

Introduction
Autism is a debilitating neuropsychiatric disorder affecting children and young adults during the most vulnerable points in psychological and social development. With an estimated incidence of one in 88 births, autism spectrum disorder is characterized by impaired social interaction and communication, and restricted interests and stereotyped behaviors that occur before the age of three. Epidemiologic studies have suggested a role for environmental factors in the development of both autism and schizophrenia (Patterson 2009). A well-documented environmental risk factor for developing these neuropsychiatric diseases is maternal infection during pregnancy.
Multiple studies have correlated a high prevalence of influenza infection with a high rate of schizophrenic births several months afterwards (Brown 2011; Mednick et al. 1988). The most direct evidence of this relationship comes from prospective studies involving banked serum from women who were pregnant in the 1960s, and follow-up work examined their offspring over 20 years later. Among mothers with serologically-verified influenza infection, schizophrenia risk was increased 3–7 fold in the offspring (Brown and Susser 2002). Similar evidence links other maternal infections, such as rubella, toxoplasma, and genital/reproductive infections, with subsequent schizophrenia in the offspring (Babulas et al. 2006; Brown et al. 2005).
A correlation between autism and maternal viral infection was recently established by a large study of the Danish medical records (Atladottir et al. 2010). Autism risk is reportedly increased almost 200-fold with a maternal rubella infection (Chess 1977). Maternal infections including toxoplasmosis, syphilis, varicella, and rubeola during pregnancy are associated with autism in small case studies, summarized in Ciaranello and Ciaranello (1995). These findings suggest a potential role for infection during pregnancy in the etiology of at least some cases of autism.
In studies aimed at modeling a viral infection during pregnancy in mice, a non-infectious Toll-like receptor 3 (TLR3) agonist, polyI:C has been injected into the mother during pregnancy and importantly this intervention altered the behavior of the adult offspring in a manner resembling autism (Patterson 2009). The mice show decreased social interaction, abnormal vocalizations, increased anxiety, and poor performance on latent inhibition and prepulse inhibition tests. Concurrent with these behavioral changes, some histologic changes reminiscent of the human disease have been documented such as fewer Purkinje cells in the cerebellum as seen in autism (Shi et al. 2009) and reduced cortical parvalbumin reactivity as seen in schizophrenia (Meyer et al. 2008). The mechanism by which maternal immune activation causes the behavioral changes has also been investigated and implicates the cytokines, particularly interleukin-6, which are produced at the site of immune stimulation but then travel throughout the maternal circulation to interfere with fetal development (Hsiao and Patterson 2011; Smith et al. 2007). Importantly, the abnormal levels of pro-inflammatory cytokines and growth factors present in the maternal immune-exposed fetal brains could potentially alter proliferation or migration of the neural progenitor cells destined to form the cerebral cortex.
Studies of the human autism brain are also consistent with the notion that autism might arise from an activation of the immune system to trigger abnormal brain development. Analyses of postmortem brain tissue and CSF from individuals with autism have established that cytokines, chemokines, and growth factors are increased (Vargas et al. 2005). Similarly, transcripts for immunity-related genes are elevated in autism postmortem brain tissues (Voineagu et al. 2011). In addition to the increase of cyto-kines, chemokines, and growth factors, individuals with autism are reported to display an increased head circumference, brain volume, and neuron number in cortex at the onset of symptoms in early childhood (Schumann and Nordahl 2010; Courchesne et al. 2011). Potentially also related to the excess of cortical neurons, minicolumns, a vertically oriented cluster of neurons spanning the cortical layers within the cerebral cortex, are more numerous and display a greater dispersion due to a decreased alignment of pyramidal cells along the core axis (Casanova and Trippe 2009). In aggregate, these studies support a model where maternal immune activity during pregnancy might promote an increase of cytokine, chemokines, and growth factors that promotes the generation of an excess number of cortical neurons in-utero.
We directly test the hypothesis that maternal immune activation during pregnancy is sufficient to trigger an expansion of the cortical neuron population by measuring cortical thickness and cell density in the neonatal cortex at postnatal day 0 (P0) following in utero maternal intraperi-toneal polyI:C injections.
Methods
Mice were housed in an AALAC-approved animal facility with ad libitum food and water on a 12:12 light cycle, and all procedures were approved by the IUCAC. Time-pregnant CD-1 mice (Charles River, Wilmington, MA) were injected intra-peritoneally daily for 5 days with 5 mL/kg of 0.9 % saline (controls) or saline with 5 mg/kg poly I:C (Sigma-Aldrich). Dosage was based on poly(I:C) content of 10 % w/w potassium salt. Two injection time points were used: mid-pregnancy (E10-14) which roughly corresponds to early human second trimester, and late-pregnancy (E14-18) corresponding roughly to late second/early third trimester of human gestation. N(control, E10-14, E14-18) = 5,6,4 pups from at least 3 independent litters.
At birth, postnatal day 0 (P0), the mouse pups were sacrificed. The brains were extracted and fixed in 10 % formalin for 2 days. They were then processed with ethanol and xylene, and embedded in paraffin. 5 μm thick coronal sections were cut and stained with hematoxylin and eosin. The same part of the somatosensory cortex (S1HL region) was examined in each mouse pup and was digitally photographed and mapped to distinguish cortical layers, I–VI. Since the six layers of the fetal mouse cortex are less distinct than in the adult, layers II and III were counted together as II–III and layers IV and V were counted as IV–V. For each animal, 2–5 slices were analyzed and then averaged prior to statistical testing.
Using ImageJ software, cells in these layers were counted using a semi-automated procedure that consisted of the following steps: (1) digital images were cropped to isolate layers of interest; (2) an intensity threshold was applied to eliminate background neuropil; (3) the “Smooth” function was applied to the image to reduce noise; (4) the “Watershed” function was used to delineate boundaries between nuclei; (5) the “Analyze Particles” function was used to count profiles approximating the size of cell nuclei. We confirmed that our counting methods were accurate by comparing a manual cell count and an automated cell count in several sections. Both methods counted similar numbers of cells (data not shown). Cortical thickness was measured using the “measure” function on straight line selections extending from the subcortical white matter to the pial surface. Differences in cell density and cortical thickness between groups were measured using one or two way ANOVA where appropriate, and all data are expressed as mean ± SEM.
Results
Poly(I:C) exposed pups were born after a normal gestation time, and appeared superficially normal. However, a marked morphologic difference in the composition of the cortical mantle was found in mouse pups exposed to maternal intra-peritoneal polyI:C during both mid- and late-pregnancy, as compared to controls. Figure 1a shows cortical sections from controls, E10-14 exposed pups, and E14-18 exposed pups. Both experimental groups show darker, denser tissue in all cortical layers. The increase in density is especially apparent in layer II–III in the third-trimester exposed group. The background neuropil is also coarser and more chromophilic in both experimental groups. The results of the semi-automated cell counting procedure is illustrated in Fig. 1b, demonstrating the ability to accurately count the number of cells present.
Fig. 1.

Maternal immune activation triggers cortical neuron overgrowth. a Pregnant mice were injected daily either on embryonic days 10–14 or 14–18. Representative H&E-stained microscopic sections (20x objective) of somatosen-sory cortex S1HL from each condition are shown. Cortical layers I, II–III, IV–V, and VI are labeled. All cell layers, but especially layers II–III, display an increased cell density in pups from polyI:C-injected compared to uninjected mothers. Scale bar 100 μm. b Upper half, layer II/III of cortex stained with H&E (top panel) compared to semi-automated cell count (bottom panel) at 60x magnification. Lower half, layer IV/V of cerebral cortex stained with H&E (top panel) compared to automated cell count (bottom panel) at a 60x magnification. Scale bar 25 μm C. The thickness of the cortex was significantly greater in polyI:C exposed animals (* p <0.05)
Cortical thickness was significantly increased in both experimental groups. Measured from the ventral border of layer VI to the pial surface, control cortices were 411± 9.7 μm thick, compared to 534 ± 38.6 μm for mid-pregnancy exposed animals (30 % increase) and 530± 9.8 μm for the late-pregnancy exposed animals (29 % increase). One way ANOVA revealed a significant difference between groups (F2,14 = 6.175 p = 0.0143) and post-hoc tests revealed a significant difference between control and mid (p<0.05) and late (p<0.05) exposed animals.
PolyI:C exposed pups also displayed an increased cell density in all layers of the cerebral cortex when compared to controls (Fig. 2). Two way ANOVA revealed a significant effect of polyI:C treatment (F2,48 = 9.76, p <0.0001), a significant difference in the neuronal density between different layers (F3,48 = 192.9, p <0.0001) and a significant polyI:C-layer interaction (F6,48 = 3.174, p = 0.0105). Post-hoc analysis revealed that layers II–III (E10-14, p<0.001; E14-18, p<0.001), IV–V (E10-14, p<0.05: E14-18, p<0.05), and VI (E10-14, p<0.001; E14-18, p<0.001) were significantly denser in both the experimental groups. Layer I did not reach statistical significance in either group after correction of multiple comparisons. The increases are similar in E10-14 and E14-18 exposed groups except in layer II–III, in which the third trimester group showed a greater increase in cell density (23 % vs. 47 %, p<0.001).
Fig. 2.
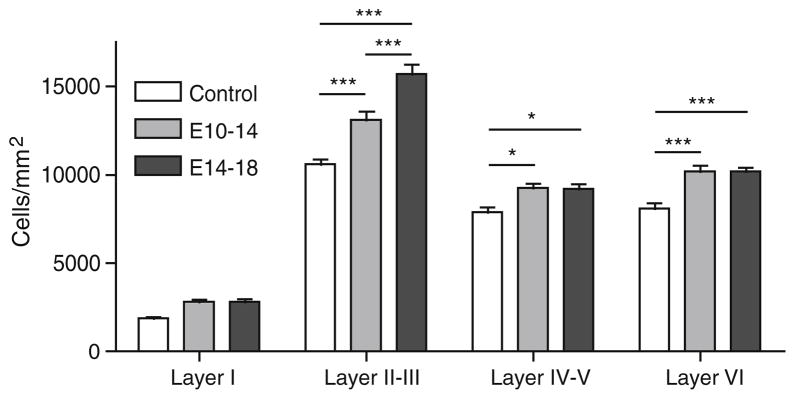
Maternal immune activation increases cell density in cortex. Cell density was significantly greater in layers II–III, layers IV–V, and layer VI in neonatal animals from mothers exposed to polyI:C, compared to control animals. * p<0.05, *** p<0.001, Bonferroni post-hoc tests
Discussion
The results of this study substantiate the hypothesis that maternal immune activation itself is sufficient to cause overgrowth of the cortical mantle in offspring. Furthermore, the characteristics of the overgrowth partially depend on the timing of the inflammatory stimulus. While both groups displayed increases in cell density, the group experiencing third trimester immune activation displayed a relatively greater increase of cell density within cortical layers II–III, but not in the deeper cortical layers. The finding that immune activation at different points in gestation affects different aspects of neuroanatomical development correlates with the results shown by Meyer et al. (2008) in which the timing of the maternal immune activation differentially affects the nature of the behavioral deficits in the offspring.
Following polyI:C injection, pro-inflammatory cytokines are found initially in the maternal circulation. These maternal cytokines can cross the placenta into the fetus directly, but cytokine mRNA is also upregulated in the placenta and in the fetal brain itself suggesting the inflammatory message is amplified and sustained in the fetus (Hsiao and Patterson 2011). Proliferation of neural progenitor cells in the fetal subventricular zone is regulated by cytokines of the CNTF family, including IL-6 and LIF. These cytokines modulate exit from the cell cycle and initiation of migration along radial glia scaffolds to the final cortical location. Increased IL-6-family signaling, originating from maternal immune response, likely interferes with cell cycle regulation resulting in the increased numbers of neurons we observe. Consistent with this hypothesis, blocking IL-6 during the polyI:C induced immune response prevents the associated behavioral deficits (Smith et al. 2007). However, the potential effects of other consequences of polyI:C, such as stress, hyperthermia, hypophagia, or inactivity, cannot be ruled out.
This study establishes an important potential mechanism for mental illness, demonstrating that maternal immune activation during pregnancy is sufficient to cause a robust morphological disturbance of fetal cortical development that in many ways matches the changes observed in autism postmortem brain tissue. The observation that maternal systemic immune activation alone is sufficient raises the possibility that other forms of maternal immune activity during pregnancy (e.g., maternal-fetal antibodies, chorioamnionitis, maternal autoimmune diseases, or maternal systemic or reproductive tract bacterial or fungal infection) might also lead to a similar outcome. These findings also raise the question of whether differences in fetal or maternal genetics might also contribute to an increased risk of immunity-induced disturbances of fetal brain development.
Acknowledgments
This work was supported in part by the National Institute of Neurological Disorders and Stroke R01 NS057444-01A2 (M.P.A.), the Nancy Lurie Marks Family Foundation (M.P.A.), Autism Speaks/NAAR (M.P.A.), and Beth Israel Deaconess Medical Center.
Footnotes
The authors declare that they have no conflict of interest.
References
- Atladottir HO, Thorsen P, Ostergaard L, Schendel DE, Lemcke S, Abdallah M, Parner ET. Maternal infection requiring hospitalization during pregnancy and autism spectrum disorders. J Autism Dev Disord. 2010;40(12):1423–1430. doi: 10.1007/s10803-010-1006-y. [DOI] [PubMed] [Google Scholar]
- Babulas V, Factor-Litvak P, Goetz R, Schaefer CA, Brown AS. Prenatal exposure to maternal genital and reproductive infections and adult schizophrenia. Am J Psychiatry. 2006;163(5):927–929. doi: 10.1176/ajp.2006.163.5.927. [DOI] [PubMed] [Google Scholar]
- Brown AS. Exposure to prenatal infection and risk of schizophrenia. Front Psychiatry. 2011;2:63. doi: 10.3389/fpsyt.2011.00063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown AS, Susser ES. In utero infection and adult schizophrenia. Ment Retard Dev Disabil Res Rev. 2002;8(1):51–57. doi: 10.1002/mrdd.10004. [DOI] [PubMed] [Google Scholar]
- Brown AS, Schaefer CA, Quesenberry CP, Jr, Liu L, Babulas VP, Susser ES. Maternal exposure to toxoplasmosis and risk of schizophrenia in adult offspring. Am J Psychiatry. 2005;162(4):767–773. doi: 10.1176/appi.ajp.162.4.767. [DOI] [PubMed] [Google Scholar]
- Casanova M, Trippe J. Radial cytoarchitecture and patterns of cortical connectivity in autism. Philos Trans R Soc Lond B Biol Sci. 2009;364(1522):1433–1436. doi: 10.1098/rstb.2008.0331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chess S. Follow-up report on autism in congenital rubella. J Autism Child Schizophr. 1977;7(1):69–81. doi: 10.1007/BF01531116. [DOI] [PubMed] [Google Scholar]
- Ciaranello AL, Ciaranello RD. The neurobiology of infantile autism. Annu Rev Neurosci. 1995;18:101–128. doi: 10.1146/annurev.ne.18.030195.000533. [DOI] [PubMed] [Google Scholar]
- Courchesne E, Mouton PR, Calhoun ME, Semendeferi K, Ahrens-Barbeau C, Hallet MJ, Barnes CC, Pierce K. Neuron number and size in prefrontal cortex of children with autism. JAMA. 2011;306(18):2001–2010. doi: 10.1001/jama.2011.1638. [DOI] [PubMed] [Google Scholar]
- Hsiao EY, Patterson PH. Activation of the maternal immune system induces endocrine changes in the placenta via IL-6. Brain Behav Immun. 2011;25(4):604–615. doi: 10.1016/j.bbi.2010.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mednick SA, Machon RA, Huttunen MO, Bonett D. Adult schizophrenia following prenatal exposure to an influenza epidemic. Arch Gen Psychiatry. 1988;45(2):189–192. doi: 10.1001/archpsyc.1988.01800260109013. [DOI] [PubMed] [Google Scholar]
- Meyer U, Nyffeler M, Yee BK, Knuesel I, Feldon J. Adult brain and behavioral pathological markers of prenatal immune challenge during early/middle and late fetal development in mice. Brain Behav Immun. 2008;22(4):469–486. doi: 10.1016/j.bbi.2007.09.012. [DOI] [PubMed] [Google Scholar]
- Patterson PH. Immune involvement in schizophrenia and autism: etiology, pathology and animal models. Behav Brain Res. 2009;204(2):313–321. doi: 10.1016/j.bbr.2008.12.016. [DOI] [PubMed] [Google Scholar]
- Schumann CM, Nordahl CW. Bridging the gap between MRI and postmortem research in autism. Brain Res. 2010;1380:175–186. doi: 10.1016/j.brainres.2010.09.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi L, Smith SE, Malkova N, Tse D, Su Y, Patterson PH. Activation of the maternal immune system alters cerebellar development in the offspring. Brain Behav Immun. 2009;23(1):116–123. doi: 10.1016/j.bbi.2008.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SE, Li J, Garbett K, Mirnics K, Patterson PH. Maternal immune activation alters fetal brain development through interleukin-6. J Neurosci. 2007;27(40):10695–10702. doi: 10.1523/JNEUROSCI.2178-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas DL, Nascimbene C, Krishnan C, Zimmerman AW, Pardo CA. Neuroglial activation and neuroinflammation in the brain of patients with autism. Ann Neurol. 2005;57(1):67–81. doi: 10.1002/ana.20315. [DOI] [PubMed] [Google Scholar]
- Voineagu I, Wang X, Johnston P, Lowe JK, Tian Y, Horvath S, Mill J, Cantor RM, Blencowe BJ, Geschwind DH. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature. 2011;474(7351):380–384. doi: 10.1038/nature10110. [DOI] [PMC free article] [PubMed] [Google Scholar]