introduction to instrumental variables for epidemiologists. Int J Epidemiol. 2000;29:1102. doi: 10.1093/oxfordjournals.ije.a019909. [DOI] [PubMed] [Google Scholar]
- 21.Staiger D, Stock JH. Instrumental variables with weak instruments. Econometrica. 1997;65:557–586. [Google Scholar]
- 22.Linsel-Nitschke P, Gotz A, Erdmann J, et al. Lifelong reduction of LDL-cholesterol related to a common variant in the LDL-receptor gene decreases the risk of coronary artery disease – a Mendelian randomisation study. PLoS ONE. 2008;3:e2986. doi: 10.1371/journal.pone.0002986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Morita M, Saeki H, Mori M, Kuwano H, Sugimachi K. Risk factors for esophageal cancer and the multiple occurrence of carcinoma in the upper aerodigestive tract. Surgery. 2002;131 (Suppl):S1–S6. doi: 10.1067/msy.2002.119287. [DOI] [PubMed] [Google Scholar]
- 24.Blot WJ. Alcohol and cancer. Cancer Res. 1992;52(Suppl):S2119–S2123. [PubMed] [Google Scholar]
- 25.Lewis SJ, Smith GD. Alcohol, ALDH2, and esophageal cancer: a meta-analysis which illustrates the potentials and limitations of a Mendelian randomization approach. Cancer Epidemiol Biomarkers Prev. 2005;14:1967–1971. doi: 10.1158/1055-9965.EPI-05-0196. [DOI] [PubMed] [Google Scholar]
- 26.Enomoto N, Takase S, Yasuhara M, Takada A. Acetaldehyde metabolism in different aldehyde dehydrogenase-2 genotypes. Alcohol Clin Exp Res. 1991;15:141–144. doi: 10.1111/j.1530-0277.1991.tb00532.x. [DOI] [PubMed] [Google Scholar]
- 27.Gutjahr E, Gmel G, Rehm J. Relation between average alcohol consumption and disease: an overview. Eur Addict Res. 2001;7:117–127. doi: 10.1159/000050729. [DOI] [PubMed] [Google Scholar]
- 28.Yokoyama A, Kato H, Yokoyama T, et al. Genetic polymorphisms of alcohol and aldehyde dehydrogenases and glutathione S-transferase M1 and drinking, smoking, and diet in Japanese men with esophageal squamous cell carcinoma. Carcinogenesis. 2002;23:1851–1859. doi: 10.1093/carcin/23.11.1851. [DOI] [PubMed] [Google Scholar]
- 29.Sacerdote C, Guarrera S, Smith GD, et al. Lactase persistence and bitter taste response: instrumental variables and Mendelian randomization in epidemiologic studies of dietary factors and cancer risk. Am J Epidemiol. 2007;166:576–581. doi: 10.1093/aje/kwm113. [DOI] [PubMed] [Google Scholar]
- 30.Grand RJ, Montgomery RK, Chitkara DK, Hirschhorn JN. Changing genes; losing lactase. Gut. 2003;52:617–619. doi: 10.1136/gut.52.5.617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Enattah NS, Sahi T, Savilahti E, Terwilliger JD, Peltonen L, Jarvela I. Identification of a variant associated with adult-type hypolactasia. Nat Genet. 2002;30:233–237. doi: 10.1038/ng826. [DOI] [PubMed] [Google Scholar]
- 32.Kim UK, Jorgenson E, Coon H, Leppert M, Risch N, Drayna D. Positional cloning of the human quantitative trait locus underlying taste sensitivity to phenylthiocarbamide. Science. 2003;299:1221–1225. doi: 10.1126/science.1080190. [DOI] [PubMed] [Google Scholar]
- 33.Weiss KM. The unkindest cup. Lancet. 2004;363:1489–1490. doi: 10.1016/S0140-6736(04)16195-3. [DOI] [PubMed] [Google Scholar]
- 34.Frayling TM, Timpson NJ, Weedon MN, et al. A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science. 2007;316:889–894. doi: 10.1126/science.1141634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zethelius B, Berglund L, Sundstrom J, et al. Use of multiple biomarkers to improve the prediction of death from cardiovascular causes. N Engl J Med. 2008;358:2107–2116. doi: 10.1056/NEJMoa0707064. [DOI] [PubMed] [Google Scholar]
- 36.Qi L, Doria A, Manson JE, et al. Adiponectin genetic variability, plasma adiponectin, and cardiovascular risk in patients with type 2 diabetes. Diabetes. 2006;55:1512–1516. doi: 10.2337/db05-1520. [DOI] [PubMed] [Google Scholar]
- 37.Ridker PM, Hennekens CH, Buring JE, Rifai N. C-reactive protein and other markers of inflammation in the prediction of cardiovascular disease in women. N Engl J Med. 2000;342:836–843. doi: 10.1056/NEJM200003233421202. [DOI] [PubMed] [Google Scholar]
- 38.Qi L, Meigs JB, Liu S, Manson JE, Mantzoros C, Hu FB. Dietary fibers and glycemic load, obesity, and plasma adiponectin levels in women with type 2 diabetes. Diabetes Care. 2006;29:1501–1505. doi: 10.2337/dc06-0221. [DOI] [PubMed] [Google Scholar]
- 39.Pischon T, Hankinson SE, Hotamisligil GS, Rifai N, Rimm EB. Leisure-time physical activity and reduced plasma levels of obesity-related inflammatory markers. Obes Res. 2003;11:1055–1064. doi: 10.1038/oby.2003.145. [DOI] [PubMed] [Google Scholar]
- 40.Frayling TM, Rafiq S, Murray A, et al. An interleukin-18 polymorphism is associated with reduced serum concentrations and better physical functioning in older people. J Gerontol A Biol Sci Med Sci. 2007;62:73–78. doi: 10.1093/gerona/62.1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Qi L, Rifai N, Hu FB. Interleukin-6 receptor gene variations, plasma interleukin-6 levels, and type 2 diabetes in U.S. women. Diabetes. 2007;56:3075–3081. doi: 10.2337/db07-0505. [DOI] [PubMed] [Google Scholar]
- 42.Keavney B, Danesh J, Parish S, et al. Fibrinogen and coronary heart disease: test of causality by”Mendelian randomization”. Int J Epidemiol. 2006;35:935–943. doi: 10.1093/ije/dyl114. [DOI] [PubMed] [Google Scholar]
- 43.Casas JP, Bautista LE, Smeeth L, Sharma P, Hingorani AD. Homocysteine and stroke: evidence on a causal link from Mendelian randomisation. Lancet. 2005;365:224–232. doi: 10.1016/S0140-6736(05)17742-3. [DOI] [PubMed] [Google Scholar]
- 44.Sesso HD, Buring JE, Rifai N, Blake GJ, Gaziano JM, Ridker PM. C-reactive protein and the risk of developing hypertension. JAMA. 2003;290:2945–2951. doi: 10.1001/jama.290.22.2945. [DOI] [PubMed] [Google Scholar]
- 45.Ridker PM, Buring JE, Cook NR, Rifai N. C-reactive protein, the metabolic syndrome, and risk of incident cardiovascular events: an 8-year follow-up of 14,719 initially healthy American women. Circulation. 2003;107:391–397. doi: 10.1161/01.cir.0000055014.62083.05. [DOI] [PubMed] [Google Scholar]
- 46.de Ferranti S, Rifai N. C-reactive protein and cardiovascular disease: a review of risk prediction and interventions. Clin Chim Acta. 2002;317:1–15. doi: 10.1016/s0009-8981(01)00797-5. [DOI] [PubMed] [Google Scholar]
- 47.Davey Smith G, Lawlor DA, Harbord R, et al. Association of C-reactive protein with blood pressure and hypertension: life course confounding and Mendelian randomization tests of causality. Arterioscler Thromb Vasc Biol. 2005;25:1051–1056. doi: 10.1161/01.ATV.0000160351.95181.d0. [DOI] [PubMed] [Google Scholar]
- 48.Zee RY, Ridker PM. Polymorphism in the human C-reactive protein (CRP) gene, plasma concentrations of CRP, and the risk of future arterial thrombosis. Atherosclerosis. 2002;162:217–219. doi: 10.1016/s0021-9150(01)00703-1. [DOI] [PubMed] [Google Scholar]
- 49.Casas JP, Shah T, Cooper J, et al. Insight into the nature of the CRP-coronary event association using Mendelian randomization. Int J Epidemiol. 2006;35:922–931. doi: 10.1093/ije/dyl041. [DOI] [PubMed] [Google Scholar]
- 50.Danesh J, Wheeler JG, Hirschfield GM, et al. C-reactive protein and other circulating markers of inflammation in the prediction of coronary heart disease. N Engl J Med. 2004;350:1387–1397. doi: 10.1056/NEJMoa032804. [DOI] [PubMed] [Google Scholar]
- 51.Kivimaki M, Lawlor DA, Smith GD, et al. Does high C-reactive protein concentration increase atherosclerosis?The Whitehall II Study. PLoS ONE. 2008;3:e3013. doi: 10.1371/journal.pone.0003013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Brunner EJ, Kivimaki M, Witte DR, et al. Inflammation, insulin resistance, and diabetes – Mendelian randomization using CRP haplotypes points upstream. PLoS Med. 2008;5:e155. doi: 10.1371/journal.pmed.0050155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Danesh J, Lewington S, Thompson SG, et al. Plasma fibrinogen level and the risk of major cardiovascular diseases and nonvascular mortality: an individual participant meta-analysis. JAMA. 2005;294:1799–1809. doi: 10.1001/jama.294.14.1799. [DOI] [PubMed] [Google Scholar]
- 54.Bailey LB, Gregory JF., 3rd Polymorphisms of methylenetetrahydrofolate reductase and other enzymes: metabolic significance, risks and impact on folate requirement. J Nutr. 1999;129:919–922. doi: 10.1093/jn/129.5.919. [DOI] [PubMed] [Google Scholar]
- 55.Reich D, Patterson N, Ramesh V, et al. Admixture mapping of an allele affecting interleukin 6 soluble receptor and interleukin 6 levels. Am J Hum Genet. 2007;80:716–726. doi: 10.1086/513206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Smith GD. Assessing intrauterine influences on offspring health outcomes: can epidemiological studies yield robust findings? Basic Clin Pharmacol Toxicol. 2008;102:245–256. doi: 10.1111/j.1742-7843.2007.00191.x. [DOI] [PubMed] [Google Scholar]
- 57.Barker DJ. Adult consequences of fetal growth restriction. Clin Obstet Gynecol. 2006;49:270–283. doi: 10.1097/00003081-200606000-00009. [DOI] [PubMed] [Google Scholar]
- 58.Phillips DI, Jones A, Goulden PA. Birth weight, stress, and the metabolic syndrome in adult life. Ann N Y Acad Sci. 2006;1083:28–36. doi: 10.1196/annals.1367.027. [DOI] [PubMed] [Google Scholar]
- 59.Martins IJ, Hone E, Foster JK, et al. Apolipoprotein E, cholesterol metabolism, diabetes, and the convergence of risk factors for Alzheimer’s disease and cardiovascular disease. Mol Psychiatry. 2006;11:721–736. doi: 10.1038/sj.mp.4001854. [DOI] [PubMed] [Google Scholar]
- 60.Eichner JE, Dunn ST, Perveen G, Thompson DM, Stewart KE, Stroehla BC. Apolipoprotein E polymorphism and cardiovascular disease: a HuGE review. Am J Epidemiol. 2002;155:487–495. doi: 10.1093/aje/155.6.487. [DOI] [PubMed] [Google Scholar]
- 61.Cardon LR, Palmer LJ. Population stratification and spurious allelic association. Lancet. 2003;361:598–604. doi: 10.1016/S0140-6736(03)12520-2. [DOI] [PubMed] [Google Scholar]
- 62.Bochud M, Chiolero A, Elston RC, Paccaud F. A cautionary note on the use of Mendelian randomization to infer causation in observational epidemiology. Int J Epidemiol. 2008;37:414–416. doi: 10.1093/ije/dym186. author reply 416–417. [DOI] [PubMed] [Google Scholar]
- 63.Temple IK. Imprinting in human disease with special reference to transient neonatal diabetes and Beckwith-Wiedemann syndrome. Endocr Dev. 2007;12:113–123. doi: 10.1159/000109638. [DOI] [PubMed] [Google Scholar]
- 64.Loos RJ, Lindgren CM, Li S, et al. Common variants near MC4R are associated with fat mass, weight and risk of obesity. Nat Genet. 2008;40:768–775. doi: 10.1038/ng.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hung RJ, McKay JD, Gaborieau V, et al. A susceptibility locus for lung cancer maps to nicotinic acetylcholine receptor subunit genes on 15q25. Nature. 2008;452:633–637. doi: 10.1038/nature06885. [DOI] [PubMed] [Google Scholar]