

Abstract
The ability to pharmacologically modulate key signaling pathways that drive tumor growth and progression, but do not negatively impact the function of lymphocytes, provides avenues for rational combinatorial approaches to improve the antitumor activity of tumor immunotherapies. Novel targeted agents can very specifically block oncogenic events in cancer cells, leading to a pro-apoptotic milieu and a potential increase in sensitivity to recognition and attack by cytotoxic T lymphocytes. Furthermore, targeted pathway modulation in lymphocytes may change their function and have activating effects in some instances. When tested together with recently developed powerful tumor immunotherapies, such combinations may exploit the highly specific targeting of oncogenes with small molecule inhibitors to lead to high frequency of tumor regressions, and merge this benefit with the durable responses achievable with effective tumor immunotherapies.
The prospects of using targeted therapies to improve immunotherapy
There is much excitement accompanying the development of effective immunotherapy for cancer, particularly given its remarkable ability to induce durable tumor regressions that may last years, even in widely metastatic cancers. Specific inhibitors of oncogenic pathways in cancer cells are being developed, in some instances with unprecedently high tumor response rates, but which tend to not be durable. In addition to their direct antitumor effects, these agents could facilitate recognition and sensitivity to effector functions by cytotoxic T lymphocytes (CTL) and natural killer (NK) cells, thereby sensitizing cancer cells to immunotherapy [1,2]. A major goal, therefore, is to develop rational combinatorial approaches that merge the significant benefits of oncogenic pathway disruption using targeted agents with the unique ability of immunotherapy to mediate long-term responses in certain metastatic cancers. Emerging experiences suggest that immune-resistant tumors can be turned into immune-sensitive ones by using targeted therapies that block pathways in tumor cells responsible for lack of recognition and/or resistance to killing by immune effector cells, while these therapies maintain the functionality of immune cells, or may even enhance it.
Ideal characteristics of targeted therapies to sensitize tumor immunotherapies
An ideal immune sensitizing targeted therapy should block a key oncogenic event in cancer cells resulting in a pro-apoptotic cancer cell milieu, inhibiting anti-apoptotic molecules and potentiating pro-apoptotic molecules. At the same time, it should ideally potentiate means of immune effector cell recognition of cancer cells, such as increasing tumor antigen presentation for T cell recognition or enhanced expression of NK activating receptors. Of particular importance, potential immune sensitizing agents should not be have cytotoxic effects or inhibit critical functions of immune cells [2]. There may even be instances where such agents could actually improve immune cell function (Figure 1). It is acknowledged that these desired features of an immune sensitizing targeted therapy may not be fulfilled by most agents, but emerging preclinical experiences are providing the proof-of-concept to translate this combinatorial approach to the clinic.
Figure 1.
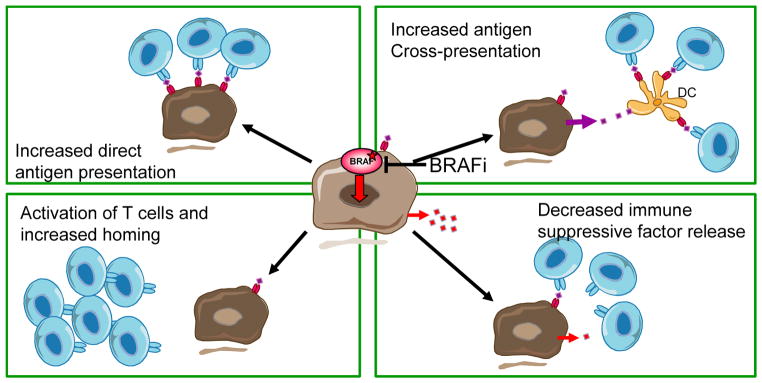
Potential mechanisms of immune sensitization by BRAF inhibition as an example of means by which targeted therapies could improve tumor immunotherapy. A) BRAF inhibitors may result in increased tumor antigen presentation directly to T cells through the increased expression of melanosomal antigens. B) The increased antigen presentation may be indirect through antigen cross-presentation by host dendritic cells taking up dying cells. C) BRAF inhibitors may have direct stimulatory effects on T cells through paradoxical MAPK activation in the setting of wild type BRAF. D) Inhibition of oncogenic BRAF with BRAF inhibitors may decrease the release of immune suppressive factors, which indirectly would result in a more permissive intratumoral milieu for T cell infiltration.
Examples of combinations of targeted therapies and immunotherapies
1. Epigenetic therapies and immunotherapy
Agents which affect epigenetics, such as demethylating agents and histone deacetylase (HDAC) inhibitors, induce profound changes in gene transcription and protein function, frequently resulting in a pro-apoptotic phenotype in malignant cells [3–5]. Demethylating agents and HDAC inhibitors activate genes of intrinsic and extrinsic pathways of apoptosis [6–11], which allows one to predict that cancer cells would be more sensitive to the antitumor effects of cytotoxic immune effector cells. These epigenetic modulating therapies have also been shown to increase the expression of MHC molecules and other molecules involved in antigen processing and presentation [11–15], improve the expression of tumor antigens [11,16], as well as ligands for NK activating receptors [17,18]. Therefore, epigenetic therapies have the potential to improve cancer cell recognition by immune cells and make them more sensitive to their antitumor cytotoxic activity.
Decitabine (5-aza-2′-deoxycytidine) is a cytosine analogue that inhibits DNA methylation and increases gene expression [19]. This agent increases the expression of NY-ESO-1 and other cancer-testis antigens, a class of tumor antigens that are expressed in a variety of tumors but, for at least some of the cancer-testis antigens (see Kvistborg et al in this issue) not in non-neoplastic normal human cells, with the exception of non-MHC expressing germ cells [11,16]. Furthermore, decitabine and other demethylating agents have the potential to re-establish functionality of apoptotic signaling in tumor cells and sensitize them to immune-mediated cell death via the Fas/Fas-ligand pathway [16].
Several preclinical experiences have provided successful testing of the combinatorial effects of HDAC inhibitors and immunotherapy. HDAC inhibitors have been shown to increase the antitumor activity of high dose IL-2 against the Renca murine kidney cancer model [20], against the modified lung cancer cell line TC-1 [15], and with several immunotherapies in the B16 murine melanoma model [21,22]. In particular, the efficacy of antigen-specific immunotherapy in the adoptive cell transfer (ACT) pmel-1 model and of a listeria-based prime-boost vaccine against B16 melanoma was improved with the addition of a HDAC inhibitor [22]. The superior combinatorial effects were mediated by an improvement in antigen presentation by tumor cells and enhanced function of immune cells, both resulting in increased anti-tumor activity [22]. The experiences with the combination of immunotherapy with HDAC inhibitors have been expanded to demonstrate that HDAC inhibitors can be combined with immune-activating antibodies such as anti-CD137 and anti-CD40, able to treat previously established subcutaneous tumors [23]. The mechanistic basis of the combined effects in this model is dependent on tumor cell apoptosis mediated by HDAC inhibitor therapy, which stimulated antigen-cross presentation to enhance the proliferation and survival of CD8+ cytotoxic T lymphocytes (CTLs) [23].
The potential beneficial effects of HDAC inhibitors in combination with immunotherapy have to be balanced with the data demonstrating evidence of HDAC inhibitors also having immune suppressive effects [24–26], in particular by increasing the suppressive effects of T regulatory (Treg) cells [20,27]. Furthermore, HDAC inhibitors are potent inhibitors of cell cycle proliferation in lymphocytes [26]. Therefore, close mechanistic analysis will be required if such combinations are tested in the clinic.
2. MAPK inhibitors and immunotherapy
Mutant BRAFV600E is an oncogene belonging to the mitogen-activated protein kinase (MAPK) pathway present in approximately 50% of cutaneous melanomas. The selective inhibition of oncogenic BRAF with type I RAF inhibitors results in unprecedented antitumor responses in this cancer [28]. Type I RAF inhibitors, such as vemurafenib (formerly PLX4032) and dabrafenib (formerly GSK2118436), exert their effects when the kinase is in the active conformation, as it is when there are activating mutations in BRAF. Since this mutation is not present in immune cells, the use of these selective BRAF inhibitors may be combinable with tumor immunotherapy (Figure 1). In fact, preclinical and early clinical evidence suggests that such combinations are feasible. BRAF inhibitors do not have significant adverse effects on human T cell functions when tested either in vitro [29] or analyzing T cells obtained from patients treated with BRAF inhibitors [30,31]. Furthermore, increased intratumoral infiltration by CD8+ T cells has been shown in some biopsies of patients treated with BRAF inhibitors soon after therapy [31,32]. This may be due to increased expression of melanosomal antigens upon BRAF inhibition [33], or because antigens released from dying tumor cells may be cross-presented by antigen-presenting cells. In addition, it is likely that the inhibition of oncogenic BRAF may result in a more immune-permissive tumor environment by decreased expression of immune suppressive factors or chemokines that otherwise limit intratumoral T cell infiltration [34].
The particular pharmacologic properties of type I RAF inhibitors may further influence how these targeted therapies interact with the immune system. RAF inhibitors induce paradoxical effects in cells that are wild type for BRAF, resulting in the activation of the MAPK pathway. The mechanism is based on the transactivation of CRAF by a partially blocked wild type CRAF-BRAF dimer when there is strong upstream activating signals [35–37]. This phenomenon of paradoxical MAPK activation is the molecular basis for the development of cutaneous squamous cell carcinomas and other RAS-induced secondary cancers in patients treated with BRAF inhibitors [38–40]. Since T cells are wild type for BRAF and TCR triggering activates the MAPK pathway, it is possible that BRAF inhibitors may directly activate lymphocytes through paradoxical MAPK activation. In a BRAFV600E-driven murine model of melanoma in fully immunocompetent mice, combined treatment with vemurafenib and TCR engineered adoptive cell therapy resulted in superior antitumor responses compared with either therapy alone [41]. In this model, vemurafenib did not increase tumor antigen expression nor did it change the expansion or distribution of the adoptively transferred cells, suggesting that the superior combined activity was not due to a direct increase in tumor antigen expression. However, vemurafenib resulted in paradoxical MAPK activation that led to an increased in vivo cytotoxic activity and intratumoral cytokine secretion by adoptively transferred cells.
The same mouse model was used to study the role of changes in chemokine signaling induced by BRAF inhibition in cancer cells and the combinatorial effects with immune modulating antibody therapy [34]. These studies showed that CD8+ T cells, but not NK cells, were partially required for the antitumor activity of BRAF inhibitors, which was related to downregulation of tumor CCL2 production upon the inhibition of oncogenic BRAF. Furthermore, combination therapy with BRAF inhibitors and agonistic anti-CD137 antibodies demonstrated significant anti-tumor activity.
A xenograft mouse model of TCR engineered ACT combined with BRAF inhibitor therapy also suggested that BRAF inhibitors may improve the tumor microenvironment by blocking the release of immune suppressive factors controlled downstream of oncogenic BRAF. Administration of a BRAF inhibitor significantly increased tumor infiltration of adoptively transferred T cells in vivo and enhanced the antitumor activity of ACT mediated by a decrease in tumor cell production of the vascular endothelial growth factor (VEGF). The same effect of decreasing VEGF was noted in patient-derived melanoma tumor biopsies during BRAF inhibitor treatment [42].
Despite this supportive data suggesting that BRAF inhibitors may have promise to combine with immunotherapy, in a mouse model of inducible BRAFV600E-driven murine melanomas the combination of BRAF inhibitor therapy and CTLA-4 blockade had no beneficial combinatorial effects. In fact, in this model BRAF inhibitor therapy decreased the intratumoral infiltration with T cells [43]. It is currently unclear the reasons for the discrepancies between models, but it is likely that the genetic constellation of oncogenic drivers in melanoma cells may influence the effect of BRAF inhibitor targeted therapies and their interplay with the immune system.
3. PI3K/AKT/mTOR inhibitors and immunotherapy
The PI3K/AKT/mTOR signaling pathway is a major target for drug development in cancer given its critical role in oncogenic signaling in multiple histologies. Agents that block this pathway at different levels are in clinical development, and inhibitors of the mammalian target of rapamycin (mTOR) have been approved for use in patients with advanced renal cell carcinoma. In addition, mTOR has important roles in the regulation of the function of immune cells, and mTOR inhibitors are used for immune suppression after organ transplantation. However, inhibiting mTOR signaling was shown to have paradoxical immune stimulating effects resulting in the generation of long-lived memory CD8+ T cells [44–46]. Additional beneficial effects of mTOR inhibition have also been observed with dendritic cells and hematopoietic stem cells, highlighting that mTOR inhibitors may work by inducing favourable immune cell changes beyond their direct oncogene targeting effects in cancer cells [47].
A further use of targeted inhibitors of the PI3K/AKT/mTOR pathway is related to the expression of the immune suppressive membrane receptor programmed death ligand-1 (PD-L1), which may have important implications for PD-1/PD-L1 blockade therapy. Loss of PTEN resulting in the activation of the PI3K pathway is a common event in glioblastoma multiforme (GBM). PTEN deficiency within GBM cells was associated with increased expression of PD-L1 and resulting immune evasion, which could be reversed with PI3K inhibitors [48]. A preclinical model where PI3K inhibitors were tested in combination with Toll-like receptor (TLR) agonists, demonstrated superior combinatorial effects against several murine tumors. These beneficial effects were mediated by the specific enhancement of polyfunctional T responses secreting IFN-γ and IL-17 [49].
4. c-kit inhibitors and immunotherapy
A clear example of the success of targeted therapy for cancer is the development of imatinib and other abl and c-kit inhibitors for the treatment of chronic myelogenous leukemia (CML) and gastrointestinal stromal tumors (GIST). Data provided by several groups suggests that part of this success may be mediated by inducing an antitumor immune response. Imatinib has been suggested to have off-target effects on immune effectors, irrespective of its effects on tumor cells. This effect was mediated by the activation of favourable cross talk between dendritic cells (DC) and natural killer (NK) cells [50,51].
In a mouse model of spontaneous development of GIST in transgenic mice carrying an activating KIT mutations, the antitumor response of imatinib was lost with CD8+ T cell depletion and was enhanced by cytotoxic T lymphocyte–associated antigen (CTLA-4) blockade with monoclonal antibody therapy [52]. In this model, imatinib reduced the expression of the immune suppressive enzyme indoleamine 2,3-dioxygenase (IDO) by GIST tumor cells, facilitating an enhanced immune response. In another mouse model, the therapeutic activity of tyrosine kinase inhibitor dasatinib (which also blocks c-kit) was strongly potentiated by immune stimulation with agonist anti-OX40 antibody therapy [53].
Conclusions
Some highly targeted therapies for cancer have potential beneficial effects in the immune system, and may result in increasing the sensitivity of cancer cells to immunotherapy. This allows the design of rational combinations for preclinical hypothesis testing and subsequent clinical translation. Such combinations may merge the benefits of the antitumor activity of targeted therapies for cancer with the durable responses induced by tumor immunotherapy.
Highlights.
Cancer targeted therapies are being tested to synergize with tumor immunotherapy
Epigenetic modulators increase tumor antigen expression
BRAF inhibitors have paradoxical activation effects on lymphocytes
BRAF inhibitors can decrease cancer-released immune suppressive factors
PI3K/AKT pathway inhibitors can directly activate T memory cells
Acknowledgments
A.R. is funded by The Seaver Institute, the Garcia-Corsini Family Fund, the Louise Belley and Richard Schnarr Fund, the Wesley Coyle Memorial Fund, the Bila Alon Hacker Memorial Fund, the Fred L. Hartley Family Foundation, the Ruby Family Foundation, the Jonsson Cancer Center Foundation, the Eli & Edythe Broad Center of Regenerative Medicine and Stem Cell Research at UCLA, the Caltech-UCLA Joint Center for Translational Medicine, and the NIH grants P50 CA086306, P01 CA132681 and U54 CA119347. J.D.W. is funded by NCI R01 CA056821, RC2 CA148468, the Ludwig Trust, Swim Across America, the Goodwin Commonwealth Fund, the Melanoma Research Alliance, the Breast Cancer Research Foundation, the Annenberg Hazen Foundation and the Live4Life Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Schumacher LY, Ribas A. Overcoming Tumor Resistance to Immunotherapy. Cancer Therapy. 2006;4:13–26. [Google Scholar]
- 2.Begley J, Ribas A. Targeted therapies to improve tumor immunotherapy. Clin Cancer Res. 2008;14:4385–4391. doi: 10.1158/1078-0432.CCR-07-4804. [DOI] [PubMed] [Google Scholar]
- 3.Johnstone RW, Licht JD. Histone deacetylase inhibitors in cancer therapy: is transcription the primary target? Cancer Cell. 2003;4:13–18. doi: 10.1016/s1535-6108(03)00165-x. [DOI] [PubMed] [Google Scholar]
- 4.Ungerstedt JS, Sowa Y, Xu WS, Shao Y, Dokmanovic M, Perez G, Ngo L, Holmgren A, Jiang X, Marks PA. Role of thioredoxin in the response of normal and transformed cells to histone deacetylase inhibitors. Proc Natl Acad Sci U S A. 2005;102:673–678. doi: 10.1073/pnas.0408732102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hadnagy A, Beaulieu R, Balicki D. Histone tail modifications and noncanonical functions of histones: perspectives in cancer epigenetics. Mol Cancer Ther. 2008;7:740–748. doi: 10.1158/1535-7163.MCT-07-2284. [DOI] [PubMed] [Google Scholar]
- 6.Zhang XD, Gillespie SK, Borrow JM, Hersey P. The histone deacetylase inhibitor suberic bishydroxamate: a potential sensitizer of melanoma to TNF-related apoptosis-inducing ligand (TRAIL) induced apoptosis. Biochem Pharmacol. 2003;66:1537–1545. doi: 10.1016/s0006-2952(03)00509-4. [DOI] [PubMed] [Google Scholar]
- 7.Zhang XD, Gillespie SK, Borrow JM, Hersey P. The histone deacetylase inhibitor suberic bishydroxamate regulates the expression of multiple apoptotic mediators and induces mitochondria-dependent apoptosis of melanoma cells. Mol Cancer Ther. 2004;3:425–435. [PubMed] [Google Scholar]
- 8.Peart MJ, Smyth GK, van Laar RK, Bowtell DD, Richon VM, Marks PA, Holloway AJ, Johnstone RW. Identification and functional significance of genes regulated by structurally different histone deacetylase inhibitors. Proc Natl Acad Sci U S A. 2005;102:3697–3702. doi: 10.1073/pnas.0500369102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Insinga A, Monestiroli S, Ronzoni S, Gelmetti V, Marchesi F, Viale A, Altucci L, Nervi C, Minucci S, Pelicci PG. Inhibitors of histone deacetylases induce tumor-selective apoptosis through activation of the death receptor pathway. Nat Med. 2005;11:71–76. doi: 10.1038/nm1160. [DOI] [PubMed] [Google Scholar]
- 10.Nebbioso A, Clarke N, Voltz E, Germain E, Ambrosino C, Bontempo P, Alvarez R, Schiavone EM, Ferrara F, Bresciani F, et al. Tumor-selective action of HDAC inhibitors involves TRAIL induction in acute myeloid leukemia cells. Nat Med. 2005;11:77–84. doi: 10.1038/nm1161. [DOI] [PubMed] [Google Scholar]
- 11.Natsume A, Wakabayashi T, Tsujimura K, Shimato S, Ito M, Kuzushima K, Kondo Y, Sekido Y, Kawatsura H, Narita Y, et al. The DNA demethylating agent 5-aza-2′-deoxycytidine activates NY-ESO-1 antigenicity in orthotopic human glioma. International journal of cancer Journal international du cancer. 2008;122:2542–2553. doi: 10.1002/ijc.23407. [DOI] [PubMed] [Google Scholar]
- 12.Magner WJ, Kazim AL, Stewart C, Romano MA, Catalano G, Grande C, Keiser N, Santaniello F, Tomasi TB. Activation of MHC class I, II, and CD40 gene expression by histone deacetylase inhibitors. J Immunol. 2000;165:7017–7024. doi: 10.4049/jimmunol.165.12.7017. [DOI] [PubMed] [Google Scholar]
- 13.Maeda T, Towatari M, Kosugi H, Saito H. Up-regulation of costimulatory/adhesion molecules by histone deacetylase inhibitors in acute myeloid leukemia cells. Blood. 2000;96:3847–3856. [PubMed] [Google Scholar]
- 14.Khan AN, Gregorie CJ, Tomasi TB. Histone deacetylase inhibitors induce TAP, LMP, Tapasin genes and MHC class I antigen presentation by melanoma cells. Cancer Immunol Immunother. 2008;57:647–654. doi: 10.1007/s00262-007-0402-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Setiadi AF, Omilusik K, David MD, Seipp RP, Hartikainen J, Gopaul R, Choi KB, Jefferies WA. Epigenetic enhancement of antigen processing and presentation promotes immune recognition of tumors. Cancer Res. 2008;68:9601–9607. doi: 10.1158/0008-5472.CAN-07-5270. [DOI] [PubMed] [Google Scholar]
- 16.Konkankit VV, Kim W, Koya RC, Eskin A, Dam MA, Nelson S, Ribas A, Liau LM, Prins RM. Decitabine immunosensitizes human gliomas to NY-ESO-1 specific T lymphocyte targeting through the Fas/Fas ligand pathway. Journal of translational medicine. 2011;9:192. doi: 10.1186/1479-5876-9-192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Skov S, Pedersen MT, Andresen L, Straten PT, Woetmann A, Odum N. Cancer cells become susceptible to natural killer cell killing after exposure to histone deacetylase inhibitors due to glycogen synthase kinase-3-dependent expression of MHC class I-related chain A and B. Cancer Res. 2005;65:11136–11145. doi: 10.1158/0008-5472.CAN-05-0599. [DOI] [PubMed] [Google Scholar]
- 18.Armeanu S, Bitzer M, Lauer UM, Venturelli S, Pathil A, Krusch M, Kaiser S, Jobst J, Smirnow I, Wagner A, et al. Natural killer cell-mediated lysis of hepatoma cells via specific induction of NKG2D ligands by the histone deacetylase inhibitor sodium valproate. Cancer Res. 2005;65:6321–6329. doi: 10.1158/0008-5472.CAN-04-4252. [DOI] [PubMed] [Google Scholar]
- 19.Watanabe Y, Maekawa M. Methylation of DNA in cancer. Advances in clinical chemistry. 2010;52:145–167. doi: 10.1016/s0065-2423(10)52006-7. [DOI] [PubMed] [Google Scholar]
- 20.Kato Y, Yoshimura K, Shin T, Verheul H, Hammers H, Sanni TB, Salumbides BC, Van Erp K, Schulick R, Pili R. Synergistic in vivo antitumor effect of the histone deacetylase inhibitor MS-275 in combination with interleukin 2 in a murine model of renal cell carcinoma. Clin Cancer Res. 2007;13:4538–4546. doi: 10.1158/1078-0432.CCR-07-0014. [DOI] [PubMed] [Google Scholar]
- 21.Murakami T, Sato A, Chun NA, Hara M, Naito Y, Kobayashi Y, Kano Y, Ohtsuki M, Furukawa Y, Kobayashi E. Transcriptional modulation using HDACi depsipeptide promotes immune cell-mediated tumor destruction of murine B16 melanoma. J Invest Dermatol. 2008;128:1506–1516. doi: 10.1038/sj.jid.5701216. [DOI] [PubMed] [Google Scholar]
- 22.Vo DD, Prins RM, Begley JL, Donahue TR, Morris LF, Bruhn KW, de la Rocha P, Yang MY, Mok S, Garban HJ, et al. Enhanced antitumor activity induced by adoptive T-cell transfer and adjunctive use of the histone deacetylase inhibitor LAQ824. Cancer research. 2009;69:8693–8699. doi: 10.1158/0008-5472.CAN-09-1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23**.Christiansen AJ, West A, Banks KM, Haynes NM, Teng MW, Smyth MJ, Johnstone RW. Eradication of solid tumors using histone deacetylase inhibitors combined with immune-stimulating antibodies. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:4141–4146. doi: 10.1073/pnas.1011037108. Histone deacetylase inhibitors induce tumor cell apoptosis resulting in tumor antigen cross-presentation, resulting in superior combinatorial antitumor effects with immune modulating antibodies. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brogdon JL, Xu Y, Szabo SJ, An S, Buxton F, Cohen D, Huang Q. Histone deacetylase activities are required for innate immune cell control of Th1 but not Th2 effector cell function. Blood. 2007;109:1123–1130. doi: 10.1182/blood-2006-04-019711. [DOI] [PubMed] [Google Scholar]
- 25.Reilly CM, Mishra N, Miller JM, Joshi D, Ruiz P, Richon VM, Marks PA, Gilkeson GS. Modulation of renal disease in MRL/lpr mice by suberoylanilide hydroxamic acid. J Immunol. 2004;173:4171–4178. doi: 10.4049/jimmunol.173.6.4171. [DOI] [PubMed] [Google Scholar]
- 26.Johnson J, Pahuja A, Graham M, Hering B, Hancock WW, Bansal-Pakala P. Effects of histone deacetylase inhibitor SAHA on effector and FOXP3+regulatory T cells in rhesus macaques. Transplantation proceedings. 2008;40:459–461. doi: 10.1016/j.transproceed.2008.01.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tao R, de Zoeten EF, Ozkaynak E, Chen C, Wang L, Porrett PM, Li B, Turka LA, Olson EN, Greene MI, et al. Deacetylase inhibition promotes the generation and function of regulatory T cells. Nat Med. 2007;13:1299–1307. doi: 10.1038/nm1652. [DOI] [PubMed] [Google Scholar]
- 28.Ribas A, Flaherty KT. BRAF targeted therapy changes the treatment paradigm in melanoma. Nature reviews Clinical oncology. 2011;8:426–433. doi: 10.1038/nrclinonc.2011.69. [DOI] [PubMed] [Google Scholar]
- 29.Comin-Anduix B, Chodon T, Sazegar H, Matsunaga D, Mock S, Jalil J, Escuin-Ordinas H, Chmielowski B, Koya RC, Ribas A. The oncogenic BRAF kinase inhibitor PLX4032/RG7204 does not affect the viability or function of human lymphocytes across a wide range of concentrations. Clin Cancer Res. 2010;16:6040–6048. doi: 10.1158/1078-0432.CCR-10-1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hong DS, Vence L, Falchook G, Radvanyi LG, Liu C, Goodman V, Legos JJ, Blackman S, Scarmadio A, Kurzrock R, et al. BRAF(V600) Inhibitor GSK2118436 Targeted Inhibition of Mutant BRAF in Cancer Patients Does Not Impair Overall Immune Competency. Clinical cancer research: an official journal of the American Association for Cancer Research. 2012;18:2326–2335. doi: 10.1158/1078-0432.CCR-11-2515. [DOI] [PubMed] [Google Scholar]
- 31.Frederick DT, Piris A, Cogdill AP, Cooper ZA, Lezcano C, Ferrone CR, Mitra D, Boni A, Newton LP, Liu C, et al. BRAF inhibition is associated with enhanced melanoma antigen expression and a more favorable tumor microenvironment in patients with metastatic melanoma. Clinical cancer research. 2013 doi: 10.1158/1078-0432.CCR-12-1630. [epub] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32**.Wilmott JS, Long GV, Howle JR, Haydu LE, Sharma RN, Thompson JF, Kefford RF, Hersey P, Scolyer RA. Selective BRAF inhibitors induce marked T-cell infiltration into human metastatic melanoma. Clinical cancer research. 2012;18:1386–1394. doi: 10.1158/1078-0432.CCR-11-2479. Metastatic melanoma lesions biopsied from patients early after starting therapy with the BRAF inhibitors dabrafenib or vemurafenib frequently have increased T cell infiltration. [DOI] [PubMed] [Google Scholar]
- 33.Boni A, Cogdill AP, Dang P, Udayakumar D, Njauw CN, Sloss CM, Ferrone CR, Flaherty KT, Lawrence DP, Fisher DE, et al. Selective BRAFV600E inhibition enhances T-cell recognition of melanoma without affecting lymphocyte function. Cancer research. 2010;70:5213–5219. doi: 10.1158/0008-5472.CAN-10-0118. [DOI] [PubMed] [Google Scholar]
- 34**.Knight DA, Ngiow SF, Li M, Parmenter T, Mok S, Cass A, Haynes NM, Kinross K, Yagita H, Koya RC, et al. Host CCR2 and CD8+ T cells contribute to the anti-melanoma activity of BRAF inhibitors. Journal of Clinical Investigation. 2013 doi: 10.1172/JCI66236. (in press). BRAF inhibitor therapy has a partial need for an intact immune system to exert antitumor activity. In part, this is resultant of a downregulation of the tumor production of CCL2 interacting with CCR2 in host immune cells. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 35.Heidorn SJ, Milagre C, Whittaker S, Nourry A, Niculescu-Duvas I, Dhomen N, Hussain J, Reis-Filho JS, Springer CJ, Pritchard C, et al. Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell. 2010;140:209–221. doi: 10.1016/j.cell.2009.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature. 2010;464:427–430. doi: 10.1038/nature08902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Halaban R, Zhang W, Bacchiocchi A, Cheng E, Parisi F, Ariyan S, Krauthammer M, McCusker JP, Kluger Y, Sznol M. PLX4032, a selective BRAF(V600E) kinase inhibitor, activates the ERK pathway and enhances cell migration and proliferation of BRAF melanoma cells. Pigment Cell Melanoma Res. 2010;23:190–200. doi: 10.1111/j.1755-148X.2010.00685.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Su F, Viros A, Milagre C, Trunzer K, Bollag G, Spleiss O, Reis-Filho JS, Kong X, Koya RC, Flaherty KT, et al. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. The New England journal of medicine. 2012;366:207–215. doi: 10.1056/NEJMoa1105358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Callahan MK, Rampal R, Harding JJ, Klimek VM, Chung YR, Merghoub T, Wolchok JD, Solit DB, Rosen N, Abdel-Wahab O, et al. Progression of RAS-mutant leukemia during RAF inhibitor treatment. The New England journal of medicine. 2012;367:2316–2321. doi: 10.1056/NEJMoa1208958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Oberholzer PA, Kee D, Dziunycz P, Sucker A, Kamsukom N, Jones R, Roden C, Chalk CJ, Ardlie K, Palescandolo E, et al. RAS Mutations Are Associated With the Development of Cutaneous Squamous Cell Tumors in Patients Treated With RAF Inhibitors. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2012;30:316–321. doi: 10.1200/JCO.2011.36.7680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41**.Koya RC, Mok S, Otte N, Blacketor KJ, Comin-Anduix B, Tumeh PC, Minasyan A, Graham N, Graeber TG, Chodon T, et al. BRAF inhibitor vemurafenib improves the antitumor activity of adoptive cell immunotherapy. Cancer research. 2012;72:3928–37. doi: 10.1158/0008-5472.CAN-11-2837. Using an implantable BRAFV600E-driven murine melanoma, the combination of a BRAF inhibitor and TCR engineered adoptive cell transfer therapy resulted in superior antitumor activity mediated by direct immune-activating effects of the BRAF inhibitor. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu C, Peng W, Xu C, Lou Y, Zhang M, Wargo JA, Chen JQ, Li HS, Watowich SS, Yang Y, et al. BRAF Inhibition Increases Tumor Infiltration by T cells and Enhances the Antitumor Activity of Adoptive Immunotherapy in Mice. Clinical cancer research. 2013;19:393–403. doi: 10.1158/1078-0432.CCR-12-1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43*.Hooijkaas A, Gadiot J, Morrow M, Stewart R, Schumacher T, Blank CU. Selective BRAF inhibition decreases tumor-resident lymphocyte frequencies in a mouse model of human melanoma. Oncoimmunology. 2012;1:609–617. doi: 10.4161/onci.20226. BRAF inhibitor therapy resulted in decreased intratumoral infiltration by CD8+ T cells in induced melanomas in mice transgenic for BRAFV600E, and the combination with anti-CTLA4 antibodies did not improve antitumor activity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44*.Li Q, Rao RR, Araki K, Pollizzi K, Odunsi K, Powell JD, Shrikant PA. A central role for mTOR kinase in homeostatic proliferation induced CD8+ T cell memory and tumor immunity. Immunity. 2011;34:541–553. doi: 10.1016/j.immuni.2011.04.006. Inhibition of mTOR results in superior T cell memory through the modulation of specific transcription factors during homeostatic proliferation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rao RR, Li Q, Odunsi K, Shrikant PA. The mTOR kinase determines effector versus memory CD8+ T cell fate by regulating the expression of transcription factors T-bet and Eomesodermin. Immunity. 2010;32:67–78. doi: 10.1016/j.immuni.2009.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Araki K, Turner AP, Shaffer VO, Gangappa S, Keller SA, Bachmann MF, Larsen CP, Ahmed R. mTOR regulates memory CD8 T-cell differentiation. Nature. 2009;460:108–112. doi: 10.1038/nature08155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Srivastava RK, Utley A, Shrikant PA. Rapamycin: A rheostat for CD8(+) T-cell-mediated tumor therapy. Oncoimmunology. 2012;1:1189–1190. doi: 10.4161/onci.20663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Parsa AT, Waldron JS, Panner A, Crane CA, Parney IF, Barry JJ, Cachola KE, Murray JC, Tihan T, Jensen MC, et al. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nature medicine. 2007;13:84–88. doi: 10.1038/nm1517. [DOI] [PubMed] [Google Scholar]
- 49.Marshall NA, Galvin KC, Corcoran AM, Boon L, Higgs R, Mills KH. Immunotherapy with PI3K inhibitor and Toll-like receptor agonist induces IFN-gamma+IL-17+ polyfunctional T cells that mediate rejection of murine tumors. Cancer research. 2012;72:581–591. doi: 10.1158/0008-5472.CAN-11-0307. [DOI] [PubMed] [Google Scholar]
- 50.Borg C, Terme M, Taieb J, Menard C, Flament C, Robert C, Maruyama K, Wakasugi H, Angevin E, Thielemans K, et al. Novel mode of action of c-kit tyrosine kinase inhibitors leading to NK cell-dependent antitumor effects. The Journal of clinical investigation. 2004;114:379–388. doi: 10.1172/JCI21102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Taieb J, Chaput N, Menard C, Apetoh L, Ullrich E, Bonmort M, Pequignot M, Casares N, Terme M, Flament C, et al. A novel dendritic cell subset involved in tumor immunosurveillance. Nature medicine. 2006;12:214–219. doi: 10.1038/nm1356. [DOI] [PubMed] [Google Scholar]
- 52**.Balachandran VP, Cavnar MJ, Zeng S, Bamboat ZM, Ocuin LM, Obaid H, Sorenson EC, Popow R, Ariyan C, Rossi F, et al. Imatinib potentiates antitumor T cell responses in gastrointestinal stromal tumor through the inhibition of Ido. Nature medicine. 2011;17:1094–1100. doi: 10.1038/nm.2438. The antitumor effects of imatinib against c-kit mutant gastrointestinal stromal tumors (GIST) are partially resultant of immune modulation through the inhibition of the immune suppressive enzyme indoleamine 2,3-dioxygenase (Ido) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yang Y, Liu C, Peng W, Lizee G, Overwijk WW, Liu Y, Woodman SE, Hwu P. Antitumor T-cell responses contribute to the effects of dasatinib on c-KIT mutant murine mastocytoma and are potentiated by anti-OX40. Blood. 2012;120:4533–4543. doi: 10.1182/blood-2012-02-407163. [DOI] [PMC free article] [PubMed] [Google Scholar]