

Abstract
Oxidative stress is recognized as one of the earliest and most intense pathological processes in Alzheimer's disease (AD), and the antioxidant vitamin E has been shown to efficiently prevent amyloid plaque formation and neurodegeneration. Plasma phospholipid transfer protein (PLTP) has a major role in vitamin E transfers in vivo, and PLTP deficiency in mice is associated with reduced brain vitamin E levels. To determine the impact of PLTP on amyloid pathology in vivo, we analyzed the vulnerability of PLTP-deficient (PLTP-KO) mice to the toxic effects induced by intracerebroventricular injection of oligomeric amyloid-β25–35 (Aβ25–35) peptide, a non-transgenic model of AD. Under basal conditions, PLTP-KO mice showed increased cerebral oxidative stress, increased brain Aβ1–42 levels, and a lower expression of the synaptic function marker synaptophysin, as compared with wild-type mice. This PLTP-KO phenotype was associated with increased memory impairment 1 week after Aβ25–35 peptide injection. Restoration of brain vitamin E levels in PLTP-KO mice through a chronic dietary supplementation prevented Aβ25–35-induced memory deficits and reduced cerebral oxidative stress and toxicity. We conclude that PLTP, through its ability to deliver vitamin E to the brain, constitutes an endogenous neuroprotective agent. Increasing PLTP activity may offer a new way to develop neuroprotective therapies.
Keywords: lipid transfer protein, oxidative stress, vitamin E, Alzheimer's disease, neuroprotection, amyloid toxicity
INTRODUCTION
Oxidative stress is widely recognized as a hallmark of natural aging and a key factor in the etiology of neurodegenerative disorders, including Alzheimer's disease (AD). Increased oxidative stress is observed in both transgenic and non-transgenic models of AD, and in humans, oxidative damage is recognized as one of the earliest changes in familial as well as sporadic forms of AD (Clark et al, 2010). Several in vitro and in vivo findings came in support of a beneficial effect of the antioxidant vitamin E in the prevention of amyloid plaque formation and neurodegeneration (Ricciarelli et al, 2007; Nishida et al, 2009), and increased plasma levels of this antioxidant have recently been associated with a reduced risk of developing AD in advanced age (Mangialasche et al, 2010). Although overexpression or targeted disruption of specific genes in transgenic animals has offered a convenient way to determine the contribution of one given factor to the processes of aging and neurodegeneration, there is still a paucity of well-suited animal models allowing to address selectively the contribution of oxidative injury to their etiology. In this context, controlled alteration in the incorporation and distribution of the lipid-soluble vitamin E antioxidant into the brain tissue is likely to constitute a relevant approach. In support of the latter view, it was recently reported that early vitamin E supplementation in young mice reduces amyloid-β peptide levels and amyloid deposition in a transgenic model of AD (Sung et al, 2004).
As a lipophilic molecule, vitamin E is transported as part of circulating lipoproteins. Although its delivery to peripheral organs was shown to involve the non-specific holoparticle uptake of lipoprotein particles through their cellular receptors (Lemaire-Ewing et al, 2010), a molecular transfer of α-tocopherol, ie, the main isomer of vitamin E, was shown to be mediated in vivo by the plasma phospholipid transfer protein (PLTP) (Kostner et al, 1995; Desrumaux et al, 1999; Tzotzas et al, 2009). Thus, genetically engineered mice with PLTP deficiency were shown to accumulate α-tocopherol in circulating apolipoprotein B-containing lipoproteins at the expense of peripheral tissues (Jiang et al, 2002; Drouineaud et al, 2006; Desrumaux et al, 2010; Ogier et al, 2007). In particular, earlier studies from our group have shown a significant, 35% decrease in the vitamin E content of the brain in PLTP-deficient (PLTP-KO) mice, with a concomitant increase in the amount of the aging pigment lipofuscin in cortex and substantia nigra (Desrumaux et al, 2005).
Although PLTP-mediated vitamin E transfers were shown to have a key role in its pro-atherogenic and pro-thrombotic potency, it has not been established whether they can affect natural and/or pathological aging. To address this question, PLTP-KO mice were used in the present study as a valuable model to study the contribution of partial vitamin E deficiency to the etiology of AD. We used an established non-transgenic model of AD, ie, the intracerebroventricular injection of an oligomeric preparation of the amyloid-β (25–35) peptide fragment (Aβ25–35). This acute Aβ25–35 injection model has been previously shown to induce within 1 week highly pertinent pathomimetic alterations, including learning and memory deficits, cholinergic cell loss, oxidative stress, neuroinflammation, Aβ1–42 seeding, and Tau hyperphosphorylation in rodents (Maurice et al, 1996; Klementiev et al, 2007; Chavant et al, 2010).
MATERIALS AND METHODS
Animals
PLTP-KO mice and wild-type (WT) mice, bred on a homogeneous C57BL/6 background, were used in the present study. PLTP-KO mice were a kind gift from Dr X. C. Jiang's laboratory (SUNY Downstate Medical Center, New York, NY, USA) (Jiang et al, 1999). Mice were fed a standard chow diet (A03 diet, Safe, Augy, France) or a vitamin E-supplemented chow diet (A03 containing 800 mg/kg α-tocopherol acetate, Safe). Male mice were used between 3 and 4 months of age and behavioral experiments were performed between 1000 and 1700 hours. All animal procedures were conducted in strict adherence of the EU Directive 86-609, modified by the decrees 87-848 and 2001-464.
Drugs, Administration Procedures, and Sample Preparation
Aβ25–35 peptide was from Genepep (St Jean-de-Vedas, France). It was solubilized in sterile distilled water at a concentration of 3 mg/ml and stored at −20 °C until use. Before injection, the peptide was incubated at 37 °C for 4 days, leading to aggregation and formation of low molecular weight oligomers, as previously described (Maurice et al, 1996; Zussy et al, 2011). It was administered intracerebroventricularly (i.c.v.) in a final volume of 3 μl per mouse. Sterile water (vehicle, V) was used as a negative control, as we previously reported that i.c.v. injection of a scrambled Aβ35–25 control peptide led to similar results as seen in V-injected animals (Zussy et al, 2011). Control groups received a 3-μl injection of the vehicle (V) only. For behavioral testing, animals were used between days 7 and 10 post-injection. For biochemical assays, animals were killed 7 days post-injection by decapitation. Brain hippocampus and temporal cortex were harvested, frozen by immersion in liquid nitrogen, and stored at −80 °C until homogenization. Immediately before the assays, brain samples were homogenized by sonication in ice-cold phosphate buffer containing anti-phosphatases (Phostop, Roche, France) and a protease inhibitor cocktail (Complete, Roche). Samples were centrifuged (10 min, 1000 g, 4 °C), and supernatants were harvested for biochemical analyses.
Behavioral Analyses
Spontaneous alternation in the Y-maze
Short-term spatial memory assessment using the Y-maze paradigm was performed as previously described (Maurice et al, 1996). The percentage of alternation was calculated as (actual alternations/maximum alternations) × 100.
Novel object recognition
Mice were placed individually in a square open-field (50 cm × 50 cm × 50 cm high) made in white Plexiglas placed over a floor equipped with infrared light-emitting diodes. Animals were first habituated to the open-field during 10 min (session 0), and total locomotion and percentage of total time spent in the center of the open-field (25 × 25 cm2) were analyzed by videotracking (Viewpoint, Champagne-au-Mont-d'Or, France). After 24 h, session 1 was performed by placing two identical objects at defined positions in the open-field (at ¼ and ¾ of one diagonal). Each mouse was placed in the open-field, and the exploratory activity was recorded during a 10-min session using the Nosetrack protocol (ViewPoint), in terms of number and duration of contacts with each object. In session 2, 24 h later, the object in position No.2 was replaced by a novel object differing in color, shape, and texture from the familiar object. Each mouse was placed again in the open-field, and the exploratory activity was recorded during 10 min. The activity was analyzed similarly. The preferential exploration index was calculated as the ratio of the number of contacts with the object No.2 over the total number of contacts with the two objects. Calculations using the duration of contacts led to strictly similar results and are not presented. Animals showing <10 contacts with objects during session 1 or 2 were considered inactive and discarded from the calculations. In the present study, this accounted for 26/105 mice (24.8% attrition) in the WT group and 33/107 mice (30.8% attrition) in the PLTP-KO group. Although these percentages appeared rather high, no genotype-related effect was measured (P>0.05, Fisher's exact test).
Lipid Assays
Cholesterol content
Cholesterol was assayed in brain homogenates by gas chromatography–mass spectrometry (GC–MS). The extraction procedure, derivatization, and GC/MS quantification were conducted as previously described (Desrumaux et al, 2005).
Phosphatidylcholine and sphingomyelin contents
Lipids were extracted according to the method of Folch et al (1957). Phosphatidylcholine and sphingomyelin were assayed by liquid chromatography–mass spectrometry (LC–MS) as described in more detail in Supplementary Materials and Methods.
α-Tocopherol contents
α-Tocopherol was extracted from brain homogenates as previously described (Katsanidis and Addis, 1999) and assayed by GC–MS. The detailed procedure is available in Supplementary Materials and Methods.
Fatty acids contents
Fatty acids were extracted from saponified brain homogenates, derivatized to pentafluorobenzyl esters, and quantified by GC–MS following the method described by Arnauld et al (2009). The detailed procedure is described in Supplementary Materials and Methods.
Assessment of Cerebral Oxidative Stress
Cerebral oxidative stress was determined in cortex homogenates by measuring the oxidation rate of dichlorofluorescein diacetate into the fluorescent product dichlorofluorescein. Homogenates were incubated for 30 min in darkness in the presence of 10 mM dichlorofluorescein diacetate. Fluorescence intensity (λexc 485 nm, λem 530 nm) was read in a Fluoroskan Ascent spectrofluorimeter (Thermo Scientific), normalized for protein concentration, and used as a measure of oxidative stress index in each sample. Results are expressed as percentage of the control V-treated WT mice group. Superoxide dismutase (SOD) activity was measured using a spectrophotometric assay kit (Sigma-Aldrich).
Western Blotting
The detailed western blotting procedure is described in Supplementary Materials and Methods. Primary monoclonal antibodies used to detect phospho-epitopes of Tau protein were AT100 and AT180 (1 : 2000, Thermo Scientific). The anti-Tau (1 : 5000, Santa Cruz, Heidelberg, Germany) antibody was used for normalization. The secondary antibodies were horseradish peroxidase-conjugated goat anti-rabbit IgG or goat anti-mouse IgG, as appropriate (1 : 2000, Sigma-Aldrich).
Synaptophysin Assay
Synaptophysin content was quantified in cortex homogenates using an ELISA kit (USCN Life Science, Euromedex, Souffelweyersheim, France). Synaptophysin levels were expressed in percentage of the vehicle-treated WT mice group.
Measurement of Cytokine Levels
Interleukin-6 (IL-6) and tumor necrosis factor alpha (TNFα) were assayed in brain homogenates by cytometric bead array using a commercially available kit (CBA mouse inflammation kit; BD Biosciences, San Diego, CA, USA) according to the manufacturer's instructions and analyzed on a Guava flow cytometer (Millipore, Billerica, MA, USA).
Protein Assay
Protein concentration was determined in brain homogenates with a protein assay kit, using bovine serum albumin as standard (BCA protein assay kit, Pierce Perbio Science, Thermo Scientific).
Statistical Analyses
Results are expressed as mean±SEM. Spontaneous alternation performances (dose–response studies) were analyzed using a two-way ANOVA (F value), with genotype and treatment as independent factors, and group comparisons made using the Bonferroni post-hoc test. In subsequent experiments, alternation performances and biochemical data were analyzed using a one-way ANOVA with Bonferroni post-hoc analysis. Object recognition data were analyzed using a two-tailed one-sample t-test using 50% as the hazardous choice level. For reading clarity, all ANOVA values are reported in the figure legends. The level of statistical significance was P<0.05.
RESULTS
Lipid Composition of WT and PLTP-KO Mouse Brain
To assess the impact of PLTP deficiency on brain lipid composition, cholesterol, phosphatidylcholine, sphingomyelin, fatty acids, and α-tocopherol were assayed in brain homogenates from WT and PLTP-KO mice. As previously reported by our group (Desrumaux et al, 2005), PLTP deficiency was associated with a selective and significant 32% decrease in brain α-tocopherol content. By contrast, levels in cholesterol, phospholipids, sphingomyelin, and the n-3/n-6 fatty-acid ratio were similar to those measured in WT mice (Supplementary Table S1).
Impact of PLTP Deficiency on Short-term Spatial Memory and Long-term Recognition Memory After Aβ25–35 Peptide Injection: Dose-response Study
We analyzed the impact of a low, an intermediate, and a high dose of Aβ25–35 peptide on short- and long-term memory in WT and PLTP-KO mice. WT and PLTP-KO mice were injected with Aβ25–35 at 1, 3, or 9 nmol. Spontaneous alternation performances in the Y-maze paradigm were similar in WT and PLTP-KO mice after injection of the vehicle solution (Figure 1a). In WT mice, only the highest dose (9 nmol) led to a highly significant alternation impairment (white columns, Figure 1a). In PLTP-KO mice, significant deficits were observed at the two highest doses tested, 3 and 9 nmol (black columns, Figure 1a). The exploratory activity, assessed through the total number of arm entries, was reduced in PLTP-KO mice as compared with WT Aβ25–35 (3 or 9 nmol)-injected mice (Figure 1b).
Figure 1.

Aβ25–35-induced learning deficits in PLTP-KO mice: dose–response study in (a, b) the spontaneous alternation test and (c, d) novel object recognition test. WT and PLTP-KO mice received an i.c.v. injection of sterile water (V) or Aβ25–35, at 1, 3, or 9 nmol). After 7 days, mice were examined in the Y maze: (a) spontaneous alternation and (b) number of arm entries. At days 8–10, they were tested in the object recognition test: (c) session 1 and (d) session 2. White bars: WT mice, black bars: PLTP-KO mice. Two-way ANOVA with Bonferroni's post-test in panels (a) and (b) (n=8–16): F(3,98)=10.3, P<0.001 for treatment, F(1,100)=1.04, P>0.05 for genotype, F(3,98)=4.17, P<0.01 for the interaction in panel (a); F(3,98)=1.71, P>0.05 for treatment; F(1,100)=22.2, P<0.0001 for genotype; F(3,98)=0.30, P>0.05 for the interaction in panel (b). *P<0.001 vs V-treated WT group; φP<0.001 vs V-treated PLTP-KO group; #P<0.05 vs WT group (same treatment). One sample t-test in panels (c) and (d) (n=9–17): t(15)=5.87 (P<0.01 vs 50%) for WT/V, t(15)=2.23 (P<0.05 vs 50%) for WT/Aβ3, t(10)=2.89 (P<0.05 vs 50%) for KO/V in (d). °P<0.05, °°P<0.01 vs the hazard level (50%).
We next assessed long-term recognition memory using the novel object recognition procedure. During session 1, with two identical objects, no preferential exploration of the object in position No.2 was observed in any group of mice (Figure 1c). During session 2, V-treated and Aβ25–35 (3 nmol)-treated WT mice were able to discriminate the novel object, with an exploration index significantly >50%, while WT animals treated with the highest dose of peptide showed a deficit in recognition memory (white columns, Figure 1d). Among PLTP-KO mice, only V-treated mice showed a preferential exploration of the novel object, whereas Aβ25–35-injected group, with either 3 or 9 nmol, showed no preference for the novel object (black columns, Figure 1d).
It must be noted that the total distance traveled during session 0 was unchanged whatever the genotype or dose of amyloid peptide injected (data not shown). Moreover, during session 2, PLTP-KO mice, regardless of treatment, showed a significant decrease in object exploration, both in terms of total number of contacts and duration of contacts (data not shown). This decrease in exploratory behavior may be related to the increased anxiety state of PLTP-KO mice compared with WT mice (Desrumaux et al, 2005).
Impact of PLTP Deficiency and Vitamin E Supplementation on Short-term Spatial Memory and Long-term Recognition Memory After Aβ25–35 Injection
We next focused on the vulnerability of PLTP-KO mice treated with the intermediate Aβ25–35 dose of 3 nmol and analyzed the impact of a chronic (2 months) dietary vitamin E supplementation. Vitamin E supplementation led to a significant 49% rise in brain α-tocopherol levels in PLTP-KO mice that reached values similar to those measured in non-supplemented WT mice (data not shown).
In accordance with our dose–response study, a significant impairment of spontaneous alternation performance was observed in PLTP-KO mice, but not in WT mice, after injection of 3 nmol Aβ25–35 (Figure 2a). In the vitamin E-supplemented PLTP-KO group, the Aβ25–35 peptide injection failed to provoke a significant alternation deficit (Figure 2a), suggesting that vitamin E supplementation efficiently prevented amyloid toxicity in PLTP-KO animals. The slight decrease in exploratory behavior in the PLTP-KO mice group was not observed in this experiment (Figure 2b).
Figure 2.

Aβ25–35-induced learning deficits in PLTP-KO mice. WT and PLTP-KO mice, supplemented in vitamin E (KOE) or not (KO), received an i.c.v. injection of sterile water (V) or Aβ25–35, 3 nmol (Aβ3). After 7 days, mice were examined in the Y maze: (a) spontaneous alternation and (b) number of arm entries. At days 8–10, they were tested in the object recognition test: (c) session 1 and (d) session 2. One-way ANOVA and Bonferroni's post-test (n=11–17): F(4,65)=7.11, P<0.0001 in panel (a); F(4,65)=2.16, P=0.08 in panel (b). φP<0.0001 vs V-treated PLTP-KO group; #P<0.001 vs Aβ3-treated WT group; §P<0.0001 vs Aβ3-treated PLTP-KO group. One sample t-test in panels (c) and (d) (n=11–22): t(14)=3.61 (P<0.01 vs 50%) for KO/Aβ3 in (c), t(16)=5.77 (P<0.01 vs 50%) for WT/V, t(21)=2.25 (P<0.05 vs 50%) for WT/Aβ3, t(10)=3.19 (P<0.01 vs 50%) for KO/V in (d). °P<0.05, °°P<0.01 vs the hazard level (50%).
We next assessed long-term recognition memory in WT, PLTP-KO, and vitamin E-supplemented PLTP-KO mice after Aβ25–35 injection. During the habituation session, without objects, PLTP-KO mice displayed a slightly reduced locomotor activity compared with WT controls, whatever the treatment. However, none of the treatments decreased the presence in center, which would have impeded object recognition in the subsequent sessions (Supplementary Table S2). During session 1, the object No.2 exploration index was close to 50% in all the groups of mice, with the exception of Aβ25–35 (3 nmol)-treated PLTP-KO mice (Figure 2c). It must be noted that no difference was observed in terms of total number of object contacts and total duration of contacts among the groups (Supplementary Figures S1A and B). During session 2, V-treated WT mice, Aβ25–35-treated WT mice, and V-treated PLTP-KO mice were able to discriminate the novel object, whereas Aβ25–35-injected PLTP-KO mice showed no preferential exploration (Figure 2d). The exploration index of Aβ25–35-injected PLTP-KO mice supplemented with vitamin E showed a tendency towards a preference for the novel object, although it was not significantly different from 50% (Figure 2d). It must be noted that, as observed earlier in the dose–response study, PLTP-KO mice, regardless of treatment, showed a significant decrease in object exploration, both in terms of total number of contacts and duration of contacts during session 2 (Supplementary Figures S1C and D).
Impact of PLTP Deficiency and Vitamin E Supplementation on Cerebral Oxidative Stress Markers After Aβ25–35 Injection
In V-injected mice, PLTP deficiency was associated with a significant, 33% increase in cerebral oxidative stress index (Figure 3a). Injection of Aβ25–35 (3 nmol) induced a slight, 16% elevation of cerebral oxidative stress in WT mice, while virtually no increase was observed in PLTP-KO animals (Figure 3a). Seven days after Aβ25–35 peptide injection, oxidative stress was significantly higher in PLTP-KO than in WT mice (Figure 3a). A significant reduction of oxidative stress was observed in vitamin E-supplemented Aβ25–35-injected PLTP-KO mice, as compared with non-supplemented mice (P<0.05), indicating that vitamin E provided an efficient antioxidant protection under our experimental conditions (Figure 3a). To address the impact of PLTP deficiency and amyloid toxicity on enzymatic antioxidant protection systems in the brain, we measured the activity of SOD in cortex homogenates of WT and PLTP-KO mice. As shown in Figure 3b, PLTP deficiency was associated with a significant, 25% reduction in SOD activity in V-injected mice (P<0.05). In WT animals, Aβ25–35 injection led to a 30% decrease in SOD activity, while in PLTP-KO mice the impact of Aβ25–35 injection on this parameter was not significant (Figure 3b).
Figure 3.

(a) Oxidative stress index and (b) SOD activity in Aβ25–35-injected PLTP-KO mice. WT and PLTP-KO mice, supplemented in vitamin E (KOE) or not (KO), received an i.c.v. injection of sterile water (V) or Aβ25–35, 3 nmol (Aβ3). They were killed after 7 days. Oxidative stress was measured in cortex homogenates using a fluorescent probe. SOD activity was measured using a spectrophotometric assay kit. One-way ANOVA: F(4,102)=5.17, P<0.001, n=17–24 in (a); F(4,27)=4.90, P<0.01, n=3–7 in (b). *P<0.05, **P<0.01 vsV-treated WT group; #P<0.05 vs Aβ3-treated WT group; §P<0.05 vs Aβ3-treated PLTP-KO group; Bonferroni's test.
Impact of PLTP Deficiency on APP Processing, Tau Phosphorylation, and Synaptophysin Levels After Aβ25–35 Injection
Recent reports indicated that i.c.v. injection of the Aβ25–35 peptide (9 nmol) stimulates APP processing in mice (Chavant et al, 2010; Lahmy et al, 2011). Seven days after injection of the intermediate, 3 nmol dose of Aβ25–35 peptide, a non-significant 15% increase in Aβ1–42 content was measured in WT mice (Figure 4a). Interestingly, Aβ1–42 content was significantly higher in V-treated PLTP-KO mice as compared with V-treated WT mice (+25%, Figure 4a), but the Aβ25–35 injection failed to further increase Aβ1–42 in PLTP-KO mice. A significant reduction in Aβ1–42 content was observed in Aβ25–35-treated PLTP-KO mice with vitamin E supplementation (Figure 4a).
Figure 4.

(a) APP processing, (b, c) Tau phosphorylation and (d) synaptophysin levels in Aβ25–35-injected PLTP-KO mice. WT and PLTP-KO mice, supplemented in vitamin E (KOE) or not (KO), received an i.c.v. injection of sterile water (V) or Aβ25–35, 3 nmol (Aβ3). They were killed after 7 days. (a) Aβ1–42 content was quantified in cortex homogenates by ELISA. ANOVA: F(4,95)=4.03, P<0.01, n=14–23. (b, c) Tau phosphorylation was measured by western blots using AT100 and AT180 monoclonal antibodies and normalized to total Tau protein. (d) Synaptophysin was quantified in cortex homogenates by ELISA. ANOVA: F(4,92)=2.77, P<0.05, n=13–23 in (b); F(4,67)=2.71, P<0.05, n=13–17 in (c); F(4,68)=2.66, P<0.05, n=12–17 in (d). *P<0.05, **P<0.01 vs V-treated WT group; φP<0.05, φφP<0.01 vs V-treated PLTP-KO group; §P<0.01 vs Aβ3-treated PLTP-KO group; Bonferroni's test.
We also examined the levels of hyper- and abnormal phosphorylation of Tau protein in the mouse hippocampus using monoclonal antibodies directed against two different epitopes. AT100 recognizes Tau protein phosphorylated at Ser212 and Thr214, ie, aberrant phosphorylation epitopes, highly specifically observed in Alzheimer's pathology (Buée et al, 2000). AT180 recognizes Tau protein phosphorylated at Thr231 and Ser235, two sites where phosphorylation occurs under physiological conditions but that undergo hyperphosphorylation under pathological conditions (Buée et al, 2000). Levels of both AT100- and AT180-positive Tau epitopes were detected at similar levels in V-injected WT and PLTP-KO mice (Figure 4b). Aβ25–35 injection induced a significant rise in AT100-positive signal in both WT (+32%) and PLTP-KO mice (+43%) (Figure 4b). AT180-positive signal was markedly increased in PLTP-KO mice after Aβ25–35 injection (+76%), while a more modest rise was observed in WT mice (+38%) (Figure 4c). A significant reduction of AT100-positive signal was observed in vitamin E-supplemented Aβ25–35-treated PLTP-KO mice, as compared with non-supplemented PLTP-KO mice (Figure 4b), while no change in AT180-positive signal was observed upon vitamin E supplementation (Figure 4c).
To evaluate the impact of amyloid toxicity on synaptic loss, the synaptic vesicle marker synaptophysin was quantified in the cortex of WT and PLTP-KO mice. A significant reduction of synaptophysin level was observed in V-treated PLTP-KO mice, as compared with WT mice (Figure 4d). Aβ25–35 injection led to a significant decrease in synaptophysin levels in WT mice but not in PLTP-KO mice, and a significant rise in synaptophysin level was observed in vitamin E-supplemented, Aβ25–35-injected PLTP-KO mice (Figure 4d).
Impact of PLTP Deficiency on Neuroinflammation Markers After Aβ25–35 Injection
The extent of brain inflammation was analyzed by measuring the contents in IL-6 and TNFα in cortex homogenates 7 days after vehicle or Aβ25–35 injection. Both IL-6 and TNFα levels were significantly increased in Aβ25–35 peptide-injected compared with V-injected WT animals (Figure 5a and b). Both cytokine levels were also increased in PLTP-KO mice after Aβ25–35 injection but to a lesser, non-significant extent (Figure 5a and b). Vitamin E supplementation in PLTP-KO mice failed to affect IL-6 and TNFα levels, as compared with Aβ25–35-treated PLTP-KO mice (Figure 5b).
Figure 5.
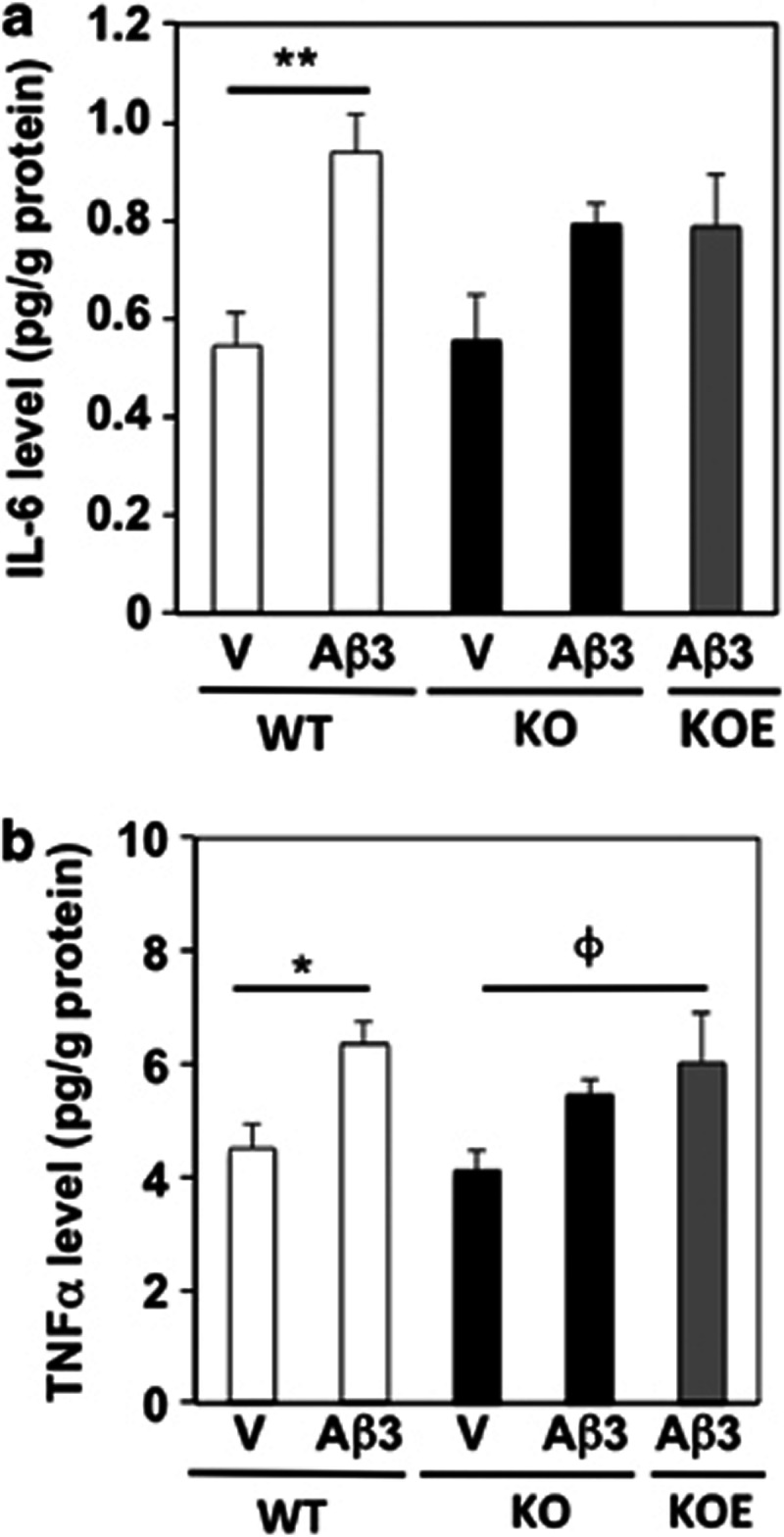
Levels of the neuroinflammatory cytokines (a) IL-6 and (b) TNFα in Aβ25–35-injected PLTP-KO mice. WT and PLTP-KO mice, supplemented in vitamin E (KOE) or not (KO), received an i.c.v. injection of sterile water (V) or Aβ25–35, 3 nmol (Aβ3). They were killed after 7 days. The contents in inflammatory cytokines IL-6 and TNFα were measured in temporal cortex homogenates by flow cytometry. One-way ANOVA: F(4,23)=3.89, P<0.05, n=4–6 in panel (a); F(4,23)=3.59, P<0.05, n=4–6 in panel (b). *P<0.05, **P<0.01 vs V-treated WT group; φP<0.05 vs V-treated PLTP-KO group; Bonferroni's test.
DISCUSSION
PLTP has emerged over the past decade as an important in vivo determinant for the biodistribution of vitamin E, the most important lipid soluble antioxidant in biological membranes (Tzotzas et al, 2009). We previously reported the role of PLTP in vitamin E delivery to the brain (Desrumaux et al, 2005). We here show that partial brain vitamin E depletion associated with PLTP deficiency results in an increased memory decline in response to the toxic effect of Aβ25–35 oligomers, which can be counteracted through dietary vitamin E supplementation. PLTP hereby appears as a previously unrecognized actor of the brain endogenous neuroprotective response.
Oxidative stress is recognized as a hallmark of natural aging as well as of neurodegenerative disorders, and the potential of antioxidants to help prevent or treat neurodegeneration has been extensively studied (for recent reviews, see Clark et al, 2010; Viña et al, 2011). In humans, a functional brain vitamin E deficiency, suggesting an impairment of its delivery to neuronal cells, has been described in ApoE4 patients with AD (Mas et al, 2006), and several epidemiological studies suggested that dietary intake of vitamin E is inversely related to the long-term risk of dementia and AD (Devore et al, 2010). Although various mouse models have been created to study the relationship between oxidative stress, antioxidants, and natural or pathological aging, most of them did not directly address the contribution of a selective reduction of vitamin E status to the initiation and/or progression of the neurological damage. Only in α-tocopherol transfer protein (α-TTP)-deficient mice, systemic vitamin E deficiency was proven to enhance the phenotype in a mouse model of AD (Nishida et al, 2006). Later on, α-TTP deficiency was shown to be associated with increased accumulation and reduced clearance of the Aβ peptide in the brain and plasma (Nishida et al, 2009). It is noteworthy that the reduction of vitamin E content in α-TTP-deficient mouse tissue is drastic and ubiquitous and does not reflect a potential dietary deficiency as observed in human populations. In this study, the PLTP-KO mouse model was used as a useful genetic tool to show that a moderate, chronic oxidative stress induced by partial brain vitamin E depletion does not by itself induce memory deficits but makes more susceptible to the development of memory impairment in response to amyloid toxicity. In accordance with our earlier report, PLTP deficiency was not associated with memory decline in young healthy mice, in contrast to α-TTP deficiency (Desrumaux et al, 2005; Gohil et al, 2004). In Aβ25–35-injected mice, however, PLTP deficiency was associated with an increased memory decline in response to the neurotoxic effect of the amyloid peptide. As previously reported (Ruan et al, 2010), Aβ25–35 injection was associated with a significant rise in oxidative stress and a decrease in SOD activity in WT mice. In PLTP-KO mice, reduced brain vitamin E content was associated with increased oxidative stress and decreased SOD activity under basal conditions, and these parameters were marginally affected by Aβ25–35. Although vitamin E supplementation effectively restored normal oxidative status parameters and prevented Aβ25–35-induced short-term memory impairment in PLTP-KO mice, the peptide effect on long-term recognition memory was not fully prevented by vitamin E supplementation. Taken together, these observations indicate that the accentuated Aβ-induced memory impairment in PLTP-KO mice may relate, at least in part, to pre-existing lower vitamin E content and higher oxidative stress in brain tissue, two related parameters that may deserve attention as potential reliable biomarkers of vulnerability to amyloid pathology. With regard to the etiology of AD, it has been reported that humans destined to develop the illness have detectable biochemical and histopathological abnormalities two decades or more before clinical symptoms, and it is increasingly recognized that a complex interplay between genetic and environmental components (among which oxidative stress) may begin early in life and act together on the mechanisms behind cognitive impairment (Selkoe, 2012). These risk factors may remain latent (putatively because of the activation of compensatory pathways), and cognitive impairment may ensue only following appearance of one or more additional traumatic events or conditions, such as amyloid toxicity or aging. The sporadic nature of 90% of AD cases, as well as the late age of onset of the disease are coherent with this theory, and results of the present study suggest that a moderate and chronic oxidative stress per se may remain silent under normal conditions but render more susceptible to the development of memory impairment in the presence of other triggering factors.
Two hypotheses can be proposed to explain the lack of effect of vitamin E supplementation on long-term recognition memory: (i) PLTP may not only have a role in vitamin E delivery to the brain, but may also be necessary for proper subcellular distribution and use of vitamin E by the cell. Vitamin E displays not only antioxidant properties but also signaling-modifying properties that may relate to its incorporation in lipid raft domains of the plasma membrane (Lemaire-Ewing et al, 2010). PLTP may be necessary for this incorporation and to explain the non-antioxidant properties of vitamin E, and vitamin E supplementation in PLTP-KO mice may not allow to recover all the functions of this vitamin. (ii) Oxidative stress/antioxidants may not be involved in long-term memory. A dichotomic effect on short- and long-term memory has been recently reported for the γ-secretase inhibitor BMS-299 897. Despite its ability to counteract oxidative stress, Aβ1–42 seeding, and short-term memory loss after Aβ25–35 injection in mice, the compound had no effect on long-term memory deficits (Meunier et al, 2011).
The significant elevation of basal Aβ1–42 level in PLTP-KO mice compared with WT mice may be a consequence of decreased vitamin E levels and elevated oxidative stress in these mice, as Aβ1–42 levels were significantly lower in vitamin E-supplemented PLTP-KO mice as compared with non-supplemented animals. Indeed, it has been reported that vitamin E can act as a regulator of APP processing and clearance of Aβ entities, both in vitro and in vivo (Nishida et al, 2009; Conte et al, 2004). In addition to Aβ1–42 seeding, Aβ25–35 injection led to the appearance of another main histopathological feature of AD, the increase in Tau phosphorylation (Klementiev et al, 2007; Lahmy et al, 2011). Interestingly, the induction of Tau phosphorylation on pathological epitopes (AT-100) by Aβ25–35 was of similar extent in WT and PLTP-KO mice but was restored after vitamin E supplementation, suggesting oxidative stress-dependent mechanisms in Aβ-induced pathological Tau phosphorylation. Such a relationship was recently proposed by Lloret et al (2011), who suggested that Aβ causes mitochondrial oxidative stress and increases production of reactive oxygen species, which results in an upregulation of the expression of the regulator of calcineurin gene RCAN1. RCAN1 proteins then both inhibit Tau dephosphorylation by calcineurin and induce expression of the GSK3β kinase, resulting in Tau hyperphosphorylation (Lloret et al, 2011). A complex interplay between Aβ and Tau involving activation of the transcription factor p53 as well as of GSK3β by oxidative stress has also been proposed by Proctor and Gray (2012). Tau hyperphosphorylation on the physiological epitope recognized by AT-180 appeared to be augmented in PLTP-KO mice as compared with WT mice but in a manner insensitive to vitamin E supplementation. This observation is concordant with in vitro findings reported by Dong et al (2009), showing that PLTP can modulate the PI3K/Akt signaling pathway, thus reducing GSK3β activity and Tau phosphorylation on the Thr231 residue in human neuroblastoma cells.
Synaptic loss in specific brain structures of AD patients significantly correlates with the severity of their cognitive symptoms (Scheff and Price, 2006), and a reduction of the synaptic density has been observed in transgenic mice overexpressing the V717F APP mutant (Games et al, 1995). Aβ25–35 injection also significantly decreased the cortical levels of synaptophysin, the major synaptic vesicle protein, in WT mice. In PLTP-KO mice, basal synaptophysin levels were reduced compared with those measured in WT mice and were not modified by Aβ25–35. Thus, a developmental compensation in terms of efficiency of the synaptic transmission may explain the preserved memory ability of PLTP-KO mice under normal conditions, while under conditions of amyloid toxicity, the decreased number of synapses may be sufficient to uncover synaptic alterations resulting in memory impairments. The fact that vitamin E efficiently prevented both Aβ25–35-induced synaptic loss and the deleterious effects of the peptide on short-term memory sustains this hypothesis. In addition, this finding is in accordance with a recent report showing the potential of vitamin E to inhibit oxidative-stress-induced denaturation of nerve terminal proteins involved in neurotransmission (Kaneai et al, 2012).
Beyond Aβ peptide accumulation, Tau hyperphosphorylation, and synaptic loss, recent evidence suggests that inflammation may be another active contributor to neurodegenerative disease progression and chronicity. In the present study, and in accordance with previous reports (Chavant et al, 2010), we observed a significant induction of the production of the pro-inflammatory cytokines IL-6 and TNFα in WT mice 1 week after Aβ25–35. Although a similar tendency was observed in PLTP-KO mice, the observed increases did not reach statistical significance. This observation may relate to the previously reported anti-inflammatory effect of PLTP deficiency in mice (Schlitt et al, 2004). Interestingly, although Aβ25–35-induced behavioral impairment was efficiently prevented by vitamin E supplementation of PLTP-KO mice, the latter had no effect on cytokine levels, which remained significantly higher than those measured in control mice. This observation does not sustain a significant impact of neuroinflammation in Aβ25–35-induced memory decline in PLTP-KO mice and particularly suggests that astroglial and, more likely, microglial activation responses are differentially regulated by oxidative stress.
Although human observational epidemiology studies are, in general, consistent with the hypothesis that there is an inverse relationship between vitamin E levels and intake, cognitive function, and ultimately the risk of developing AD, most randomized clinical trials with antioxidants did not fulfill the promises of those studies (Usoro and Mousa, 2010). The latter observation does not necessarily mean that the oxidative stress hypothesis of AD is not valid, but might rather be explained, at least in part, by an insufficient knowledge and understanding of the actors that govern vitamin E metabolism and bioavailability, among which we point out here the role of PLTP. Interestingly, negative clinical trials most of the time lack important informations such as surrogate markers for an in vivo therapeutic effect of the antioxidant. Thus, it would be important to get new information on the endogenous antioxidant levels of the participating subjects, as well as to design new markers to assess the ability of individuals to respond to vitamin E treatment. Our findings suggest that PLTP activity should be evaluated as such a marker.
Acknowledgments
This work was supported by INSERM external ressources, University of Montpellier 2 external ressources, and by a French Government grant managed by the French National Research Agency under the program ‘Investissements d'Avenir' with reference ANR-11-LABX-0021. Funding sources were not involved in any other way in this study.
JM and VV are employees of AMYLGEN. TM is member of the scientific boards of AMYLGEN and ANAVEX Life Sciences. The companies have no decisional or funding role in this work. All the other authors have no conflict of interest to declare.
Footnotes
Supplementary Information accompanies the paper on the Neuropsychopharmacology website (http://www.nature.com/npp)
Supplementary Material
References
- Arnauld S, Fidaleo M, Clémencet MC, Chevillard G, Athias A, Gresti J, et al. Modulation of the hepatic fatty acid pool in peroxisomal 3-ketoacyl-CoA thiolase B-null mice exposed to the selective PPARalpha agonist Wy14,643. Biochimie. 2009;91:1376–1386. doi: 10.1016/j.biochi.2009.09.004. [DOI] [PubMed] [Google Scholar]
- Buée L, Bussière T, Buée-Scherrer V, Delacourte A, Hof PR.2000Tau protein isoforms, phosphorylation and role in neurodegenerative disorders Brain Res Brain Res Rev 3395–130.Review. [DOI] [PubMed] [Google Scholar]
- Chavant F, Deguil J, Pain S, Ingrand I, Milin S, Fauconneau B, et al. Imipramine, in part through tumor necrosis factor alpha inhibition, prevents cognitive decline and beta-amyloid accumulation in a mouse model of Alzheimer's disease. J Pharmacol Exp Ther. 2010;332:505–514. doi: 10.1124/jpet.109.162164. [DOI] [PubMed] [Google Scholar]
- Clark TA, Lee HP, Rolston RK, Zhu X, Marlatt MW, Castellani RJ, et al. Oxidative stress and its implications for future treatments and management of Alzheimer disease. Int J Biomed Sci. 2010;6:225–227. [PMC free article] [PubMed] [Google Scholar]
- Conte V, Uryu K, Fujimoto S, Yao Y, Rokach J, Longhi L, et al. Vitamin E reduces amyloidosis and improves cognitive function in Tg2576 mice following repetitive concussive brain injury. J Neurochem. 2004;90:758–764. doi: 10.1111/j.1471-4159.2004.02560.x. [DOI] [PubMed] [Google Scholar]
- Desrumaux C, Deckert V, Athias A, Masson D, Lizard G, Palleau V, et al. Plasma phospholipid transfer protein prevents vascular endothelium dysfunction by delivering alpha-tocopherol to endothelial cells. FASEB J. 1999;13:883–892. doi: 10.1096/fasebj.13.8.883. [DOI] [PubMed] [Google Scholar]
- Desrumaux C, Deckert V, Lemaire-Ewing S, Mossiat C, Athias A, Vandroux D, et al. Plasma phospholipid transfer protein deficiency in mice is associated with a reduced thrombotic response to acute intravascular oxidative stress. Arterioscler Thromb Vasc Biol. 2010;30:2452–2457. doi: 10.1161/ATVBAHA.110.207654. [DOI] [PubMed] [Google Scholar]
- Desrumaux C, Risold PY, Schroeder H, Deckert V, Masson D, Athias A, et al. Phospholipid transfer protein (PLTP) deficiency reduces brain vitamin E content and increases anxiety in mice. FASEB J. 2005;19:296–297. doi: 10.1096/fj.04-2400fje. [DOI] [PubMed] [Google Scholar]
- Devore EE, Grodstein F, van Rooij FJ, Hofman A, Stampfer MJ, Witteman JC, et al. Dietary antioxidants and long-term risk of dementia. Arch Neurol. 2010;67:819–825. doi: 10.1001/archneurol.2010.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong W, Albers JJ, Vuletic S. Phospholipid transfer protein reduces phosphorylation of tau in human neuronal cells. J Neurosci Res. 2009;87:3176–3185. doi: 10.1002/jnr.22137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drouineaud V, Lagrost L, Klein A, Desrumaux C, Le Guern N, Athias A, et al. Phospholipid transfer protein deficiency reduces sperm motility and impairs fertility of mouse males. FASEB J. 2006;20:794–796. doi: 10.1096/fj.05-5385fje. [DOI] [PubMed] [Google Scholar]
- Folch J, Lees M, Sloane Stanley GH. A simple method for the isolation and purification of total lipids from animal tissues. J Biol Chem. 1957;226:497–509. [PubMed] [Google Scholar]
- Games D, Adams D, Alessandrini R, Barbour R, Berthelette P, Blackwell C, et al. Alzheimer-type neuropathology in transgenic mice overexpressing V717F beta-amyloid precursor protein. Nature. 1995;373:523–527. doi: 10.1038/373523a0. [DOI] [PubMed] [Google Scholar]
- Gohil K, Godzdanker R, O'Roark E, Schock BC, Kaini RR, Packer L, et al. Alpha-tocopherol transfer protein deficiency in mice causes multi-organ deregulation of gene networks and behavioral deficits with age. Ann NY Acad Sci. 2004;1031:109–126. doi: 10.1196/annals.1331.012. [DOI] [PubMed] [Google Scholar]
- Jiang XC, Bruce C, Mar J, Lin M, Ji Y, Francone OL, et al. Targeted mutation of plasma phospholipid transfer protein gene markedly reduces high-density lipoprotein levels. J Clin Invest. 1999;103:907–914. doi: 10.1172/JCI5578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang XC, Tall AR, Qin S, Lin M, Schneider M, Lalanne F, et al. Phospholipid transfer protein deficiency protects circulating lipoproteins from oxidation due to the enhanced accumulation of vitamin E. J biol Chem. 2002;277:31850–31856. doi: 10.1074/jbc.M205077200. [DOI] [PubMed] [Google Scholar]
- Kaneai N, Arai M, Takatsu H, Fukui K, Urano S. Vitamin E inhibits oxidative stress-induced denaturation of nerve terminal proteins involved in neurotransmission. J Alzheimers Dis. 2012;28:183–189. doi: 10.3233/JAD-2011-111133. [DOI] [PubMed] [Google Scholar]
- Katsanidis E, Addis PB. Novel HPLC analysis of tocopherols, tocotrienols, and cholesterol in tissue. Free Radic Biol Med. 1999;27:1137–1140. doi: 10.1016/s0891-5849(99)00205-1. [DOI] [PubMed] [Google Scholar]
- Klementiev B, Novikova T, Novitskaya V, Walmod PS, Dmytriyeva O, Pakkenberg B, et al. A neural cell adhesion molecule-derived peptide reduces neuropathological signs and cognitive impairment induced by Abeta25–35. Neuroscience. 2007;145:209–224. doi: 10.1016/j.neuroscience.2006.11.060. [DOI] [PubMed] [Google Scholar]
- Kostner GM, Oettl K, Jauhiainen M, Ehnholm C, Esterbauer H, Dieplinger H. Human plasma phospholipid transfer protein accelerates exchange/transfer of alpha-tocopherol between lipoproteins and cells. Biochem J. 1995;305 (Part 2:659–667. doi: 10.1042/bj3050659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahmy V, Meunier J, Malmström S, Villard V, Vamvakides A, Maurice T.2011The novel tetrahydrofuran derivative ANAVEX2-73 attenuated GSK-3ß activation, Tau hyperphosphorylation and endogenous Aß1-42 seeding in a nontransgenic Alzheimer's disease model in mice. Program No. 352.17. 2011 Neuroscience Meeting Planner Society for Neuroscience: Washington, DC; Online. [Google Scholar]
- Lemaire-Ewing S, Desrumaux C, Néel D, Lagrost L.2010Vitamin E transport, membrane incorporation and cell metabolism: is alpha-tocopherol in lipid rafts an oar in the lifeboat Mol Nutr Food Res 54631–640.Review. [DOI] [PubMed] [Google Scholar]
- Lloret A, Badia MC, Giraldo E, Ermak G, Alonso MD, Pallardó FV, et al. Amyloid-β toxicity and tau hyperphosphorylation are linked via RCAN1 in Alzheimer's disease. J Alzheimers Dis. 2011;27:701–709. doi: 10.3233/JAD-2011-110890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangialasche F, Kivipelto M, Mecocci P, Rizzuto D, Palmer K, Winblad B, et al. High plasma levels of vitamin E forms and reduced Alzheimer's disease risk in advanced age. J Alzheimers Dis. 2010;20:1029–1037. doi: 10.3233/JAD-2010-091450. [DOI] [PubMed] [Google Scholar]
- Mas E, Dupuy AM, Artero S, Portet F, Cristol JP, Ritchie K, et al. Functional Vitamin E deficiency in ApoE4 patients with Alzheimer's disease. Dement Geriatr Cogn Disord. 2006;21:198–204. doi: 10.1159/000090868. [DOI] [PubMed] [Google Scholar]
- Maurice T, Lockhart BP, Privat A. Amnesia induced in mice by centrally administered beta-amyloid peptides involves cholinergic dysfunction. Brain Res. 1996;706:181–193. doi: 10.1016/0006-8993(95)01032-7. [DOI] [PubMed] [Google Scholar]
- Meunier J, Malmstrom S, Villard V, Givalois L, Maurice T.2011Impact of gamma-secretase inhibitor, BMS-299897, on Aβ1-42 seeding, oxidative stress and memory deficits in the Aβ25-35 nontransgenic mouse model of Alzheimer's disease. Program No. 352.18. 2011 Neuroscience Meeting Planner Society for Neuroscience: Washington, DC; Online. [Google Scholar]
- Nishida Y, Ito S, Ohtsuki S, Yamamoto N, Takahashi T, Iwata N, et al. Depletion of vitamin E increases amyloid beta accumulation by decreasing its clearances from brain and blood in a mouse model of Alzheimer disease. J Biol Chem. 2009;284:33400–33408. doi: 10.1074/jbc.M109.054056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishida Y, Yokota T, Takahashi T, Uchihara T, Jishage K, Mizusawa H. Deletion of vitamin E enhances phenotype of Alzheimer disease model mouse. Biochem Biophys Res Commun. 2006;350:530–536. doi: 10.1016/j.bbrc.2006.09.083. [DOI] [PubMed] [Google Scholar]
- Ogier N, Klein A, Deckert V, Athias A, Bessède G, Le Guern N, et al. Cholesterol accumulation is increased in macrophages of phospholipid transfer protein-deficient mice: normalization by dietary alpha-tocopherol supplementation. Arterioscler Thromb Vasc Biol. 2007;27:2407–2412. doi: 10.1161/ATVBAHA.107.151753. [DOI] [PubMed] [Google Scholar]
- Proctor CJ, Gray DA. A unifying hypothesis for familial and sproadic Alzheimer's disease. Int J Alzheimers Dis. 2012;2012:978742. doi: 10.1155/2012/978742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricciarelli R, Argellati F, Pronzato MA, Domenicotti C.2007Vitamin E and neurodegenerative diseases Mol Aspects Med 28591–606.Review. [DOI] [PubMed] [Google Scholar]
- Ruan CJ, Zhang L, Chen DH, Li Z, Du GH, Sun L. Effects of trans-2,4-dimethoxystibene against the neurotoxicity induced by Abeta(25-35) both in vitro and in vivo. Neurosci Res. 2010;67:209–214. doi: 10.1016/j.neures.2010.03.009. [DOI] [PubMed] [Google Scholar]
- Scheff SW, Price DA.2006Alzheimer's disease-related alterations in synaptic density: neocortex and hippocampus J Alzheimers Dis 9(3 Suppl101–115.Review. [DOI] [PubMed] [Google Scholar]
- Schlitt A, Liu J, Yan D, Mondragon-Escorpizo M, Norin AJ, Jiang XC. Anti-inflammatory effects of phospholipid transfer protein (PLTP) deficiency in mice. Biochim Biophys Acta. 2004;1733:187–191. doi: 10.1016/j.bbalip.2004.12.011. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Preventing Alzheimer's disease. Science. 2012;337:1488–1492. doi: 10.1126/science.1228541. [DOI] [PubMed] [Google Scholar]
- Sung S, Yao Y, Uryu K, Yang H, Lee VM, Trojanowski JQ, et al. Early vitamin E supplementation in young but not aged mice reduces Abeta levels and amyloid deposition in a transgenic model of Alzheimer's disease. FASEB J. 2004;18:323–325. doi: 10.1096/fj.03-0961fje. [DOI] [PubMed] [Google Scholar]
- Tzotzas T, Desrumaux C, Lagrost L.2009Plasma phospholipid transfer protein (PLTP): review of an emerging cardiometabolic risk factor Obes Rev 10403–411.Review. [DOI] [PubMed] [Google Scholar]
- Usoro OB, Mousa SA.2010Vitamin E forms in Alzheimer's disease: a review of controversial and clinical experiences Crit Rev Food Sci Nutr 50414–419.Review. [DOI] [PubMed] [Google Scholar]
- Viña J, Lloret A, Giraldo E, Badia MC, Alonso MD.2011Antioxidant pathways in Alzheimer's disease: possibilities of intervention Curr Pharm Des 173861–3864.Review. [DOI] [PubMed] [Google Scholar]
- Zussy C, Brureau A, Delair B, Marchal S, Keller E, Ixart G, et al. Time-course and regional analyses of the physiopathological changes induced after cerebral injection of an amyloid β fragment in rats. Am J Pathol. 2011;179:315–334. doi: 10.1016/j.ajpath.2011.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.