

Abstract
In Salmonella enterica serovar Typhimurium, oxidoreductases of the thioredoxin superfamily contribute to bacterial invasiveness, intracellular replication and to the virulence in BALB/c mice as well as in the soil nematode Caenorhabditis elegans. The scsABCD gene cluster, present in many but not all enteric bacteria, codes for four putative oxidoreductases of the thioredoxin superfamily. Here we have analyzed the potential role of the scs genes in oxidative stress tolerance and virulence in S. Typhimurium. An scsABCD deletion mutant showed moderate sensitization to the redox-active transition metal ion copper and increased protein carbonylation upon exposure to hydrogen peroxide. Still, the scsABCD mutant was not significantly affected for invasiveness or intracellular replication in respectively cultured epithelial or macrophage-like cells. However, we noted a significant copper chloride sensitivity of SPI1 T3SS mediated invasiveness that strongly depended on the presence of the scs genes. The scsABCD deletion mutant was not attenuated in animal infection models. In contrast, the mutant showed a moderate increase in its competitive index upon intraperitoneal challenge and enhanced invasiveness in small intestinal ileal loops of BALB/c mice. Moreover, deletion of the scsABCD genes restored the invasiveness of a trxA mutant in epithelial cells and its virulence in C. elegans. Our findings thus demonstrate that the scs gene cluster conditionally affects virulence and underscore the complex interactions between oxidoreductases of the thioredoxin superfamily in maintaining host adaptation of S. Typhimurium.
Introduction
Salmonella enetrica serovar Typhimurium (S. Typhimurium) is a facultative intracellular enteric pathogen that traditionally has been used as a model organism for studying typhoid fever caused by the human adapted serovar Typhi [1], [2]. In the murine model for typhoid fever, S. Typhimurium invades the intestinal epithelium after per oral challenge. Invasion is followed by dissemination and intracellular bacterial replication in phagocytic cells of the liver and spleen.
Virulence of S. Typhimurium in mammalian cell culture and in mice, as well as in alternative host infection models, strongly relies on horizontally acquired DNA regions termed as Salmonella pathogenicity islands (SPI:s) [3]. Two of the SPI:s, SPI1 and SPI2, code for protein type III secretion systems (T3SS) that translocate bacterial effector proteins into host cells. The effector proteins act to manipulate central host cell functions, such as actin polymerization, vesicular trafficking, and signal transduction [4], [5], [6]. In mice, SPI1 and SPI2 enable the bacteria to transcytose the intestinal epithelial barrier, and disseminate to replicate in macrophages of the liver and spleen respectively [7], [8]. Moreover, SPI1 and SPI2 T3SS also modulate pro-inflammatory responses of the host during Salmonella infection [9], [10], [11].
Reactive oxygen species (ROS) generated by Salmonella-induced inflammatory responses most evidently contribute to the clearance and pathogenesis of salmonellosis. Mice (phox −/−) lacking a functional phagocyte oxidase rapidly succumb upon infection doses well controlled by wild type littermates [12]. Furthermore, individuals suffering from chronic granulomatous disease, a condition marked by a defective oxidative phagocyte response, show an increased prevalence of extraintestinal infections caused by non-typhoidal serovariants of Salmonella [13].
The genome sequence of S. enterica reveals a number of enzymes that potentially provide protection against ROS, such as H2O2. These enzymes include super oxide mutases, catalases, thiol peroxidases and methionine sulphoxide reductases [14], [15], [16], [17]. In addition, oxidoreductases of the thioredoxin superfamily contribute to the assembly or activities of many bacterial virulence factors. SPI2 activity in part relies on the periplasmic DsbA and SrgA proteins, which both belong to the thioredoxin superfamily of oxidoreductases [18]. SrgA is in fact coded for by a plasmid-carried fimbrial operon pef, and moreover is required for the assembly of Pef fimbriae [19]. The activity of SPI2 also depends on the cytoplasmic thioredoxin 1 (TrxA, trxA), whereby trxA mutants are severely attenuated for replication in macrophage-like cells and in mice [20], [21]. TrxA additionally mediates a redox-associated lethality caused by S. Typhimurium in an infection model based on the soil nematode C. elegans [22].
Many of the oxidoreductases such as TrxA, DsbA and thiol peroxidase Tpx implicated in virulence of S. Typhimurium and in redox tolerance of E. coli, are highly conserved within enterobacteria [15], [18], [23]. In contrast to laboratory E. coli strains, S. Typhimurium includes the scsABCD gene cluster that encodes four proteins each with a Cys-X-X-Cys motif characteristic for the thioredoxin superfamily [24] (Figs. 1A and 1B). When cloned into E. coli, the scs genes restore copper tolerance of selected copper-sensitive mutants [24]. However, the actual role of the scs genes for redox tolerance in S. Typhimurium and their contribution to virulence, if any, has remained enigmatic. Here we demonstrate that a S. Typhimurium scsABCD deletion mutant shows moderate sensitization to copper chloride and, surprisingly, a conditional enhanced invasiveness in epithelial cells and virulence in C. elegans.
Figure 1. In silico analyses and localization of the ScsABCD proteins.
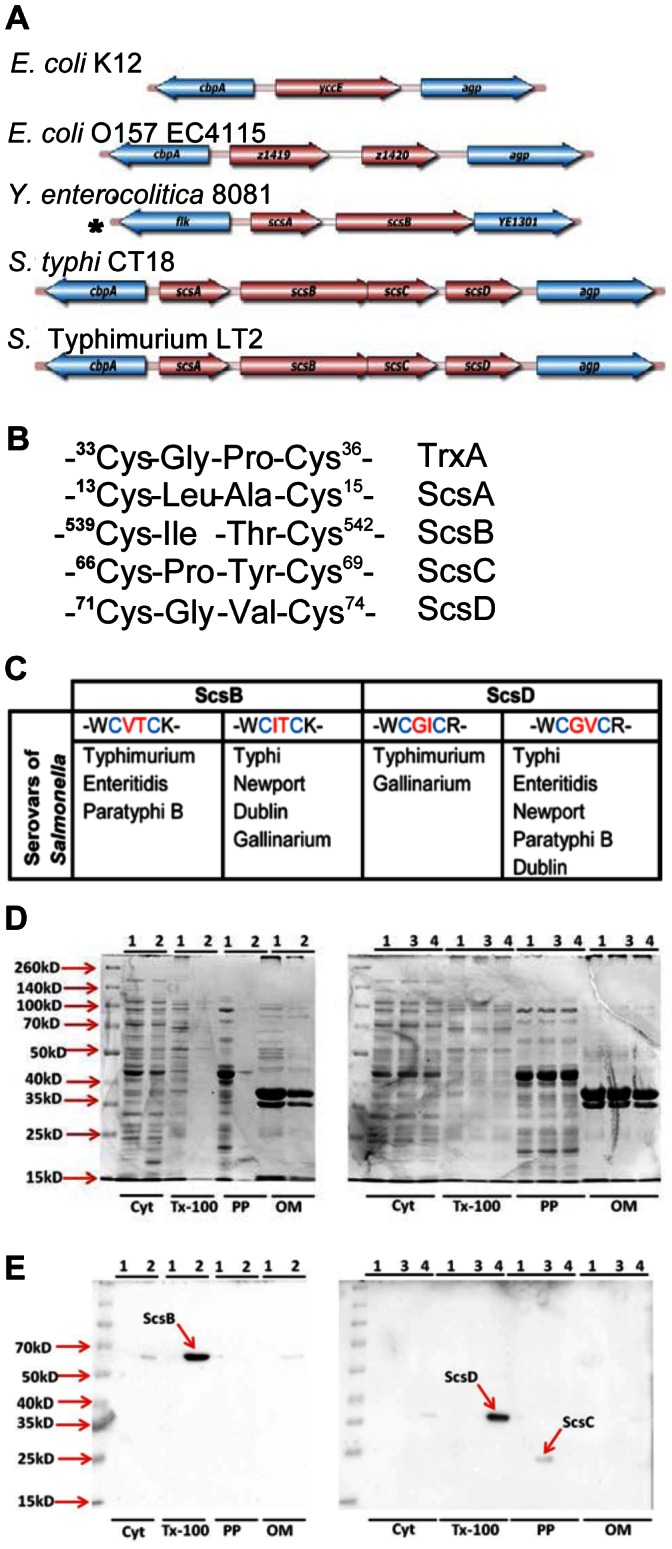
A) Illustrates the cbpA-agp genomic region in selected strains of Eschericia coli (E. coli), Yersinia enterocolitica (Y. enterocolitica) and Salmonella enterica (S. enterica); * No cbpA or agp present. B) and C) detail and compare the Cys-X-X-Cys motives of Scs proteins with the prototype TrxA and their variability in predicted SccB and ScsD proteins. D) shows Coomassie blue stained 12%SDS-polyacrylamide gels presenting sub-cellular fractionation of the proteins E) presents immunoblot with anti-His to show the localization of Scs proteins in different subcellular fractions. In (D) and (E), cyt = cytoplasmic fraction; Tx-100 = Triton X-100 soluble fraction; PP = periplasmic fraction and OM = Tx-100 insoluble integral outer membrane fraction. 1 = WT/pET32a; 2 = scsB clone (pNA15), 3 = scsC clone (pNA16) and 4 = scsD clone (pNA17).
Results
The scsABCD Gene Cluster
The predicted primary sequences of the S. Typhimurium ScsABCD proteins were presented in 1997 [24]. Since, a number of new genome sequences and protein prediction algorithms have been annotated, whereby homologues of Scs proteins have been identified in a number of diverse bacterial species [25]. Hence, we revisited the S. enterica scsABCD genes and protein predictions for the ScsABCD protein sequences in terms of in silico analyses using more recent public databases.
In the genome sequence of S. Typhimurium LT2, the scsABCD genes position between cbpA and agp genes [26]. The scs genes are contained in two transcriptional units read from the same DNA strand [24]; one consistsing of scsA followed by a second including the scsBCD genes. In contrast, E. coli K-12 and many other members of Enterobacteriacae and related organisms lack scs gene sequences, or contain only selected scs genes positioned between cbpA and agp genes (Fig.1A).
Each of the scs genes codes for a predicted protein with a single Cys-X-X-Cys motif and an enriched containment of hydrophobic amino acids between the Cys residues (Fig. 1B). Such a motif fits the hallmark of the catalytic site of the thioredoxin/glutaredoxin family of oxidoreductases. The Cys-X-X-Cys motifs for ScsA and ScsC are conserved in all annotated S. enterica genome sequences, whereas for ScsB and ScsD either one of the two residues between the cysteines is variable (Fig. 1C) potentially leading to alterations in the redox potential of the proteins [27], [28].
According to signal sequence predictions for the S. Typhimurium Scs proteins, only ScsC would contain a classical signal sequence. This would also fit with its suggested role as a periplasmic disulphide isomerase in Caulobacter cresentus [25]. ScsA and ScsB both contain a putative lipobox at their N-termini that predict the proteins to be outer membrane lipoproteins. However, for ScsA any processing at the predicted lipobox sequence would delete the Cys-X-X-Cys motif, while for ScsB the putative lipobox sequence positions at only 13 residues from the initiation methionine. Still, ScsB as well as ScsD were predicted to be integral membrane proteins.
Expression and Localization of Recombinant Scs Proteins
To get indications for the actual localization of the Scs proteins we first cloned them individually in the pBAD30 vector that allows for L-arabinose assisted inductions of recombinant proteins in S. Typhimurium. Each scs gene was amplified individually including its putative ribosome binding site at the 5′-end, and coding for C-terminal 6xHis tag. Attempts to express the proteins in S. Typhimrium only revealed signal for ScsC upon immunobloting for the His-tag at the expected mass position (data not shown). However, detection of plain ScsA, ScsB or ScsD proteins in S. Typhimurium appeared difficult.
One possibility could be extreme sensitivity to proteolytic degradation, or suboptimal induction of ScsA, ScsB or ScsD in S. Typhimurium from our recombinant plasmid constructs. In a next attempt we tried to express all Scs proteins individually in E. coli BL21 strain, deficient in the cytoplasmic Lon protease and expressing the phage T7 RNA polymerase, using the T7 promoter-based expression vector pET32a. The recombinant BL21 E.coli strain was next fractionated into a detergent insoluble cell wall fraction (representing integral outer membrane proteins), detergent soluble cell wall fraction, periplasmic protein fraction and cytosolic fraction. The fractions were separated on SDS-PAGE gels (Fig. 1D) and used for immunobloting of the His-tag. In this scenario, a band corresponding to calculated molecular mass of ScsC-6xHis clearly appeared enriched in the periplasmic fraction at 23-kDa as was detected in S. Typhimurium (Fig. 1E). This would be consistent with the predicted periplasmic localization of the protein.
Similarly, cloning of scsB with His-tag coding region into pET32a allowed for detection of a 67-kDa band in the detergent soluble fraction (Fig. 1E). This would be consistent with the predicted localization of ScsB within the inner membrane, the localization predictions recently presented for the ScsB superfamily of redox transporters [25] and with the predicted apparent mass of the fusion protein.
In selected cases, introduction of an N-terminal thioredoxin 1 tag results in stabilization of recombinant proteins. Such a strategy was considered inappropriate for ScsA as a concomitant fusion protein would be severely affected for any subsequent signal peptide processing. However, predicted to be devoid of the signal sequence, for ScsD we generated a translational thioredoxin-ScsD-His-tag fusion protein as coded for by pET32a. Upon induction in E. coli BL21, we noted an additional protein band of 36-kDa molecular mass in the detergent soluble fraction in Coomassie stained protein gel (data not shown). This matched the predicted size of a TrxA-ScsD-His fusion protein. A band at the same position was detected upon immunoblotting for the His-tag (Fig 1E). When excised from the gel, and subjected to mass-spectrometric analyses, it revealed both thioredoxin and ScsD peptides. Admittedly, this fusion protein clearly is artificial, but the analyses would be in line with an assumed cytoplasmic membrane association of ScsD, and assuming that ScsD contains a strong enough internal topological membrane insertion domain(s).
Construction of scs Deletion Mutants
In order to study any functional aspect of the scs genes in S. Typhimurium, we set out to delete each reading frame using recombinase-assisted site-directed mutagenesis [29]. For scsA, the deletion comprised the entire reading frame, but not extending into the intergenic region between scsA and scsB (for primer details, see Table S2). The open reading frames (ORF) of scsB and scsC partially overlap [24]. Therefore the deletions of the individual scsB, scsC and scsD genes were designed not to interfere with the ORFs of the rest. We also created a strain construct, a ΔscsABCD quadruple deletion mutant, lacking all the four scs genes from the 5′-end of scsA to the 3′-end of scsD ORFs. Finally, to exclude any polar effects caused by the resistance cassette used for tagging the mutations, the cassettes were removed from all deletion mutants.
The scsBCD Genes Equally Contribute to Copper Chloride Tolerance in S. Typhimurium
Copper chloride (CuCl2) sensitivity in E. coli mutants is reversed when provided with the cloned scsABCD genes from S. Typhimurium [24]. Therefore we started to probe for the contribution of scs genes to the intrinsic copper tolerance in S. Typhimurium using the deletion mutants defined above.
The ΔscsA mutant did not reveal CuCl2 sensitivity while deleting either the scsB, scsC or scsD gene resulted in a moderate but equal and highly reproducible sensitization to CuCl2 (Fig. 2A). Similarly, the ΔscsABCD quadruple deletion mutant showed the same sensitization as the individual ΔscsB, ΔscsC and ΔscsD mutants (Fig. 2A).
Figure 2. Sensitivity to copper chloride, zinc chloride and hydrogen peroxide.

A) Optical densities of overnight wild type S. Typhimurium and different scs mutants grown in the presence of CuCl2. This sensitivity is reversed by trans-complementation using the cloned scsABCD genes on plasmid pNA10 for ΔscsABCD mutant. B) shows equal sensitization to ZnCl2 for wild type S. Typhimurium and its Δscs mutants. C) Loss in viable count of the wild type S. Typhimurium strain after exposure to H2O2. Values are given as percentage relative to a hydrogen peroxide deficient control. *** = p≤0.001 D) Relative indices of viable counts of different scs knockouts compared to wild type reveals sensitization only for the ΔscsA mutant. ** = p≤0.01, *** = p≤0.001. Error bars indicate standard error of the mean.
Attempts to complement individual Δscs mutants with a cloned corresponding gene failed (data not shown). However, all copper sensitive single Δscs mutants as well the quadruple deletion mutant, were fully complemented for CuCl2 tolerance by the scsABCD genes cloned in cloning vector pSU41 (pNA10) (Fig. 2A and data not shown). None of the mutants revealed increased sensitization to another antibacterial transition metal in the form of zinc chloride (Fig. 2B).
The scsABCD Genes Protect against Protein Carbonylation
As CuCl2 mediates cysteine disulphide bond formation and potentially promotes generation of ROS, we next tested for any sensitization of the scs mutants to hydrogen peroxide (H2O2). In doing this we neither observed H2O2 sensitization for the ΔscsB, ΔscsC and ΔscsD single mutants nor for the ΔscsABCD quadruple mutant (Fig. 2D). Notwithstanding the fact, the ΔscsA single mutant showed increased sensitivity to H2O2 (Fig. 2D).
Apart from mediating disulphide bond formation, ROS also results in protein carbonylation in S. Typhimurium [17]. Thus, we assayed protein carbonylation in cultures of S. Typhimurium exposed to 0.75 mM H2O2 to detect any discernible differences. To exclude any confounding effects by the ΔscsA single mutant, we continued using the ΔscsABCD mutant only. The rationale for this was that this mutant lacked all the scs genes yet did not reveal sensitization to H2O2. In this experiment we detected a more pronounced accumulation of carbonylated proteins in the ΔscsABCD mutant (Fig. 3A), notably in the periplasmic fraction (Fig. 3C). Expressing the cloned scsABCD genes from pNA10 in the quadruple mutant reduced the H2O2-mediated protein carbonylation (Fig. 3B). That rather few proteins became increasingly carbonylated in the ΔscsABCD mutant could explain why this scs mutant did not display increased loss of viability upon exposure to H2O2. Still, these series of experiments strongly implicated that the Scs proteins are involved in balancing periplasmic oxidative stress in S. Typhimurium.
Figure 3. Carbonylated protein profiles after exposure to hydrogen peroxide.

Protein carbonylation was detected by immunoblotting after coupling to dinitrophenol and protein separation on 12% SDS-PAGE gels. A) Whole cell lysate obtained from the ΔscsABCD mutant reveals a higher concentration of carbonylated proteins after exposure to 0.75 mM hydrogen peroxide (B). Trans-complementation with cloned scsABCD (pNA10) reduces the level of protein carbonylation in the whole cell fraction, as well as in the periplasmic fraction (C). DNPH = Dinitrophenyl hydrazine (+) = presence (−) = absence. p+ indicates the trans-complementing scsABCD genes in pNA10. p- indicates the vector control pSU41.
S. Typhimurium Still Relies on trxA for Copper Chloride Tolerance
TrxA also contributes to copper tolerance in E. coli [30]. We thereby set out to test whether the presence of scsABCD genes made trxA dispensable for CuCl2 tolerance in S. Typhimurium. We noted a decreased CuCl2 tolerance of a S. Typhimurium ΔtrxA mutant that in magnitude equaled to that of the ΔscsABCD mutant (Fig. 4A). S. Typhimurium lacking trxA as well as the scsABCD genes did not show any further sensitization to CuCl2 (Fig. 4A).
Figure 4. Sensitivity to copper chloride and invasion in MDCK epithelial cell line, with or without copper chloride stress.

A) Optical density of overnight wild type S. Typhimurium, ΔscsABCD and different ΔtrxA mutants grown in the presence of CuCl2. B) The compromised invasion of a ΔtrxA deletion mutant can be trans-complemented with cloned trxA (pFA3) but not with a plasmid coding for catalytically inactive TrxA (pFA8). C) The scsABCD deletion acts as a suppressor mutation for the decreased invasion of the ΔtrxA mutant. D) and E) The viability of ΔscsABCD and ΔtrxA under CuCl2 stress (3 mM) is compromised, yet the invasiveness is highly enhanced at this concentration. F) shows fold changes in invasion of 3 mM CuCl2 treated culture relative to the untreated culture. ns = non-significant; * = p≤0.05; ** = p≤0.01; *** = p≤0.001.The error bars indicate standard error of the mean.
Redox Sensitivity and Dependency on scsABCD Genes for in vitro Invasiveness of S. Typhimurium
Full invasiveness of S. Typhimurium in mammalian cell cultures relies on trxA [20]. As trxA and the scsABCD mutants revealed an equal sensitization to CuCl2, we set testing that to what extent the invasiveness was affected in our mutants, and whether invasiveness would be influenced by CuCl2. For this, the cultures were grown to induce SPI1 expression in the absence or presence of CuCl2 for 4 hours, and subsequently exposed to MDCK epithelial cells without CuCl2 for one hour.
When cultures were propagated in the absence of CuCl2, we corroborated the decreased invasion reported for a ΔtrxA mutant [20] (Fig. 4B). This decrease in invasiveness was retained when the mutant was provided with the cloning vector pBAD33 or when provided with a pBAD33-derivative coding for a catalytically inactive TrxA. When complemented with the wild type trxA in pBAD33, invasiveness was enhanced above wild type levels (Fig. 4B). These observations clearly implicate a role for TrxA in invasiveness of S. Typhimurium.
The ΔscsABCD quadruple mutant invaded as efficiently as the parental wild type strain (Fig. 4B). Surprisingly though, deleting the scsABCD genes from the ΔtrxA mutant resulted in increased invasiveness (Fig. 4C).
When the cultures were grown to induce SPI1-mediated invasiveness at 3 mM of CuCl2 we noted a moderate decrease in colony forming units in relation to the optical density of the cultures for the ΔscsABCD and ΔtrxA mutants (Fig. 4D). Still, while not reduced for viability, the invasiveness of the wild type S. Typhimurium strain was virtually lost upon growth under CuCl2 stress (Fig. 4E and 4F). In contrast, even though the ΔtrxA mutant showed decreased invasion under ordinary growth conditions, the invasion index for the mutant became increased by pre-exposure to CuCl2 (Fig. 4E and 4F). Also, the relative invasiveness of both the ΔscsABCD and ΔscsABCD/ΔtrxA mutant was much retained upon growth in the presence of CuCl2 (Fig. 4E and 4F). These results showed that invasiveness of S. Typhimurium is strongly reduced by CuCl2, yet that this chemical attenuation depended on the Scs and TrxA oxidoreductases.
As the invasiveness of S. Typhimurium in cell cultures strongly relies on SPI1 T3SS and the fact that scsABCD mutant retained invasiveness at CuCl2 stress evoked the question whether this enhanced invasion of the scsABCD relied on SPI1 T3SS. Therefore, we inserted a polar Tn5::lacZY mutation in the scsABCD mutant inactivating the main SPI1 transcriptional activator gene hilA, and another in the prgH gene coding for a central component of the SPI1 T3SS apparatus. When exposed to CuCl2, the accompanying hilA and prgH mutants failed to reveal invasiveness in the MDCK cell based assay, to an extent that approached our detection limit (data not shown). Thus, the retained invasiveness of the scsABCD mutant at CuCl2 exposure continued to be SPI1-dependent.
The scsABCD and SPI1 Gene Expression
That the scsABCD mutant retained invasiveness upon CuCl2 stress in a SPI1-mediated manner led us to test whether the scs gene cluster affected SPI1 gene expression. For this we measured SPI1 gene expression using the two forth mentioned SPI1 Tn5::lacZY constructs. When grown over night in LB we noted clear reading of the hilA::lacZY and prgH::lacZY transcriptional fusions by assaying beta-galactosidase activities (Fig. 5A and B). Addition of CuCl2 to the culture prior to the inoculation resulted in an overall decreased reporter activity with a slight relative decrease in both fusions for the scsABCD mutant (Fig. 5A and B). However, this difference disappeared when bacteria were grown for invasiveness in cull culture medium supplemented with CuCl2 (Fig 5C and D).
Figure 5. β-galactosidase and β-lactamase activities and expression measurements.

A) and B) β-galactosidase activities for hilA and prgH promoter fusions from overnight LB culture are slightly reduced for scsABCD mutant in the presence of CuCl2. C) and D) β-galactosidase activities for hilA and prgH promoter fusions from 4 hrs DMEM-based invasive culture are unaffected irrespective of scsABCD mutation. β-lactamase activities from supernatants and cell lysates of the invasive cultures of wild type and in ΔscsABCD mutant bacteria in the absence (E) and presence (F) of CuCl2. G) β-lactamase as assayed from the same cultures by immunoblotting for β-lactamase. ns = non-significant; ** = p≤0.01; *** = p≤0.001. Error bars indicate standard error of the mean.
The scsABCD Genes and SPI1 Activity
To address whether scsABCD genes affected SPI1 activity, we followed the expression and secretion of a SPI1 effector fusion protein. For this we used plasmid pAUN1 that carries the 5′-end of the effector protein gene sipB translationally fused to TEM beta-lactamase gene devoid of its region coding for the signal sequence [31]. We chose to use cultures propagated in cell culture medium as this is the medium used to generate invasion competent bacteria, and as the lacZ reporter activities did not differ between the wild type and mutant in this medium (Fig. 5C and D). When grown to generate invasiveness, we noted a clear presence of the SipB-Bla fusion protein in the culture supernatant and cell lysates from the wild type bacteria carrying pAUN1 as indicated by the enzymatic activity of the SipB-Bla fusion protein (Fig. 5E). For the scsABCD mutant we could likewise detect enzymatic activity from both fractions. However, as compared to the activities expressed by the wild type, the proportion of the extracellular activity was reduced and the cell-bound activity increased in the scsABCD mutant (Fig. 5E). The enzymatic activity became much reduced when the medium was supplemented with CuCl2 at 2 mM (Fig. 5F), and decreased below detection limit at 3 mM (data not shown). Interestingly though, at 2 mM CuCl2, the proportions of enzymatic activities for cell-bound and secreted fusion protein appeared the same for the wild type and scsABCD mutant (Fig. 5F).
To exclude any potential distractions emerging from alternations in specific enzyme activity generated by CuCl2, we also assayed for the SipB-Bla fusion protein by immunoblotting. In the absence of CuCl2, the amounts of the fusion protein in the supernatant appeared much higher in the wild type, and with a substantial accumulation in cell lysate for the ΔscsABCD mutant (Fig. 5G). At 2 mM CuCl2 the signals for the fusion protein decreased, but remained detectable upon longer exposure. Still, the proportions of the fusion protein in the extracellular versus intracellular fraction of the wild type appeared more pronounced compared to the ΔscsABCD mutant (Fig. 5G).
Combined, these observations indicated that while SPI1 expression was redox sensitive, the scs gene cluster did affect prgH expression in LB. Under conditions where the scsABCD deletion mutant did not reveal difference in prgH expression, the deletion still affected secretion of a SipB fusion protein.
The scsABCD Genes, Intracellular Replication and in vivo Virulence of S. Typhimurium
Apart from affecting invasion, TrxA strongly affects bacterial intracellular replication in RAW264.7 macrophage-like cells, as well as virulence in BALB/c mice [20]. Regarding intracellular replication in RAW264.7 cells we did not note any significant deviation for the ΔscsABCD mutant compared to the wild type (Fig. 6A).
Figure 6. Replication in RAW264.7 macrophages and mice, and invasion in ileal loops of mice.
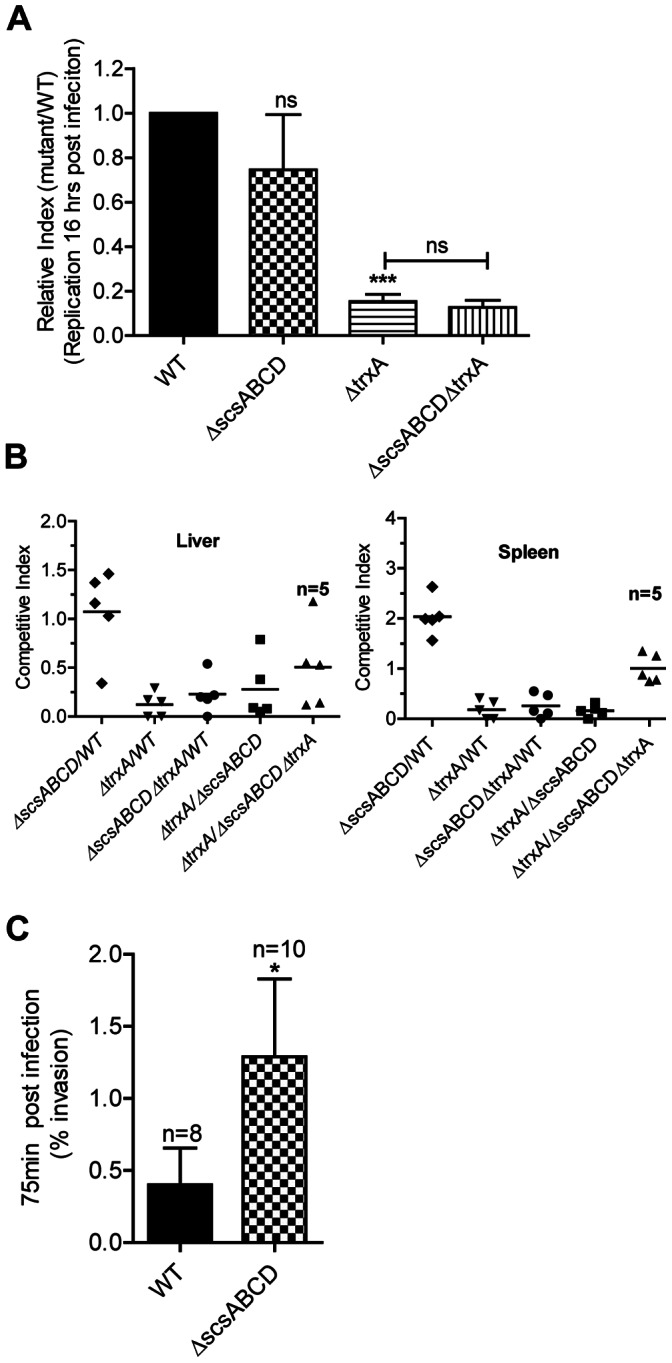
A) The ΔscsABCD does not restore replication of ΔtrxA in macrophage cell line RAW 264.7 B) The ΔscsABCD mutation shows a moderate increase in spleenic load in BALB/c mice on intraperitoneal challenge; n = number of mice C) The ΔscsABCD invades more efficiently in ileal loop ligation experiment. n = number of loops. ns = non-significant; * = p≤0.05; *** = p≤0.001. Error bars indicate standard error of the mean.
Upon intraperitoneal challenge the ΔscsABCD mutant showed a moderate relative increase in spleenic loads three days post challenge (Fig. 6B). When comparing the ratios of wild type and mutant bacterial ratios by a nonparametric test, the difference did not breach statistical significance. However, we also extracted the actual viable counts of individual strains from the data to allow the use of a parametric test. In this, the differences in viable counts became statistically significant (P<0.05). Despite this enhancement in the replication of scsABCD mutant, the ΔscsABCD deletion neither restored the strong attenuation exhibited by the ΔtrxA mutant background in RAW264.7 nor in mice (Figs. 6A and 6B). In other words, in contrast to the decreased invasiveness, the attenuation of the ΔtrxA mutant in RAW264.7 cells and BALB/c mice evidently did not depend on the scs genes.
SPI1 is important in mediating invasion of S. Typhimurium from the intestine of mice. However, the redox conditions in the intestine are likely to deviate from the ones prevailing in cell culture settings. Hence we continued by comparing invasiveness of wild type and ΔscsABCD mutant of S. Typhimurium in ileal loops of BALB/c mice. In these experiments, the ΔscsABCD mutant indeed revealed an increase in numbers of invading bacteria (Fig. 6C).
The scsABCD Gene Cluster Conditionally Suppresses Virulence in C. elegans
Feeding C. elegans with S. Typhimurium results in a significant shortening of the nematode life span, which in part relies on a massive hypodermal oxidative stress response in the nematode [22]. This stress response is initiated at the intestine some 24 hours post infection, and at 36 to 48 hours post infection fills the nematode hypodermal space, as revealed by staining with the fluorescent ROS indicator 2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA) [22]. Nematodes fed with a ΔtrxA mutant of S. Typhimurium lack this oxidative response and gain a significant restoration of their life span [22]. Therefore, we set out to test whether scsABCD genes would bear any role in the pathogenesis of S. Typhimurium in C. elegans.
C. elegans infected with the ΔscsABCD mutant of S. Typhimurium behaved as nematodes infected with the wild type regarding life span (Fig. 7A). Still, transfer of the scsABCD deletion into the ΔtrxA mutant resulted in retrieved virulence as evidenced by a shortening of the nematode life span (Fig. 7A).
Figure 7. Virulence and ROS dissemination are restored in C. elegans by removing ΔscsABCD in the ΔtrxA mutant.

A) Survival of wild-type N2 nematodes was compared when infected with S. Typhimurium 14028 wild-type, ΔtrxA, ΔscsABCD or the ΔscsABCD/ΔtrxA mutant. B to I). Nematodes were harvested 48 or 72 hours post infection and stained with H2DCFDA to detect intracellular ROS. In these images, ROS is shown in green and intestinal autofluorescence in blue. Images are shown at 40x magnification and are representative of at least 20 nematodes from 2 independent assays. J) Florescence intensities are compared with wild type and between different mutants. ns = non-significant, ** = p≤0.01, **** = p≤0.0001. Error bars indicate the standard error of the mean.
Nematodes infected with the wild type or ΔscsABCD mutant evoked an apparently equal hypodermal ROS staining with H2DCFDA (wild type, Figs 7B and C; ΔscsABCD mutant, Figs. 7F and G). This observation was corroborated by quantification of oxidative stress induced fluorescence of H2DCFDA (Fig. 7J). To corroborate our previous finding [22], the oxidative stress response was much reduced upon infection with the ΔtrxA mutant (Figs. 7D and E, Fig. 7J). Surprisingly, infection with the ΔscsABCD/ΔtrxA mutant accompanied with a substantial restoration in the hypodermal oxidative stress such that the H2DCFDA staining reached wild type levels at 72 h post infection (Figs. 7H and I, Fig. 7J).
Thereby, the scsABCD genes affected the ΔtrxA mutant phenotype not only regarding invasiveness of epithelial cells, but also regarding the virulence and pathogenesis of the ΔtrxA mutant in C. elegans.
Discussion
Reactive oxygen species play an important role in the pathogenesis of many bacterial species both in cell culture setting and for virulence in man, mice and nematodes [12], [13], [14], [15], [16], [17]. In E. coli selected oxidoreductases of the thioredoxin superfamily, including thioredoxin 1 (TrxA) itself, are not only needed for tolerance to H2O2 but also for resistance to the redox active transition metal copper [30], [32], [33]. Copper ions mediate non-enzymatic catalysis of protein disuplhide formation [34], [35], which mimics a facet of oxidative stress that interferes with periplasmic disulphide bond formation. S. enterica carries a gene cluster, the scsABCD genes which, when expressed in copper sensitive laboratory strains of E. coli, enhances their copper tolerance [24]. Recently, copper was coupled to the ability of phagocytes to control infection with S. Typhimurium [36]. Furthermore, when S. Typhimurium replicates in macrophages, the scs genes are significantly induced for their expression [37]. Given the coupling between tolerance to oxidative stress and copper [30], [32], [33], we have searched for a role of the ScsABCD proteins, all putative members of the thioredoxin superfamily of oxidoreductases, for oxidative stress tolerance and virulence in S. Typhimurium.
Individual ΔscsB, ΔscsC, ΔscsD and ΔtrxA mutants showed equal sensitization to copper chloride, and that deleting the ΔscsABCD genes in a ΔtrxA mutant did not cause further sensitization. This deduced that the scsABCD and trxA genes contributed to copper tolerance through a common pathway. This conclusion would fit well with recent analyses in C. cresentus where ScsB represented a DsbD-like cytoplasmic membrane electron transporter and ScsC, a putative periplasmic disulphide isomerase and a reduction substrate for ScsB [25]. TrxA is also known to provide DsbD with electrons [38], which could adopt a connection between the ScsABCD system and TrxA in mediating copper tolerance. Exposure to H2O2 resulted in protein carbonylation both in the wild type S. Typhimurium and in the strain lacking the scsABCD genes. Still, the level of protein carbonylation was higher in the ΔscsABCD mutant as compared to wild type, notably in the periplasmic sub-cellular fraction. Such results further implicate a role of the Scs proteins in balancing redox stress in S. Typhimurium.
ScsA did not add to CuCl2 tolerance despite the presence of a Cys-X-X-Cys motif. However, this motif resides within its predicted signal sequence. Hence a processed ScsA would be missing a thioredoxin-like catalytic motif. Such a scenario invites for speculating that processing of ScsA could rely on disulfide formation in the putative signal sequence, and hence depend on periplasmic redox status for processing and transport. Still the ΔscsA mutant showed increased sensitization to H2O2, while no such sensitization was noted for any of the other scs deletion mutants or for the ΔscsABCD mutant. That is, an imbalance in the Scs protein content rather than lack of ScsA might explain the selective sensitization to H2O2. Such reports have already been published with periplasmic protein DsbA, where overexpression of DsbA protein suppresses the motility contrary to the requirement of DsbA for proper motility [39]. This said, we could not at present functionally distinguish ScsA from the rest of the Scs proteins or TrxA. The strong contribution of thiol peroxidase and methionine sulfoxide reductases for protection from H2O2 mediated killing in S. Typhimuirum [17] could further explain the redundancy of ScsBCD proteins in mediating any H2O2 stress tolerance in vitro.
When the ΔscsABCD mutant was tested in cell culture based virulence assays we did not note any alterations in its ability to invade epithelial cells or to replicate in macrophage-like cells. We did however observe that CuCl2 blocked invasion with extreme efficacy. Furthermore, this decrease in invasion strongly relied on the scsABCD and trxA genes. When given intraperitoneally to BALB/c mice, the ΔscsABCD mutant showed a moderate enhancement in fitness as compared to the wild type. Also, when assaying invasion in ileal loops of mice, we noted an enhanced invasion efficacy for the ΔscsABCD mutant.
Apart from exhibiting decreased invasiveness in vitro, a ΔtrxA mutant of S. Typhimurium also reveals strong virulence attenuation in the soil nematode C. elegans [22]. Here we demonstrate that both these attenuations depended on the scs genes. That is, selected attenuations caused by CuCl2 or TrxA-deficiency are “conditionally” dependent on the scs genes.
Conditional phenotypes for oxidoreductase mutants of the thioredoxin superfamily have previously been described in E. coli. For example, H2O2 tolerance of E. coli relies on TrxA [30]. Surprisingly though a thioredoxin reductase (TrxB) deficient mutant while sensitive to H2O2 in stationary phase, shows even an increased tolerance to H2O2 in logarithmic phase of growth. This conditionality with H2O2 sensitivity comes from the fact that TrxB-deficiency associates with increased catalase expression evidently induced by accumulated oxidized TrxA [33]. A second example comes from the conditional contribution of oxidoreductases to copper chloride. In E.coli, lack of the periplasmic disulfide isomerase DsbC results in copper chloride sensitization, while deleting the disulfide oxidase DsbA does not [40]. Still, CuCl2 sensitivity is enhanced in strains lacking both DsbA and DsbC. The explanation to this apparent contradiction may come from CuCl2 itself; CuCl2 is an efficient but evidently a non-specific oxidant that introduces non-native disulfide bonds in periplasmic proteins such as alkaline phosphatase [40]. Thus in the absence of a proteins catalyzing and proof-reading disulphide bond formation, CuCl2 might cause accumulation of wrongly oxidized proteins to the extent of compromising bacterial fitness.
Assuming the Scs system participates in periplasmic redox shuffling, its conditional effects on virulence could relate to mechanisms similar to that proposed for the TrxA-TrxB and DsbA-DsbC interactors; presence of CuCl2 or TrxA-deficiency could cause differential Scs-mediated accumulation of oxidized periplasmic proteins. Such accumulating proteins, being the Scs proteins themselves or their substrates, could subsequently relate to altered T3SS activity, and thus explain the containment of the scsABCD genes in S. enterica. Indeed, the DsbA activity of S. Typhimurium has been connected to both SPI1 gene expression and in the SPI1 T3SS apparatus functionality [41]. Such connections led us to probe for the impact of the scsABCD gene cluster for SPI1 gene expression and secretion potential. When applying CuCl2 stress in cell culture medium, the transcriptional activity of the hilA and prgH promoters were reduced, as was the expression of the SipB-Bla fusion protein. Still, whether applying copper stress or not, the ΔscsABCD mutant appeared defective in secreting the SipB-Bla fusion protein. Clearly, the fusion protein does not represent an original T3SS effector protein, but the results still implicate a role for the Scs system in balancing SPI1 T3SS activity.
The above findings also provide a plausible mechanistic explanation for the invasiveness of the mutant. Under ordinary conditions the expression of SPI1 T3SS mediated invasion genes in wild type is highly active but leaky allowing secretion of effector proteins even in the absence of host cell contact. In the ΔscsABCD mutant the apparatus is less active, or stricter. Effector proteins accumulating inside mutant bacteria create a secretion competent pool applicable even after copper induced down-regulation of SPI1 gene expression, and notably, only used for translocation. Effectors secreted by SPI1 activate host JNK and p38 kinases, as well as nuclear localization of transcription factor NF-kB [9]. These kinases and transcription factors are described to potentiate expression of the inducible nitric oxide synthase [42], [43] as well as the phagocyte oxidase subunit gp91 [44]. Thereby a rational for S. enterica maintaining the scs genes could relate to the ability of regulating SPI1 secretion activity.
The redox-dependency of T3SS is not restricted to S. Typhimurium. The activity of the virulence-associated T3SS of Pseudomonas aeruginosa and Shigella flexneri relies on their DsbA homologues [45], [46]. Furthermore, expression of fimbrial adhesins in uropathogenic and enterotoxogenic E. coli [47], [48] as well as secretion of cholera toxin in Vibrio choleare [49] rely on corresponding DsbA homologues. Such reports in combination with our data on the scs genes implicate dependence and a highly balanced interaction of virulence factors with evolutionary conserved bacterial oxidoreductases belonging to the thioredoxin superfamily. Thereby, phenotypes caused by mutations in genes for oxidoreductases could be conditional in more general terms, and not restricted to scs genes in S. Typhimurium. If indeed so, then one must consider that mutating oxidoreducates would not only cause alterations in virulence but also act as suppressor mutations.
Materials and Methods
Ethical Statement
The work did not involve human subjects or non-human primates. However, the mice were used in this study and the ethical permits were obtained from Stockholm Norra djurförsöksetiska Nämd with N491/11 for intraperitoneal challange and N172/08 for ileal loop infections. The animals were housed and monitored according to the national and international guidelines.
In silico Analyses
The S. Typhimurium strain LT2 and 14028s genome sequences annotated in 2001 [26] and 2010 [50] respectively, were used as reference for all the in silico analyses and primer design. Sequences from additional genome annotations were searched and compared in NCBI protein cluster for Cys-X-X-Cys motives. Search for Lipobox, classical signal peptide sequences and cleavage sites were conducted using the LipP 1.0 and SignalP servers [51], while protein localization and topologies were predicted using PSORTb vs3.0.2 and the Phyre2 server respectively [52].
Bacterial Strains, Plasmids, Phages and Nematodes
S. Typhimurium 14028 (ATCC, Manassas, VA, USA) was used as the wild type S. Typhimurium strain throughout the study. The plasmids used for site-directed mutagenesis [29] were pDK3, pDK4 and pDK46. For cloning purposes, the pBAD derivative pBAD33, the pACYC-184 derivative pSU41 and T7 promoter based expression vector pET32a (Novagen) were used [53], [54], [55]. The primers for cloning are given in Table S1. Cultures were propagated in Luria broth or on Luria agar plates with 10g sodium chloride per liter (Duchefa Biochemie, The Netherlands). For generating invasive bacterial cultures, bacteria were propagated in defined cell culture medium as given below. When necessary, growth media were supplemented with ampicillin (100 µg/ml), chloramphenicol (10 µg/ml), kanamycin sulfate (50 µg/ml) or tetracycline (10 µg/ml). To activate the arabinose promoter in pBAD33, cultures were supplemented with L-arabinose to a final concentration of 1% (w/v). All antibiotics and L-arabinose were from Sigma-Aldrich (St. Louis, MO, USA). Phage P22 int transduction was used to transfer mutations between strains [56].
The C. elegans strain used in this study was wild-type variant Bristol N2. Nematodes were cultured and maintained at 20°C on modified nematode growth media (NGM, 0.35% peptone) agar plates and fed with E. coli strain OP50, as described [57]. Both strains were obtained from the Caenorhabditis Genetics Center (Minneapolis, USA).
Bacterial strains and plasmids used and generated in this study are summarized in Table 1.
Table 1. Strains and plasmid.
| Strains | Genotype/Property | Reference |
| MC5 | Wild Type | ATCC 14028 |
| Fia-1280 | Wildtype::Tetr | [63] |
| NA48 | 14028(ΔscsA) | This study |
| NA49 | 14028(ΔscsB) | This study |
| NA50 | 14028(ΔscsC) | This study |
| NA51 | 14028(ΔscsD) | This study |
| NA91 | 14028(ΔscsABCD) | This study |
| NA198 | 14028(ΔscsABCD/ΔtrxA) | This study |
| Fia-1195 | 14028(ΔtrxA) | [20] |
| Fia-1381 | 14028(ΔprgH::Tn5 lacZY) | [63] |
| Fia-1199 | 14028(ΔhilA::Tn5 lacZY) | [63] |
| Plasmids | ||
| pSU41 | pACYC-184 derivative vector control, Kanr | [54] |
| pNA10 | The scsABCD gene cluster cloned in pSU41 with 209 bp upstream region of scsA, Kanr | This study |
| pAUN1 | sipB-β-lactamase fusion cloned in pSU41, Kanr | [64] |
| pET32a | T7 promoter based expression vector | [55] |
| pNA14 | The scsA cloned in BamHI and XhoI site of pET32a, Ampr | This sutdy |
| pNA15 | The scsB cloned in EcoRI and XhoI site of pET32a, Ampr | This sutdy |
| pNA16 | The scsC cloned in BamHI and XhoI site of pET32a, Ampr | This sutdy |
| pNA17 | The scsD cloned in BamHI and XhoI site of pET32a, Ampr | This sutdy |
| pBAD33 | pBAD series vector control, Cmr | [53] |
| pFA3 | Full length trxA cloned in pBAD33, Cmr | [20] |
| pFA8 | Catalytically inactive trxA cloned in pBAD33, Cmr | [20] |
Generation of Knockout Mutants
Individual scs genes, or the entire scsABCD locus, were deleted in the wild type S. Typhimurium using phage recombinase-assisted homologous recombination [29]. Briefly, the resistance genes from pDK3 or pDK4 were PCR-amplified with primer extensions homologous to individual scs genes, as defined in Table S2. PCR products were electroporated into the recipients containing pDK46 and recombinants selected using proper antibiotic selection. To exclude possible secondary mutations, the inserted cassettes were transduced by the phage P22 int into a fresh S. Typhimuirum 14028 background. Finally, the antibiotic resistance cassettes were removed with the aid of plasmid pCP20 coding for recombinase [29].
All mutants were verified by PCR amplification of inserted resistance cassette with primers designed 100 bp up- and downstream of the target genes respectively, as listed in Table S3.
Protein Expression and Cell Fractionation
Individual scs genes with C-terminal His-tags were cloned into the cloning site of pET32a expression vector. The clones were transformed to E.coli BL21 strain. The cultures were grown overnight in 2 ml LB and were subcultured as 1∶100 in 20 ml of LB with aeration at 37°C. The cultures were either induced with 2 mM IPTG or left uninduced and samples were collected after 3 hrs. Harvested cultures were fractionated into cytoplasmic, Triton X-100 (Tx-100) soluble, Tx-100 insoluble and periplasmic proteins as described in [58], [59] and [60] respectively.
For generatio of Tx-100 soluble, Tx-100 insoluble fractions, the bacteria were pelleted and the pellets were resuspended in PBS on ice. Chilled suspensions were sonicated in 4 ml volume using the Soniprep 150 SANYO at 12 micron amplitude for 2×1.5 min on ice. Whole Cells were removed by centrifugation at 5000 rmp for 10 min. Supernatants were collected and centrifuged for 15 min at 14000 rpm and 4°C. The supernatant was collected as cytoplasmic soluble fraction. The pellet was solubilized in cytoplasmic membrane solution; 50 mM Tris-HCl, 10 mM MgCl2, 1% (v/v) Triton X-100 buffer (pH 8.0), and centrifuged aforesaid. The supernatant containing cytoplasmic membranes was collectd as the TX-100 soluble fraction. The pellet containing integral outer membrane proteins was solubilized in reducing SDS sample buffer.
For periplasmic proteins, the pellet from 5 ml culture was resuspended in 50 µl of chloroform, vortexed and kept at room temperature (22°C) for 15 minutes. Five hundred microlitre of 0.01 M Tris hydrochloride (pH = 8.0) was added and mixed. After centrifugation at 6000xg for 20 minutes, the aqueous supernatant containing periplasmic proteins was collected [60].
Fractions were separated using SDS-polyacrylamid gel electrophoresis [61] and transferred to PVDF membrane by using iblot system (invitrogen) The proteins were detected by using HRP conjugated monoclonal anti-His antibody and ECL substrate (SuperSignal West Pico Chemiluminescent Substrate, Thermo Scientific). Biorad Gel doc machine was used for signal capture.
For mass-spectrometry, protein bands were cut from the gel, washed with water and reduced by DTE and alkylated with iodoacetamide. Proteins were digested with trypsin (Promega) in 50 mM NH4HCO3 at 37°C overnight. Extracted peptides were analyzed with MALDI-Tof analysis using a Bruker Ultraflex-Tof/Tof instrument. Identification was done by using the peptide maps for search in the NCBInr database using the Mascot search engine.
Copper Chloride, Zinc Chloride and Hydrogen Peroxide Tolerance
Bacteria were grown overnight on Luria agar (LA) plates and re-suspended in phosphate buffered saline (PBS, pH = 7.4). For testing sensitivity to either CuCl2 or ZnCl2 (Sigma Aldrich), bacteria were inoculated from PBS into 2 ml of LB to contain 103 bacteria per ml. Cultures were supplemented with CuCl2 to a final concentration of 0 mM, 2 mM, 4 mM or 6 mM. For ZnCl2, the final concentration was adjusted to 0 mM, 0.25 mM, 0.5 mM, 0.75 mM, 1.0 mM, 1.25 mM, 1.5 mM, 1.75 mM and 2.0 mM respectively.Tubes were incubated at 37°C overnight on Brunswick roller. Overnight cultures were diluted 1∶10 in PBS and OD600 were recorded, taking respective reaction mixture as blank. For H2O2 test, 103 bacteria in 100 µl of PBS were mixed with 100 µl of H2O2 (Sigma) solution in 96-well microtitre plate, in triplicate, giving final concentration of H2O2 as 0 mM, 1 mM, 2 mM and 4 mM in PBS respectively. Plates were incubated at 37°C for 2 hours and 100 µl from each well was spread on LA plate. The seeded plates were incubated overnight at 37°C. Viable counts were determined by counting CFU on plates and percent viability as related to wells devoid of H2O2 were calculated.
Detection of Protein Carbonylation
The protocol detailed by [17] was followed with slight modifications. Overnight 2 ml LB cultures of S. Typhimurium were diluted 1∶100 in fresh 25 ml LB and grown for 3.5 hrs at 37°C at 200 rpm. Cultures were exposed to H2O2 at a final concentration of 0.75 mM and incubated for further 3 hours at 37°C without shaking. Cells were then centrifuged at 5000 rpm and pellet was resuspended in 1 ml of 10 mM Tris-HCl, 25 mM NaCl and 50 mM DTT (pH 7.5) and transferred to 2 ml Eppendorf tube. Cells were mechanically lysed using the Soniprep 150 SANYO at 20 micron amplitude for 3 min on ice. The cell debris was removed by centrifugation at 14000 rpm for 10 minutes. After another centrifugation step, the clear supernatant was collected.
The periplasmic proteins were isolated according to [60] and as described above.
Oxidized proteins content was detected by following the manual provided with OxyBlot™ protein oxidation detection kit (Millipore). Briefly, 5 µl from each culture supernatant was mixed with 5 µl of 12% SDS. After 5 min 10 µl of the derivatizing reagent 1 x DNPH (dinitrophenyl hydrazine) was added and incubated at room temperature (22°C) for 15 min. Reaction was stopped by adding 7.5 µl neutralization solution provided in the kit. The samples were analyzed on a 12% SDS-PAGE.
The proteins were blotted onto a PVDF membrane (Amersham Hybond™-P, GE Healthcare), and subsequently were blocked overnight in blocking buffer (PBS with 1% BSA and 0.05% Tween 20). The membrane was incubated in rabbit anti-DNP-antibody (1∶150), diluted in blocking buffer, for 1 hr at room temperature with shaking. After subsequent washing with PBS-T (0.05%) goat anti-rabbit IgG horseradish peroxidase conjugate (1∶300), diluted in blocking buffer, was applied for 1h. Oxidized proteins were detected by using the ECL substrate (SuperSignal West Pico Chemiluminescent Substrate, Thermo Scientific) and Biorad Gel doc machine.
In vitro Invasion Assays
To generate invasive cultures of S. Typhimrium 14028, overnight cultures propagated in Luria broth were diluted (1∶10) in 4 ml D-MEM (Gibco) supplemented with 10 mM HEPES and 10 mM L-Glutamine as described in [20]. The cultures were grown in 15 ml polystyrene plastic tubes under shaking for 4 hours lacking or being exposed to 3 mM CuCl2. MDCK epithelial cell line was maintained and infected as described pereviously [20]. Briefly, the confluent cells in 24 well plates were exposed to 1×106 bacteria (MOI 10∶1) diluted from the invasion competent culture in 1 ml buffered D-MEM devoid of serum and gentamicin. Invasion was synchronized by briefly centrifuging the plates, where after plates were incubated for 1 hour at 37°C. After washing the monolayers twice with PBS, cells were covered with 10%FBS supplemented D-MEM containing 50 µg/ml of gentamicin for 45 minutes. Subsequently, the cells were washed twice with PBS and bacteria were released by lyzing cells in 0.5% sodium deoxycholate in PBS (Merck, Darmstadt, Germany). Invasion efficacy was defined as the ratio of recovered CFU in relation to the bacteria enumerated from the inoculum.
Promotor-fusions and β-galactosidase Measurements
The transcriptional activities for hilA::lacZY and prgH::lacZY promotor fusions were determined as described previously [62] from overnight cultures in LB, or from bacteria grown to induce invasiveness in cell culture medium. The hilA::lacZY and prgH::lacZY constructs, received from Catherine A. Lee [63], were transduced in scsABCD mutant and wild type 14028 background.
SipB-β-lactamase Fusion Proteins Determination
Activity of the fusion protein was assayed either enzymatically or by immunoblotting. For determining the β-lactamase activities, pAUN1 plasmid carrying sipB-β-lactamase fusions were transformed in respective strains and enzymatic activities from the supernatants and whole cell lysates were determined as described by Negrea et al. [64]. Briefly, the invasive cultures, optically normalized, were centrifuged at 5000 rpm to collect the clear supernatant and the cells pelleted. Pellets were resuspended in PBS and were sonicated as described above. The proteins from the supernatant as well as from cell lysate were precipitated with 10% final concentration of trichloro acetic acid and resuspended in reducing SDS sample buffer. Equal volumes of protein preparations were run on SDS-polyacryamide gel and blotted to PVDF membrane with iblot-system (Invitrogen). The proteins were detected by using Rabbit-anit-β-lactamase antibody (Abcam) in 1∶3000 dilutions. After treating with anti-rabbit HRP-conjugated secondary antibody, the bands were detected as described earlier.
Intracellular Replication in RAW264.7 Cells
Murine macrophage-like RAW264.7 cells were grown in RPMI (Gibco) supplemented with FBS, HEPES, L-glutamine and gentamicin as described by [20], [65]. To assay for possible alterations in intracellular replication, bacteria were diluted from overnight plate cultures in PBS, opsonized with mouse serum (10% v/v final concentration) for 30 minutes, and diluted in supplemented RPMI (Gibco) devoid of serum and gentamicin to represent 1×106 bacteria per ml. The macrophages were exposed to 1 ml of inoculum in 24-well plates. To synchronize phagocytosis, plates were briefly centrifuged and incubated at 37°C for 1 hr. Cells where after washed twice with PBS and exposed to serum supplemented RPMI containing 50 µg/ml of gentamicin for 45 minutes. At this point, a 1 hr sample was collected after washing the cells in PBS and releasing bacteria by hypotonic lysis of cells. The second sample was collected at 16 hrs post infection. Growth yields were defined as the ratio in viable counts from the 16 and one-hour samples.
Infection Experiments in Mice
Female BALB/c J mice at the age of 6 to 8 weeks were purchased from Taconic Europe (Denmark) and housed at Microbiology Tumor and Cell biology (MTC) animal facility, Karolinska Institutet, Stockholm, Sweden, under normal conditions in accordance with both institutional and national guidelines. For competition experiment, each group of mice (n = 5) was infected intra-peritoneal by 1×104 bacteria/100 µl of infection dose consisting of wild-type, tagged with a tetracycline resistance marker inserted at a neutral genomic position, and mutant mixture in 1∶1 ratio [66]. The mice were sacrificed on third day post infection and livers and spleens were excised, homogenized in PBS and appropriate dilutions were placed on L-agar plates. Both members in each infection mixture were separated by appropriate resistance conferred by each of them [67]. Competitive indices (CI) were calculated as described previously [68].
The ileal loop infection experiment was carried out as described earlier [69]. The mice were deprived of water 4 hrs prior to the experiment. Mice were anaesthetized with isofluorane and were operated to expose the bowl. The ileum was located and near cecum, minimum 1.5 cm long piece was ligated at both ends taking care not to cut the ileum. 108 bacteria in a dose of 100 µl were injected into the ligated part. The bowl of the mouse was stitched aseptically and mice were allowed to wake up. The mice were closely observed for 75 min and then sacrificed by cervical dislocation. The bowl was re-opened and ligated loop was cut apart and taken out. The loops were cut open, washed in PBS and extracellular bacteria were killed with the application of gentamicin at 100 µg/ml in PBS for 1 hr. The washed ileum tissue was smashed in PBS and appropriate dilutions were plated to get the viable counts.
Infection Experiments in C. elegans
Bacterial strains were grown overnight in LB broth at 37°C and lawns were prepared by spreading 200 µl of overnight culture on modified NGM agar. 20 L4-staged wild-type N2 C. elegans were subsequently infected as described [70]. Briefly, nematodes were set down on unseeded agar before transferring to bacterial lawns to reduce, as much as possible, the transfer of E. coli. Nematode survival was scored at 24°C and nematodes were considered dead when not responding to gentle touch by a platinum wire. Nematodes were also transferred to fresh bacterial lawns every day. Results are representative of triplicates from 3 independent assays.
Detection of Reactive Oxygen Species in C. elegans
2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA; Sigma-Aldrich) was used to visualize intracellular ROS in nematodes. Stock aliquots of H2DCFDA (2 mM) were prepared in dimethyl sulfoxide and stored in the dark at −80°C. L4-staged N2 nematodes were infected as described above. At each time point, infected nematodes were harvested into tubes and washed twice with M9 buffer [57]. Nematodes were subsequently incubated with 25 µM H2DCFDA in 250 µl M9 buffer, in the dark for 30 min in a 20°C water bath. Nematodes were subsequently washed thrice with M9 buffer and mounted for microscopy in PBS with 25 mM sodium azide (NaN3, Merck). Slides were visualized on a LEICA DMRE microscope and images were analyzed by GNU Image Manipulation Program. Total nematode ROS fluorescence intensities were quantified using ImageJ software and expressed as a ratio relative to wild-type ATCC 14028 as described previously [71]. Images are representative of at least 20 nematodes from 2 independent assays.
Statistical Analyses
All the experiments were repeated at least three times and data were analyzed using the PRISM (version 5.0) software. The data for Figs. 2A to 2C were analyzed by linear regression and Fig. 2D further analyzed by t–test on the raw percent survival values. The data for Fig. 4, Fig. 5, Fig. 6A and Fig. 6C were analyzed by t-test. Data for Fig. 6B were analyzed by Wilcoxin signed-rank test. Fig. 7A data were analyzed by Kaplan–Meier estimation and Fig. 7J data were analyzed by Wilcoxin signed-rank test.
Supporting Information
Primer sequences for cloning of scs genes. S. Typhimurium LT2 genome sequence was used as reference for designing of all the primers for cloning.
(DOC)
Primers used for mutagenesis of scs genes. Homologous overhangs in the mutagenesis primers are designed by taking S. Typhimurium LT2 genome sequence as reference.
(DOC)
PCR verification primers for mutants. The primers were designed 100 bp up- and downstream of ORFs of scs genes to amplify the inserted antibiotic cassette. The reference genome sequence for primer designing was of S. Typhimurium LT2 strain.
(DOC)
Acknowledgments
We are thankful to KI Core Visualization Facility at MTC for microscopy, Birgitta Henriques-Normark laboratory for use of equipment and Catherine A. Lee for providing SPI1 transcription lacZ-fusion constructs. We extend our thanks to Åke Engström for carrying out Mass Spectrometric analyses of the proteins and to Ute Römling laboratory for helping in mice ileal loop experiments.
Funding Statement
This study was supported by grants from Vetenskapsrådet (Swedish Research Council) to Mikael Rhen with grant number VR-2011-3379. Naeem Anwar is a Ph.D. fellow of Higher Education Commission of Pakistan. The funders had no role in the study design, data collection and analysis,decision to publish, or preparation of the manuscript.
References
- 1. Mastroeni P, Sheppard M (2004) Salmonella infections in the mouse model: host resistance factors and in vivo dynamics of bacterial spread and distribution in the tissues. Microbes Infect 6: 398–405. [DOI] [PubMed] [Google Scholar]
- 2. Ohl ME, Miller SI (2001) Salmonella: a model for bacterial pathogenesis. Annu Rev Med 52: 259–274. [DOI] [PubMed] [Google Scholar]
- 3. Hansen-Wester I, Hensel M (2001) Salmonella pathogenicity islands encoding type III secretion systems. Microbes and Infection 3: 549–559. [DOI] [PubMed] [Google Scholar]
- 4. Agbor TA, McCormick BA (2011) Salmonella effectors: important players modulating host cell function during infection. Cell Microbiol 13: 1858–1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Galan JE, Zhou D (2000) Striking a balance: modulation of the actin cytoskeleton by Salmonella. Proc Natl Acad Sci U S A 97: 8754–8761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Srikanth CV, Mercado-Lubo R, Hallstrom K, McCormick BA (2011) Salmonella effector proteins and host-cell responses. Cell Mol Life Sci 68: 3687–3697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hapfelmeier S, Stecher B, Barthel M, Kremer M, Muller AJ, et al. (2005) The salmonella pathogenicity island (SPI)-2 and SPI-1 type III secretion systems allow Salmonella serovar typhimurium to trigger colitis via MyD88-dependent and MyD88-independent mechanisms. Journal of Immunology 174: 1675–1685. [DOI] [PubMed] [Google Scholar]
- 8. Hernandez LD, Hueffer K, Wenk MR, Galan JE (2004) Salmonella modulates vesicular traffic by altering phosphoinositide metabolism. Science 304: 1805–1807. [DOI] [PubMed] [Google Scholar]
- 9. Hobbie S, Chen LM, Davis RJ, Galan JE (1997) Involvement of mitogen-activated protein kinase pathways in the nuclear responses and cytokine production induced by Salmonella typhimurium in cultured intestinal epithelial cells. J Immunol 159: 5550–5559. [PubMed] [Google Scholar]
- 10. Lyons S, Wang L, Casanova JE, Sitaraman SV, Merlin D, et al. (2004) Salmonella typhimurium transcytoses flagellin via an SPI2-mediated vesicular transport pathway. J Cell Sci 117: 5771–5780. [DOI] [PubMed] [Google Scholar]
- 11. Miao EA, Alpuche-Aranda CM, Dors M, Clark AE, Bader MW, et al. (2006) Cytoplasmic flagellin activates caspase-1 and secretion of interleukin 1beta via Ipaf. Nat Immunol 7: 569–575. [DOI] [PubMed] [Google Scholar]
- 12. Mastroeni P, Vazquez-Torres A, Fang FC, Xu Y, Khan S, et al. (2000) Antimicrobial actions of the NADPH phagocyte oxidase and inducible nitric oxide synthase in experimental salmonellosis. II. Effects on microbial proliferation and host survival in vivo. J Exp Med 192: 237–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mastroeni P, Ugrinovic S, Chandra A, MacLennan C, Doffinger R, et al. (2003) Resistance and susceptibility to Salmonella infections: lessons from mice and patients with immunodeficiencies. Reviews in Medical Microbiology 14: 53–62. [Google Scholar]
- 14. Hebrard M, Viala JP, Meresse S, Barras F, Aussel L (2009) Redundant hydrogen peroxide scavengers contribute to Salmonella virulence and oxidative stress resistance. J Bacteriol 191: 4605–4614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Horst SA, Jaeger T, Denkel LA, Rouf SF, Rhen M, et al. (2010) Thiol peroxidase protects Salmonella enterica from hydrogen peroxide stress in vitro and facilitates intracellular growth. J Bacteriol 192: 2929–2932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Aussel L, Zhao W, Hebrard M, Guilhon AA, Viala JP, et al. (2011) Salmonella detoxifying enzymes are sufficient to cope with the host oxidative burst. Mol Microbiol 80: 628–640. [DOI] [PubMed] [Google Scholar]
- 17. Denkel LA, Horst SA, Rouf SF, Kitowski V, Bohm OM, et al. (2011) Methionine sulfoxide reductases are essential for virulence of Salmonella typhimurium. PLoS One 6: e26974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Miki T, Okada N, Danbara H (2004) Two periplasmic disulfide oxidoreductases, DsbA and SrgA, target outer membrane protein SpiA, a component of the Salmonella pathogenicity island 2 type III secretion system. J Biol Chem 279: 34631–34642. [DOI] [PubMed] [Google Scholar]
- 19. Bouwman CW, Kohli M, Killoran A, Touchie GA, Kadner RJ, et al. (2003) Characterization of SrgA, a Salmonella enterica serovar Typhimurium virulence plasmid-encoded paralogue of the disulfide oxidoreductase DsbA, essential for biogenesis of plasmid-encoded fimbriae. J Bacteriol 185: 991–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bjur E, Eriksson-Ygberg S, Aslund F, Rhen M (2006) Thioredoxin 1 promotes intracellular replication and virulence of Salmonella enterica serovar Typhimurium. infection and Immunity 74: 5140–5151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Peters SE, Paterson GK, Bandularatne ESD, Northen HC, Pleasance S, et al. (2010) Salmonella enterica Serovar Typhimurium trxA Mutants Are Protective against Virulent Challenge and Induce Less Inflammation than the Live-Attenuated Vaccine Strain SL3261. Infection and Immunity 78: 326–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sem X, Rhen M (2012) Pathogenicity of Salmonella enterica in Caenorhabditis elegans Relies on Disseminated Oxidative Stress in the Infected Host. PLoS One 7: e45417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Harrison JJ, Tremaroli V, Stan MA, Chan CS, Vacchi-Suzzi C, et al. (2009) Chromosomal antioxidant genes have metal ion-specific roles as determinants of bacterial metal tolerance. Environmental Microbiology 11: 2491–2509. [DOI] [PubMed] [Google Scholar]
- 24. Gupta SD, Wu HC, Rick PD (1997) A Salmonella typhimurium genetic locus which confers copper tolerance on copper-sensitive mutants of Escherichia coli. Journal of Bacteriology 179: 4977–4984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cho SH, Parsonage D, Thurston C, Dutton RJ, Poole LB, et al.. (2012) A new family of membrane electron transporters and its substrates, including a new cell envelope peroxiredoxin, reveal a broadened reductive capacity of the oxidative bacterial cell envelope. MBio 3. [DOI] [PMC free article] [PubMed]
- 26. McClelland M, Sanderson KE, Spieth J, Clifton SW, Latreille P, et al. (2001) Complete genome sequence of Salmonella enterica serovar Typhimurium LT2. Nature 413: 852–856. [DOI] [PubMed] [Google Scholar]
- 27. Mossner E, Huber-Wunderlich M, Glockshuber R (1998) Characterization of Escherichia coli thioredoxin variants mimicking the active-sites of other thiol/disulfide oxidoreductases. Protein Sci 7: 1233–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mossner E, Huber-Wunderlich M, Rietsch A, Beckwith J, Glockshuber R, et al. (1999) Importance of redox potential for the in vivo function of the cytoplasmic disulfide reductant thioredoxin from Escherichia coli. J Biol Chem 274: 25254–25259. [DOI] [PubMed] [Google Scholar]
- 29. Datsenko KA, Wanner BL (2000) One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97: 6640–6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rietsch A, Belin D, Martin N, Beckwith J (1996) An in vivo pathway for disulfide bond isomerization in Escherichia coli. Proc Natl Acad Sci U S A 93: 13048–13053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Negrea A, Bjur E, Ygberg SE, Elofsson M, Wolf-Watz H, et al. (2007) Salicylidene Acylhydrazides That Affect Type III Protein Secretion in Salmonella enterica Serovar Typhimurium. Antimicrobia Agents and Chemotherapy 51: 2867–2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Holmgren A (2000) Antioxidant function of thioredoxin and glutaredoxin systems. Antioxid Redox Signal 2: 811–820. [DOI] [PubMed] [Google Scholar]
- 33. Takemoto T, Zhang QM, Yonei S (1998) Different mechanisms of thioredoxin in its reduced and oxidized forms in defense against hydrogen peroxide in Escherichia coli. Free Radic Biol Med 24: 556–562. [DOI] [PubMed] [Google Scholar]
- 34. Hurme R, Namork E, Nurmiaho-Lassila EL, Rhen M (1994) Intermediate filament-like network formed in vitro by a bacterial coiled coil protein. J Biol Chem 269: 10675–10682. [PubMed] [Google Scholar]
- 35. Lehrer SS (1975) Intramolecular crosslinking of tropomyosin via disulfide bond formation: evidence for chain register. Proc Natl Acad Sci U S A 72: 3377–3381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Achard ME, Stafford SL, Bokil NJ, Chartres J, Bernhardt PV, et al. (2012) Copper redistribution in murine macrophages in response to Salmonella infection. Biochem J 444: 51–57. [DOI] [PubMed] [Google Scholar]
- 37. Eriksson S, Lucchini S, Thompson A, Rhen M, Hinton JCD (2003) Unravelling the biology of macrophage infection by gene expression profiling of intracellular Salmonella enterica. Molecular Microbiology 47: 103–118. [DOI] [PubMed] [Google Scholar]
- 38. Chung J, Chen T, Missiakas D (2000) Transfer of electrons across the cytoplasmic membrane by DsbD, a membrane protein involved in thiol-disulphide exchange and protein folding in the bacterial periplasm. Mol Microbiol 35: 1099–1109. [DOI] [PubMed] [Google Scholar]
- 39. Lee Y, Oh S, Park W (2009) Inactivation of the Pseudomonas putida KT2440 dsbA gene promotes extracellular matrix production and biofilm formation. FEMS Microbiology Letters 297: 38–48. [DOI] [PubMed] [Google Scholar]
- 40. Hiniker A, Collet JF, Bardwell JC (2005) Copper stress causes an in vivo requirement for the Escherichia coli disulphide isomerase DsbC. J Biol Chem 280: 33785–33791. [DOI] [PubMed] [Google Scholar]
- 41. Lin D, Rao CV, Slauch JM (2008) The Salmonella SPI1 type three secretion system responds to periplasmic disulfide bond status via the flagellar apparatus and the RcsCDB system. J Bacteriol 190: 87–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gao YL, Jiang WL, Dong CH, Li CM, Fu XJ, et al. (2012) Anti-inflammatory effects of sophocarpine in LPS-induced RAW 264.7 cells via NF-kappa B and MAPKs signaling pathways. Toxicology in Vitro 26: 1–6. [DOI] [PubMed] [Google Scholar]
- 43. Xie QW, Kashiwabara Y, Nathan C (1994) Role of transcription factor NF-kappa B/Rel in induction of nitric oxide synthase. Journal of Biological Chemistry 269: 4705–4708. [PubMed] [Google Scholar]
- 44. Anrather J, Racchumi G, Iadecola C (2006) NF-kappaB regulates phagocytic NADPH oxidase by inducing the expression of gp91phox. J Biol Chem 281: 5657–5667. [DOI] [PubMed] [Google Scholar]
- 45. Ha UH, Wang YP, Jin SG (2003) DsbA of Pseudomonas aeruginosa is essential for multiple virulence factors. infection and Immunity 71: 1590–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Watarai M, Tobe T, Yoshikawa M, Sasakawa C (1995) Disulfide oxidoreductase activity of shigella flexneri is required for release of Ipa proteins and invasion of epithelial cells. Proceedings of the National Academy of Sciences of the United States of America 92: 4927–4931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Donnenberg MS, Zhang HZ, Stone KD (1997) Biogenesis of the bundle-forming pilus of enteropathogenic Escherichia coli: reconstitution of fimbriae in recombinant E-coli and role of DsbA in pilin stability - a review. Gene 192: 33–38. [DOI] [PubMed] [Google Scholar]
- 48. Jacobdubuisson F, Pinkner J, Xu Z, Striker R, Padmanhaban A, et al. (1994) PapD chaperone function in pilus biogenesis on oxidant and chaperon like activities of DsbA. Proceedings of the National Academy of Sciences of the United States of America 91: 11552–11556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yu J, Kroll JS (1999) DsbA: a protein-folding catalyst contributing to bacterial virulence. Microbes and Infection 1: 1221–1228. [DOI] [PubMed] [Google Scholar]
- 50. Jarvik T, Smillie C, Groisman EA, Ochman H (2010) Short-Term Signatures of Evolutionary Change in the Salmonella enterica Serovar Typhimurium 14028 Genome. Journal of Bacteriology 192: 560–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Juncker AS, Willenbrock H, Von Heijne G, Brunak S, Nielsen H, et al. (2003) Prediction of lipoprotein signal peptides in Gram-negative bacteria. Protein Sci 12: 1652–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kelley LA, Sternberg MJ (2009) Protein structure prediction on the Web: a case study using the Phyre server. Nat Protoc 4: 363–371. [DOI] [PubMed] [Google Scholar]
- 53. Guzman LM, Belin D, Carson MJ, Beckwith J (1995) Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J Bacteriol 177: 4121–4130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Yanisch-Perron C, Vieira J, Messing J (1985) Improved M13 phage cloning vectors and host strains: nucleotide sequences of the M13mp18 and pUC19 vectors. Gene 33: 103–119. [DOI] [PubMed] [Google Scholar]
- 55. Lavallie ER, Diblasio EA, Kovacic S, Grant KL, Schendel PF, et al. (1993) A Thioredoxin Gene Fusion Expression System That Circumvents Inclusion Body Formation in the Escherichia-Coli Cytoplasm. Bio-Technology 11: 187–193. [DOI] [PubMed] [Google Scholar]
- 56. Schmieger H (1972) Phage P22-mutants with increased or decreased transduction abilities. Mol Gen Genet 119: 75–88. [DOI] [PubMed] [Google Scholar]
- 57. Brenner S (1974) The genetics of Caenorhabditis elegans. Genetics 77: 71–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Rhen M, Sukupolvi S (1988) The Role of the Tra-T Gene of the Salmonella-Typhimurium Virulence Plasmid for Serum Resistance and Growth within Liver Macrophages. Microbial Pathogenesis 5: 275–285. [DOI] [PubMed] [Google Scholar]
- 59. Palva ET (1978) Major outer membrane protein in Salmonella typhimurium induced by maltose. J Bacteriol 136: 286–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ames GF, Prody C, Kustu S (1984) Simple, rapid, and quantitative release of periplasmic proteins by chloroform. J Bacteriol 160: 1181–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227: 680–685. [DOI] [PubMed] [Google Scholar]
- 62.Miller JH, editor (1972) Experiments in molecular genetics. New York: Cold Spring Harbor Laboratory Press,U.S. 325–355 p.
- 63. Bajaj V, Hwang C, Lee CA (1995) hilA is a novel ompR/toxR family member that activates the expression of Salmonella typhimurium invasion genes. Mol Microbiol 18: 715–727. [DOI] [PubMed] [Google Scholar]
- 64. Negrea A, Bjur E, Ygberg SE, Elofsson M, Wolf-Watz H, et al. (2007) Salicylidene acylhydrazides that affect type III protein secretion in Salmonella enterica serovar typhimurium. Antimicrob Agents Chemother 51: 2867–2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Negrea A, Bjur E, Puiac S, Ygberg SE, Aslund F, et al. (2009) Thioredoxin 1 Participates in the Activity of the Salmonella enterica Serovar Typhimurium Pathogenicity Island 2 Type III Secretion System. Journal of Bacteriology 191: 6918–6927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Eriksson S, Bjorkman J, Borg S, Syk A, Pettersson S, et al. (2000) Salmonella typhimurium mutants that downregulate phagocyte nitric oxide production. Cell Microbiol 2: 239–250. [DOI] [PubMed] [Google Scholar]
- 67. Bjorkman J, Rhen M, Anderson DI (1996) Salmonella typhimurium cob mutants are not hyper-virulent. FEMS Microbiol Lett 139: 121–126. [DOI] [PubMed] [Google Scholar]
- 68. Beuzon CR, Holden DW (2001) Use of mixed infections with Salmonella strains to study virulence genes and their interactions in vivo. Microbes Infect 3: 1345–1352. [DOI] [PubMed] [Google Scholar]
- 69. Jones BD, Ghori N, Falkow S (1994) Salmonella-Typhimurium Initiates Murine Infection by Penetrating and Destroying the Specialized Epithelial M-Cells of the Peyers-Patches. Journal of Experimental Medicine 180: 15–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Powell JR, Ausubel FM (2008) Models of Caenorhabditis elegans infection by bacterial and fungal pathogens. Methods Mol Biol 415: 403–427. [DOI] [PubMed] [Google Scholar]
- 71. Burgess A, Vigneron S, Brioudes E, Labbe JC, Lorca T, et al. (2010) Loss of human Greatwall results in G2 arrest and multiple mitotic defects due to deregulation of the cyclin B-Cdc2/PP2A balance. Proceedings of the National Academy of Sciences of the United States of America 107: 12564–12569. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Primer sequences for cloning of scs genes. S. Typhimurium LT2 genome sequence was used as reference for designing of all the primers for cloning.
(DOC)
Primers used for mutagenesis of scs genes. Homologous overhangs in the mutagenesis primers are designed by taking S. Typhimurium LT2 genome sequence as reference.
(DOC)
PCR verification primers for mutants. The primers were designed 100 bp up- and downstream of ORFs of scs genes to amplify the inserted antibiotic cassette. The reference genome sequence for primer designing was of S. Typhimurium LT2 strain.
(DOC)