

Abstract
Chemotherapeutics target rapidly dividing cancer cells by directly or indirectly inducing DNA damage. Upon recognizing DNA damage, cells initiate a variety of signaling pathways collectively referred to as the DNA damage response (DDR). Interestingly, the pathways used to elicit this response are as varied as the types of DNA damage induced. However, the activation of these various pathways has similar results including DNA repair, suppression of global general translation, cell cycle arrest and, ultimately, either cell survival or cell death. This review will focus on a series of chemotherapy-induced DNA lesions and highlight recent advances in our understanding of the DDR, the DNA repair pathways it activates and the cellular consequences of these converging pathways.
Keywords: ATM, ATR, DNA damage, DNA-PK, PIKK, chemotherapy, cisplatin, signaling
Introduction
The DDR is a crucial signaling pathway that serves to coordinate the necessary series of biochemical and cellular events in response to both exogenous and endogenous induced DNA damage. The complexity of the DDR is in part a function of the requirement to detect and respond to a wide variety of DNA damage events and to regulate the numerous potential outcomes of the genetic insult. The clinical use of DNA damage-inducing therapies remains a mainstay in the treatment of cancer. Targeting the rapidly dividing cancer cells with genotoxic agents has demonstrated clinical utility and more recently, it has become apparent that the DDR impacts the response to these therapies both in terms of anti-cancer activity and toxicity to non-cancer cells. Thus, to begin to understand why different cancer types respond to various DNA damage therapies, the detailed mechanisms involved in the initiation of the DDR in response to these therapies is essential and recently has begun to be addressed.
Initiators of the DDR
Phosphatidylinositol-3 kinase- like protein kinases (PIKKs)
The PIKK family of protein kinases includes Ataxia-Telangiectasia Mutated (ATM), ATM and Rad3 Related (ATR) and the DNA-Dependent Protein Kinase Catalytic Subunit (DNA-PKcs) (Table 1). These kinases are relatively large in size and show a target preference for serine or threonine residues that are followed by glutamines. As discussed below, however, some important targets have recently been identified in non-consensus sequences. Despite the fact that the PIKKs are involved in different repair pathways, their respective activation involves some common themes. They are all initial responders to DNA damage and as far as we know the first kinases to initiate the DDR signaling cascade. In addition they are all activated at the site of DNA damage but cannot bind DNA, damaged or undamaged, without the assistance of DNA scaffolding proteins. While some early work suggested that ATM might bind directly to DNA, no new studies have confirmed or supported this. Even DNA-PKcs which has clearly defined DNA binding domains, does not bind DNA by itself under physiological salt concentrations.1 Evidence suggests that the scaffolding proteins Ku80, Nbs1 and ATRIP not only recruit the kinases to the sites of DNA damage but also play a major role in activation of DNA-PKcs, ATM and ATR respectively. Interestingly these scaffolding proteins also share significant sequence similarity at their extreme C-termini, a feature essential for complex formation and DDR signaling.2 The scaffolding proteins themselves must also be in complexes in order to efficiently activate the signaling kinase. Ku80 must be in complex with Ku70 while Nbs1 interacts with Mre11 and Rad50. ATRIP interacts with the RPA bound ssDNA complex and TOPBP1.3 While similarities certainly exist within the PIKKs family, their differences become apparent upon examination.
Table 1.
| DNA-PKcs | ATM | ATR | |
|---|---|---|---|
| DNA tethering complex |
Ku70/80 |
MRN |
RPA, Rad9/Rad1/Hus1 |
| H2AX/MDC1 | |||
| Activating protein |
Ku80 |
Nbs1 |
ATRIP |
| Activating DNA substrates |
ds-DNA termini, higher activation with 5′ ss overhangs |
Long 3′ ss regions |
Short 3′ ss regions near ds junctions |
| Unique phosphorylation targets |
Multiple sites of autophosphorylation |
Chk2 |
Chk1 |
| RPA phosphorylation |
Ser 4, Ser 8, Ser 12 |
Ser 33 |
|
| Ser 21 | |||
| Quaternary structure | Dimer when active | Dimer when inactive | Multimer upon activation in complex wih RPA |
ATM
Ataxia-telangiectasia mutated (ATM) is a 315kDa protein that plays a major role in initiating the DDR. ATM remains a homodimer while inactive, but upon activation undergoes trans-autophosphorylation at serine 1981, leading to disruption of the dimer and allowing monomeric ATM to be recruited to dsDNA via an interaction with the MRN complex.4 While this phosphorylation event may be necessary for disruption of the dimmer, data suggest that it is not sufficient (See next paragraph). How this initial autophosphorylation event is stimulated is not well understood but may rely on chromatin relaxation.4 The nuclease activity of the MRN complex results in 3′ssDNA which along with its interaction with the C-terminus of Nbs1 stimulates ATM kinase activity and ultimately promotes homologous recombination (HR) (Fig. 1A).5 In an independent activation pathway, ATM has been shown to be activated by ATMIN under hypotonic stress which is independent of Nbs1 interactions.6 Interestingly, HR is restricted to S and G2 phases of the cell cycle, yet ATM is activated following DSBs regardless of cell cycle stage.7 Some data suggests that DNA resection is a major component of whether ATM activation promotes HR or NHEJ. ATM activation following damage occurring in G1 leads to a minute amount of DNA resection due to low levels of cyclin dependent kinases and promotes NHEJ. ATM activation in S or G2, when cyclin dependent kinase levels are high, promotes DNA resection by MRN leading to HR promotion via ATR signaling chapman.5 Regardless S checkpoint cell cycle arrest is a hallmark of ATM activation.8 Upon recruitment of ATM to DSBs via the MRN complex, monomeric ATM undergoes autophosphorylation at additional sites including the recently identified Serine 367 and Serine 2996.9 Importantly, when these sites were mutated to phosphor-ablating alanines ATM was unable to arrest the cell cycle at the S checkpoint, suggesting these phosphorylation events are essential in the DDR.9
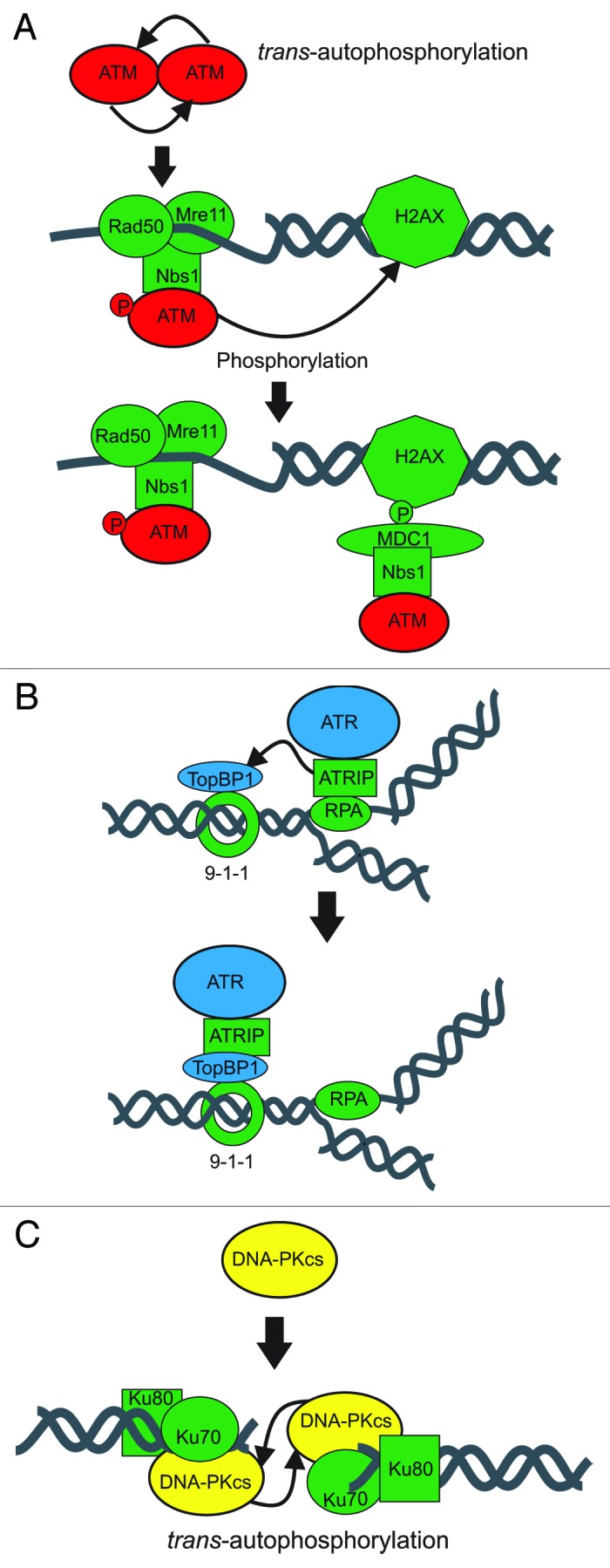
Figure 1. Summary of PIKK Activation pathways. Preferential DNA substrates and recognition complexes are presented. (A) ATM responds to long 3′ single stranded regions via the MRN complex. (B) ATR is activated by short 3′ regions near duplex junctions via RPA:ATRIP and TopBP1. Protein complexes that tether the PIKKs to DNA are colored green while proteins involved in activating the PIKKs are indicated by the squares. (C) DNA-PKcs recognizes double stranded DNA termini via Ku.
Unlike ATR and DNA-PK, ATM seems to be activated independently of DNA damage through direct exposure to reactive oxygen species (ROSs). Guo and colleagues have demonstrated and defined a distinct mechanism for activation that is independent of DNA or MRN.10 In this pathway, oxidized ATM becomes activated and retains autophosphorylation at 1981 but remains a dimer. Indeed dimerization via intermolecular disulfide bonds involving Cystine 2991, which is near the kinase domain of ATM, is essential for this mechanism of activation. Interestingly, oxidatively-activated ATM and DNA damage dependent activated ATM share some, but not all, downstream targets. For example both pathways lead to phosphorylation of p53 at Ser15 and Chk2 at Thr68 but oxidatively activated ATM does not phosphorylate H2AX or Kap1. Guo et al. suggest that the specificity of targets stems from their stable association with DNA: i.e., H2AX and Kap1 phosphorylation is restricted to ATM activated by DNA damage. This seems logical in that DNA damage dependent activation of ATM involves a close association of ATM with DNA through MRN interactions. While the distinction between the activation pathways is apparent both lead to the DDR as evidenced by the fact that major downstream signaling factors including Chk2 and p53 are induced. This point is apparent in the context of cancer therapies which can produce a tremendous amount of reactive oxygen species (ROSs) that, in turn, may cause DNA damage in vivo, leading to both ATM activation pathways and the DDR (see below).
ATR
The last member to be identified in the PIKKs is ATR. ATR has been demonstrated to respond to DNA replication stress and signal to CHK1 via phosphorylation of ser345 in an RPA:ATRIP dependent process.11 Consistent with the model ATR activation is restricted to S and G2 phases of the cell cycle. Current models involve RPA detecting and binding the single-stranded DNA generated as a function of disrupted DNA replication. What distinguishes normal DNA replication-associated RPA from RPA associated with stalled replication forks is a combination of the unique gapped DNA structures associated with stalled replication and the proteins bound to these structures. The association of ATRIP and ultimate activation of ATR required a combination of DNA- protein and protein-protein interactions. DNA damage-dependent ATM phosphorylation of TOPBP1, mediated in part by CtIP and the MRN complex, stimulates association of TOPBP1 with the ATR-ATRIP complex. This association has been demonstrated to activate ATR leading to downstream target phosphorylation (Fig. 1C). The various protein complexes formed by ATR and ATRIP are further evidenced by the finding that both ATR and ATRIP exist as oligomers. This oligomeric state is not changed as a function of DNA damage and ATR oligomers are not dependent on ATRIP. Similarly ATRIP oligomers are not dependent on ATR.12 This is very different from the ATM dimer which when disrupted by phosphorylation leads to ATM activation. Part of the difficulty in assessing these differences in activation lies in the indirect measures used to measure ATR activation. To date the best measure of ATR activation is measurement of downstream target phosphorylation though many of the targets are substrates for other kinases, thus complicating the analyses. More recently, a putative ATR autophosphorylation, site Thr1989, was characterized that could potentially be a useful marker for ATR activation,13 perhaps enabling the mechanism of ATR activation to be more completely elucidated.
DNA-PK
The largest member of the PIKKs is the DNA-dependent protein kinase catalytic subunit (DNA-PKcs). Indeed this protein with the staggering size of 469kDa is believed to be the largest single subunit protein in mammalian cells. It also seems to be the most abundant member of the kinase family with approximately 500,000 molecules per nucleus in human cells (Meek personal communication). Unlike ATM and ATR, DNA-PKcs plays a major, direct role in DNA repair and also initiates the DDR. Similar to ATM, DNA-PKcs forms homodimers; however the nature and role of these dimerization events are completely different. Dissimilar to the inactive ATM dimer and active monomer form, DNA-PKcs exists as a monomer in the cell when inactive. Following a DSB, the Ku70/80 protein binds to both termini of the break and recruits monomeric DNA-PKcs to both sides.14 Together, Ku70/80 and DNA-PKcs form the heterotrimeric DNA-PK. The DNA-PKcs molecules dimerize and interact across the DNA termini forming the synaptic complex (Fig. 1B). As part of these interactions, DNA-PK undergoes trans-autophosphorylation at over 40 sites.15 A substantial amount of data suggests that Ku70/80 is required for DNA-PK formation and activation.16-18 Several groups including our own have presented evidence for a direct protein/protein interaction between the carboxy-terminus of Ku80 and DNA-PKcs.19 While some early work suggested that these interactions are necessary for kinase activity, more recent work has questioned this, concluding that the c-terminus is dispensable for activation.16,18 Bridging the divide in the contrasting conclusions of previous studies, work from our group shows that the influence of the c-terminus of Ku80 on DNA-PK activation varies depending on the structure of the DNA cofactor to which DNA-PK is bound (data not published). Keeping with this theme SAXS structural studies have shown that the nature of DNA-PK dimerization across the synapse is different depending on the structure of the dsDNA termini to which the complex is bound.20 Thus, it seems that the structure of the DNA termini induce different protein/protein and protein/DNA interactions. Further, structural studies have revealed an extensive interface between the Ku and DNA-PKcs that does not involve the C-terminus of Ku80.20 It is possible that these interactions contribute to Ku80 C-terminus independent DNA-PK activation; however, it is also possible that the binding of Ku to the DNA changes the conformation of the DNA which subsequently promotes DNA-PK activation. Distinguishing between these two possibilities has proven difficult.
Similar to ATM and ATR regulation, DNA-PKcs activity seems tunable. Two important components of the control of this activity are the proteins with which DNA-PK interacts and the structure of the DNA to which DNA-PKcs is bound. A probable result of the DNA substrate specific tuning is its regulation of DNA termini processing and facilitating DNA repair. Our group was the first to show that DNA-PKcs is activated by the 5′ end of the DNA terminus while the 3′ end is involved in mediating microhomology pairing across the synapse.21 Additionally, DNA-PK autophosphorylation at the ABCDE cluster promotes DNA processing, while phosphorylation at the PQR cluster limits processing.22 More recently, related work has shown that DNA-PK autophosphorylation at the JK cluster and threonine 3950 promotes DNA double strand break repair through homologous recombination (HR) and inhibits NHEJ.23 This same study identified a novel phosphorylation site at the N-terminus of DNA-PKcs which seems to ablate DNA-PK activity. Whether the phosphorylation status of this site is regulated by ATM, ATR, DNA-PKcs itself or some combination therein is yet to be seen.
Interplay of PIKKs
A theme in the initiation of DDR by the PIKK is the interdependence of the kinases in regulation. Among the 40 sites of DNA-PKcs that become phosphorylated only a few have been shown to significantly alter repair. Of these sites the ABCDE cluster mentioned above is perhaps the most significant and is phosphorylated by ATM following DSBs and ATR following UV irradiation.24,25 DNA-PKcs, on the other hand, seems to be involved in a feedback loop regulating the expression of ATM as evidenced by the fact that cells with knocked down DNA-PKcs have reduced levels of ATM. Further, it was shown that when cell lines that were DNA-PKcs null were transfected with DNA-PKcs plasmids, ATM levels rose.26 The control of both of these events was shown to be at the transcriptional level suggesting that DNA-PKcs regulates the transcription of ATM. Additionally, the autophosphorylation of DNA-PKcs at the JK and N sites promotes HR, which activates ATR and ATM.23 Recently, MRN and CtIP, whose resection activity has been shown to promote HR and stimulate ATM activity, were shown to promote NHEJ following etoposide treatment of cells in G1.27 This is seemingly in contrast to work in yeast which suggests that Mre11 and CtIP are responsible for the release of Ku from dsDNA termini and promoting HR.28 Whether this functional relationship is also retained in mammals remains untested. Because Ku activates and regulates DNA-PKcs activity, it is important that the influence of Mre11 and CtIP on displacing Ku from DNA be investigated further. This model however, is consistent with data demonstrating that ATM phosphorylation of TOPbp1 in conjunction with NBS1 stimulated ATR activity. These and other data provide convincing evidence that the PIKKSs regulate each other directly through phosphorylation and transcription while indirectly regulating each other by promoting the pathways which in turn activate other PIKKs (Fig. 2).
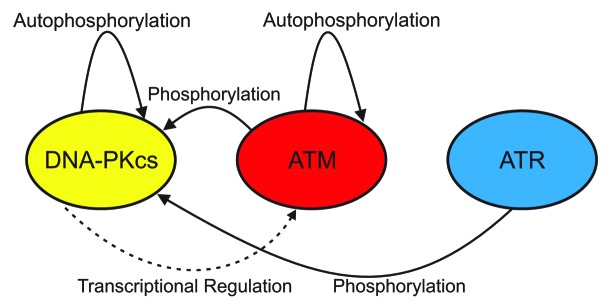
Figure 2. Summary of intra- and inter- PIKK regulation. DNA-PKcs and ATM undergo autophosphorylation. DNA-PKcs is a phosphorylation target of ATM and ATR while DNA-PKcs promotes ATM transcription.
While not as clearly defined as signaling in DSB repair, the repair of bulky lesions such as those resulting from platinum damage may trigger the DDR as well.29 These lesions are primarily repaired by the nucleotide excision repair (NER) pathway which removes bulky, helical distorting intrastrand lesions via endonuclease activity on both sides of the insult.30 Unlike DSB repair, NER does not necessarily activate the DDR during normal repair scenarios. However, dysfunctional NER may lead to longer resection via ExoI, activity which in turn may provide an adequate ssDNA substrate to initiate the DDR.31 Supporting DDR initiation, phosphorylation of serine 317 of ChK1 was shown to increase in this system. Because this is a specific downstream target of ATR, it is likely that ATR initiates the DDR following dysfunctional NER. This model is attractive because RPA plays an essential role in initiating NER and also is implicated in activating ATR (see above). It will be interesting in the next few years to see if any direct evidence will show that ATR initiates the DDR when NER is dysfunctional.
Other important regulators of the DDR are the Poly (ADP-ribose) polymerases including PARP-1 and PARP-2. These enzymes modulate signaling by producing poly ADP-ribose chains on target proteins and become activated as part of the single strand break repair and base-excision repair. In recent years, these proteins have been investigated extensively in the context of targeting DDR in cancer therapies, particularly in those cancers with defective BRCA1 and BRCA2. For more information concerning the role of PARPs in DDR and DNA repair we refer you to the excellent reviews by Gibson and Kraus32 and Jackson.33
Downstream Targets in the DDR
The PIKKs are known to phosphorylate at least 700 downstream targets upon activation.14 This section will highlight recent work on a few of the targets to which a significant advance in our mechanistic understanding has been made .
H2AX
The phosphorylation status of H2AX is one of the most monitored targets of the PIKKs in the DDR. Upon phosphorylation of serine 139, H2AX is referred to as γ-H2AX and has been shown by several groups to be important in DSB repair and, more recently, in response to UV induced damage.34,35 Interestingly, while each of the PIKKs has been shown to phosphorylate serine 139, dysfunction in any of the members leads to prolonged γ-H2AX35,36 suggesting that repair is compromised (Table 1). Tyrosine 142 is constitutively phosphorylated under basal conditions but is dephosphorylated by the protein tyrosine phosphatase EYA.37 This dephosphorylation event is crucial for MDC1 binding to γ-H2AX and foci formation.38 In ATM-mediated γ-H2AX signaling, the MRN complex recruits ATM to DNA where it is activated and subsequently phosphorylates H2AX at serine 139. Following MDC1 binding to γ-H2AX, ATM by way of an interaction to Nbs1 which interacts with MDC1 becomes tethered to the focus (Fig. 1A). The active ATM can propagate γ-H2AX foci formation from 2–30 Mbp surrounding the break.39 Similarly, evidence suggests that the TopBP1/ATR complex, which is activated following stalled DNA replication, can be recruited to γ-H2AX foci by MDC1 and ATR and propagates γ-H2AX foci formation.40
Replication protein A (RPA)
RPA is a heterotrimeric, ssDNA binding protein consisting of the 70, 32 and 14 kDa subunits.41 Because of its ability to bind ssDNA, RPA has been shown to be involved in several nuclear pathways including DNA replication and DNA repair. RPA is an important target of the PIKKs particularly on the 32kDa subunit. There are at least 7 sites which undergo phosphorylation during the DDR at the N-terminus of RPA 32. The regulation of the phosphorylation status of each of these sites and their influence on the neighboring sites within the N-terminus is complex. Recently, however, Oakley and colleagues reported some elegant work that demystifies the process.42 Using in vivo and in vitro data they convincingly show that Ser4 and Ser8 are phosphorylated by DNA-PKcs and, to a lesser extent by ATM. The phosphorylation of these serines seems to moderately stimulate the phosphorylation of Ser33 by ATR and significantly stimulates the phosphorylation of Ser12 by ATM and DNA-PKcs. Interestingly, ATR activation was shown to be influenced by RPA phosphorylation at Ser4 and Ser8, as RPA S4A/S8A mutants caused a decrease in phosphorylation of the ATR-activating protein TopBP1 at sites essential in stimulating ATR activity (Table 1).
Besides regulating ATR activity, there are several important cellular consequences of RPA hyperphosphorylation following the initiation of the DDR (Table 1). A recent study showed that RPA interacts with the tumor suppressor p53 and that this interaction is ablated upon hyperphosphorylation of RPA32 at the N-terminus and phosphorylation of p53 at Ser37 and Ser46.43 Interestingly, this regulation requires participation of DNA-PK, ATM and ATR, as DNA-PK primarily phosphorylates RPA and ATM and ATR phosphorylate p53 at Ser46 and Ser37, respectively. In this study, the authors suggest that this dissociation may be important for RPA to function in DNA repair pathways. Others have shown that RPA32 hyperphosphorylation facilitates NHEJ by suppressing sister chromatid exchange.44 Ser4/6, Thr21 and Ser33 phosphorylation mark checkpoint arrest and S12 phosphorylation marks replication recovery. While hyperphosphorylation of RPA is critical in cell cycle arrest and DNA repair, removing these posttranslational modifications is important in relieving these effects. For example, PPA2-mediated RPA32 dephosphorylation of Thr21 and Ser33 is required for checkpoint release and cell cycle re-entry.45
Chk1 and Chk2
A major downstream target of ATR that elicits the DDR is Chk1 (Table 1). ATR phosphorylates Chk1 at Ser317 and Ser345, which results in stimulation of Chk1’s kinase activity.46 This event requires the scaffolding protein Claspin, whose recruitment itself requires ATR-mediated phosphorylation of Rad17.47 Further, even though Chk1 is a major target of ATR through direct phosphorylation, Oakley et al. show that this event is dependent on RPA32 S4 and S8 phosphorylation via DNA-PKcs.42 Chk2, on the other hand, is a major downstream target of ATM and perhaps DNA-PKcs, which phosphorylate Chk2 at Thr6842,48 (Table 1). This event is believed to stimulate Chk2 kinase activity but does not appear to be essential, as the Thr38Ala mutation does not completely abolish IR-induced Chk2 activation.49 Thr68 phosphorylation does, however, appear to prime trans-autophosphosphorylation activity at Thr 383 and 387 and in cis at Ser516, ultimately leading to maximum activation, likely through oligomerization.50
Following activation, the Chk1 and Chk2 arms of the DDR converge on the regulation of the Cdc25 family of dual-specificity phosphatases. Chk1 and Chk2 inactivate these proteins and keep them from removing important inhibitory phosphorylation signals on Cdk/Cyclin complexes ultimately leading to cell cycle arrest.51 Cdc25A has been shown to be an important substrate of Chk1, which leads to Cdc25A degradation and limits cell cycle progression at the S-phase checkpoint and the G2/M checkpoint.52,53 Further, oncogene Survivin was recently reported to be regulated by Chk2, where Chk2 targets Survivin for degradation leading to cell cycle arrest.54 Besides targeting other signaling proteins, Chk2 may directly influence cell cycle progress by regulating replication enzymes. Recently Chk2 was shown to inhibit the CMG replicative helicase complex in Drosophila melanogaster.55 Similarly, it was recently observed in fission yeast that the Chk2 homolog Cds1 targets and activates the nuclease Dna2 which is necessary to prevent stalled replication forks from regressing.56 In summary the activation of Chk1 and Chk2 are crucial events in the DDR and regulate cell cycle arrest directly through the regulation of DNA repair and replication proteins and indirectly by propagating DDR signaling cascades.
DNA Damaging Cancer Therapeutics
DNA damaging cancer therapeutics can be divided into groups based on their mechanism of action and type of damage induced though there is considerable crossover between classes (Table 2). Alkylating agents directly modify DNA and often induce bulky DNA damage that is repaired via the nucleotide excision repair pathway (NER). Platinum-based agents also induce bulky DNA damage repaired by the NER pathway and are effective in treating a wide array of cancers. Other non-traditional alkylators include direct methylating agents. Aberrant methylation of DNA bases such as that induced by temozolomide is repaired via the base excision repair pathway (BER). The induction of DNA DSB via radiation or radiomimetics is also an effective method to induce cellular death as DSB are considered the most toxic form of DNA damage. The NHEJ and HR pathways are involved in repair DNA DSBs. A large class of agents ultimately target DNA metabolism and include DNA intercalating agents, topoisomerase poisons and antimetabolites. Eventually these result in DNA adducts, strand breaks or stalled/collapsed DNA replication forks, the repair or restart of which often requires HR and Fanconi anemia proteins. In addition, treatment with many of these agents results in the generation of ROSs in the cell. ROSs can directly induce a wide array of DNA damage including base oxidation, sugar fragmentation and single strand DNA breaks. Clearly, cancer therapy presents a plethora of insults to chromosomal DNA and a considerable challenge for DDR.
Table 2.
| Agents | Indication | DNA damage | DDR pathways engaged | DNA repair pathways engages | |
|---|---|---|---|---|---|
| Alkylating agents |
Bendamustine |
Lymphoma |
predominantly monofunctional |
ATM ATR/Chk1 |
DER |
| Melphalan |
Multiple myeloma |
monofunctional and bifunctional |
ATR/Chk1 |
NER/BER |
|
| Platinums |
Cisplatin |
Various |
Intra-strand and interstrand |
ATM/ATR/DNA-PK |
NER/HR |
| Carboplatin |
Lung ovarian |
Intra-strand and interstrand |
|
NER/HR |
|
| Oxaliplatin |
Colon |
Intra-strand and interstrand |
ATR-DNA-PK |
NER/HER |
|
| Replication disrupting agents |
Gemcitibine |
Pancreatic |
Chain termination/stalled replication |
ATR |
HR |
| Ara-C |
Hematologic |
Stalled replication |
ATR |
HR |
|
| Etoposide |
SCLC |
DSB topo-DNA adducts |
ATR |
HR |
|
| Doxorubicin |
Breast |
DSB topo-DNA adducts |
ATR |
HR |
|
| Radiomimetics | Bleomycin |
Testicular |
DSB, SSB, oxidized bases |
ATM/DNA-PK |
NHEJ/HR |
| C-1027 | Pre-clinical | DSB, SSB, oxidized bases | ATM/ATR/DNA-PK | NHEJ | |
Alkylating agents
Alkylating agents are perhaps the oldest class of agents used to treat cancer and result in the covalent transfer of alkyl-groups to DNA resulting in DNA damage and includes nitrogen mustards and nitrosoureas (Fig. 3A and B). There has been considerable recent excitement over bendamustine, an alkylating agent originally developed in the 1960s and approved in 2008 for treatment of lymphoma.57 Despite the recent clinical implementation, there is dearth information concerning its mechanism of action.58 While bendamustine forms mono adducts on purine bases as a function of the 2-cloroethylamine moiety, there is only limited evidence for the formation of purine intra or interstrand crosslinks.59 Significant levels of DNA double strand breaks have been observed when compared with comparable alkylating agents.60 COMPARE analysis revealed limited similarity with other agents including similar alkylating agents like melphalan.58 Gene ontology analyses of Bendamustine treated cells indicated DDR as the top regulated pathway with some evidence that the BER pathway is in part responsible for processing bendamustine induced DNA adducts.58 While there is also evidence that melphalan mono and di-adducts melphalan are repaired via the NER pathway as demonstrated by the hypersensitivity of NER deficient cells to treatment,61 this has not been addressed for bendamustine. Bendamustine activation of the DDR has been studied in a few models. Initial studies in a myeloma cancer model revealed activation of ATM and Chk2, but not Chk1.62 However this study was limited by use of non-specific DDR inhibitors which led to the conclusion that inhibition of ATM/ATR Chk1/2 does not alter bendamustine sensitivity. More recently in a nice series of experiments, a more specific inhibitor of Chk1 (AZD7762) was shown to potentiate the activity of both melphalan and bendamustine which did so with an accompanying increase Chk2 phosphorylation.63 The authors suggested that this sensitivity was a function of the generation of more DNA DSBs, though this was not directly measured. A study of the effects of differing doses of Bendamustine revealed differential activation of cell cycle checkpoints, consistent with the multiple mechanisms of bendamustine action.64 The same study also revealed increased sensitivity to the Chk1 inhibitor UCN-01 but not a Chk2 inhibitor consistent with an ATR Chk1 driven DDR pathway. Clearly it will be of interest to determine if ATR inhibitors alter sensitivity to Bendamustine and melphalan treatment.
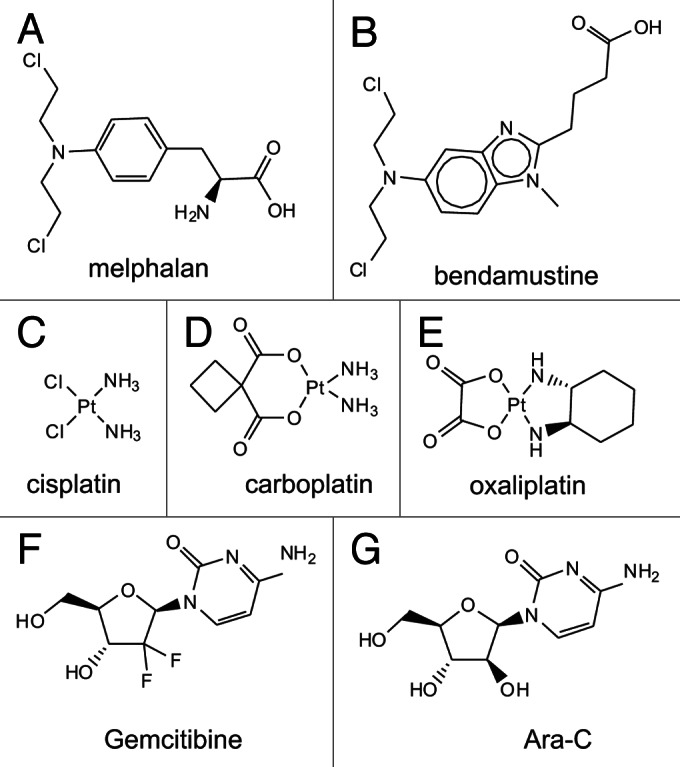
Figure 3. Chemical structures of DNA damaging chemotherapeutics.
Platinum-based drugs, cisplatin and carboplatin induce bulky DNA adducts through coordinate–covalent bonds between DNA and the platinum moiety (Fig. 3C–E). Technically, platinum agents are therefore not alkylators in that no carbon groups are transferred. It is well established that the intrastrand adducts formed by cisplatin are repaired via NER and can be tolerated via HRR dependent mechanisms or via by-pass DNA polymerases.65 The DDR as a function of platinum treatment has also been studied in a variety of model systems and yielded varying results. DNA-PK, ATM and ATR have each been implicated in mediating the response to cisplatin-induced DNA damage. In some cases, the response is dependent on genetic defects in specific repair pathways. Cells defective in by-pass polymerase pol eta display increased DNA-PK dependent signaling, as measured by RPA2 hyperphosphorylation following cisplatin treatment.66 Similar effects were observed with oxaliplatin treatment, though ATM was not implicated in the response to cisplatin and oxaliplatin, consistent with Chk1 phosphorylation via ATR. More recently, an examination of RPA phosphorylation in response to DNA damage and replication stress revealed a complex coordination of RPA phosphorylation via ATM, ATR and DNA-PK to initiate replication arrest and recovery after cisplatin induced DNA damage.42 These data are consistent with earlier studies suggesting that activation of ATR signaling pathway results in both Chk1 and Chk2 phosphorylation in response to cisplatin,67 though the Chk2 phosphorylation is likely to be indirect. While a direct link was suggested, an alternative possibility is that the temporal events in cisplatin-induced DNA damage and processing lead to activation of in vivo activation of DNA-PK and ATM that may not be accurately measured in pull-down assays of cell extracts prepared relatively soon < 8 h after cisplatin treatment.67 ATM has been implicated in the response to cisplatin and proposed to impact NER-catalyzed repair.68 This study using normal human fibroblasts and XPA and XPG mutants demonstrated decreased ATM phosphorylation and increased Chk1 phosphorylation in the NER mutants in response to cisplatin treatment.68 In the absence of measuring DNA-PK or ATR activity, expression was unchanged; it is difficult to pinpoint the pathways involved in the presence of an active NER system. The data do suggest that an active NER system increases the potential that ATM is activated as measured by S1981 phosphorylation. This is consistent with a model for a low level of aberrant repair resulting in the generation of DNA DSB’s which could activate ATM directly.
The role of ATR in response to cisplatin has gained considerable attention with the demonstration and genetic knockdown of ATR-sensitized p53 null cancer cells to cisplatin.69 The effect of ATR-induced S-phase arrest was found to be a significant contributor to cisplatin-induced apoptosis, though oxaliplatin-induced apoptosis was less sensitive to ATR inhibition.70 The recent development of ATR inhibitors has reinvigorated the field and notable synergy with cisplatin has been observed in conjunction with ATR inhibition.71,72 Analysis of the VE-821 ATR inhibitor revealed impressive synergy with cisplatin over a wide range of drug concentrations.71 The demonstration of synthetic lethality with ATM in a cisplatin treatment model suggests that aberrant or reduced ATR signaling leads to the generation of collapsed replication forks in S-phase and the activation of ATM-dependent signaling and S-phase arrest. How ATR inhibition impacts NER catalyzed repair is an interesting question that has yet to be directly addressed. The canonical pathway from ATR to Chk1 first demonstrated to be induced by cisplatin73 has led to a series of studies of assessing the effect of Chk1 inhibition on cisplatin sensitivity and clinical trials of putative Chk1 inhibitors.74 Modest results led to the development of more potent and specific Chk1 inhibitors, though similar lack of synergy has been observed, calling into question the utility of targeting Chk1 as a mechanism to sensitize cell to cisplatin based therapy.75
DNA replication targets
Blockage of DNA replication is often an effective therapy for rapidly dividing cells and can be achieved either through direct DNA damage or indirectly through inhibition of replication proteins such as that observed with topoisomerase inhibitors, camptothecin and etoposide (Fig. 4A). Numerous anti-metabolites target DNA metabolism for instance thymidylate synthase, ribonucleotide reductase and dihydrofolate reductase, via agents including 5-fluorouracil, gemcitabine and methotrexate respectively and are often used in combination (Fig. 3F and G).76 These agents along with the laboratory workhorse for inducing replication stress, HU, are strong activators of the ATR pathway. Similar to HU, gemcitabine has been demonstrated to inhibit ribonucleotide reductase; thus, the activation of ATR is not unexpected.77 However, gemcitabine can also be incorporated into a growing DNA chain acting as a chain terminator like cytosine arabinoside (Ara-C). Interestingly Ara-C sensitivity is increased in cells with defective ATR signaling77 suggesting that in the ATR activation via gemcitabine is, in part, a function of both its activities, inhibition of ribonucleotide reductase and acting as a chain terminator. This data are consistent with a more recent finding that the small molecule ATR VE82 sensitizes cells to gemcitibine71 treatment suggesting that ATR inhibition could be an effective strategy to increase the anti-cancer activity of gem and other DNA damaging therapeutics that activates the ATR pathway.

Figure 4. Chemical structures of natural product-based DNA damaging therapeutics.
Topoisomerase II poisons doxorubicin and etoposide (Fig. 4) also are effective anticancer agents and act through the inhibition of topoisomerase II which is necessary for relieving the positive supercoiles associated with both DNA replication and transcription. Etoposide stabilizes the covalent DNA-protein complex and imparts significant replication stress, while doxorubicin intercalates into DNA to inhibit topo II. This general mechanism of action is consistent with the strong activation of the DDR. Similar to the effect of cisplatin, etoposide induces RPA hyperphosphorylation via DDR kinases42 as well as histone H2AX phosphorylation.78 Interestingly, while etoposide has been demonstrated to activate ATM and ATR42 doxorubicin showed dose dependent activation of ATM and no ATR activation as determined by Ser428 phosphorylation.78 However, there is no data to suggest that p-428 is in fact a measure of ATR activation. This remains an open questions and the use of a phosphospecific antibody should be avoided until there is convincing demonstration that the specific phosphorylation site is a true measure of activation. In fact the demonstration that the ATR inhibitor Nu6027 sensitized cells to doxorubicin argues that ATR is a contributor to the cellular response to doxorubicin treatment.72 Similarly, activation of ATR by the topoisomerase I poison camptothecin and increase sensitivity observed in ATR or CHK1 knockdown has been demonstrated.79 The NU6027 inhibitor also potentiated the effect of camptothecin in breast cancer cell culture models.72 One would expect considerable synergy with small molecule ATR inhibitors with second generation topo I inhibitors including irinotecan and topotectan although this has not yet been demonstrated.
Direct DNA DSB inducing therapies
DNA damage in the form of DNA double strand breaks can be induced by IR therapy as well as radiomimetic agents, including bleomycin and enediyne compounds (Fig. 4C). There have been a number of excellent reviews on the roles of NHEJ and HR in the response to IR. The cellular response to radiomimetic agents appears to be somewhat more variable and clear exceptions to the canonical DDR pathways have been demonstrated. These differences may be a function of either the temporal differences in DNA damage induction or in the variability or complexity of the damage induced. The anti-tumor enediyne antibiotic C-1027 (Fig. 4D) induced direct DNA DSB and was demonstrated to activate ATM as assessed by Ser1981 phosphorylation and downstream target phosphorylation of Chk1 and 2 and p53.80 Interestingly, in the absence of ATM, the same downstream targets were effectively phosphorylated suggesting ATR or DNA-PK dependent signaling. Knockdown of ATR decreased Chk1 phosphorylation, but not phosphorylation of Chk2 or p53 and only with combined ATM and ATR deficiency was downstream signaling reduced. This is in direct contrast to IR-induced DNA DSB’s where in the absence of ATM, Chk2 and p53 phosphorylation has been demonstrated to be significantly reduced.49 Interestingly, the ATM-independent signaling in response to C-1027 was attributed to the formation of interstrand crosslinks and activation of ATR, suggesting a replication stress model.81 With the development of effective and specific inhibitors of ATM, ATR and DNA-PK it should be possible to rationally combine these agents with individual DNA damaging therapies to maximize anti-cancer efficacy.
Conclusion
DNA damaging agents have been and will continue to be a mainstay in numerous cancer therapies. These drugs induce a variety of different DNA lesions which the cell must recognize and counter in order to survive. The pathways utilized in this endeavor converge on the DDR. Originally thought to be three separate pathways, we are just beginning to elucidate the network of interactions and regulations that impact the outcome following DNA damage. The dysregulation of DNA repair and damage response a contributing factor in carcinogenesis, can be exploited for cancer therapy. Effectively targeting the DDR and DNA repair requires the thorough understanding of the interactions between the pathways being induced. An important consideration is how one pathway compensates for the decreased activity of another as a function transient inhibition induced by therapies targeting the DDR. With this knowledge there are unlimited opportunities for targeting the DDR to in combination with DNA damaging therapies to enhance cancer treatment.
Acknowledgments
We thank the members of the Turchi Lab, past and present, for their insightful discussions. This research was supported by grant CA82741 from the National Institutes of Health to J.J.T.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest are disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/cbt/article/23761
References
- 1.Meek K, Dang V, Lees-Miller SP. DNA-PK: the means to justify the ends? Adv Immunol. 2008;99:33–58. doi: 10.1016/S0065-2776(08)00602-0. [DOI] [PubMed] [Google Scholar]
- 2.Falck J, Coates J, Jackson SP. Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature. 2005;434:605–11. doi: 10.1038/nature03442. [DOI] [PubMed] [Google Scholar]
- 3.Shiotani B, Zou L. Single-stranded DNA orchestrates an ATM-to-ATR switch at DNA breaks. Mol Cell. 2009;33:547–58. doi: 10.1016/j.molcel.2009.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 2003;421:499–506. doi: 10.1038/nature01368. [DOI] [PubMed] [Google Scholar]
- 5.Chapman JR, Taylor MR, Boulton SJ. Playing the end game: DNA double-strand break repair pathway choice. Mol Cell. 2012;47:497–510. doi: 10.1016/j.molcel.2012.07.029. [DOI] [PubMed] [Google Scholar]
- 6.Kanu N, Behrens A. ATMIN defines an NBS1-independent pathway of ATM signalling. EMBO J. 2007;26:2933–41. doi: 10.1038/sj.emboj.7601733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jazayeri A, Falck J, Lukas C, Bartek J, Smith GCM, Lukas J, et al. ATM- and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat Cell Biol. 2006;8:37–45. doi: 10.1038/ncb1337. [DOI] [PubMed] [Google Scholar]
- 8.Houldsworth J, Lavin MF. Effect of ionizing radiation on synthesis of sub-replicon size DNA in ataxia telangiectasia cells. Biochem Int. 1983;6:349–56. [PubMed] [Google Scholar]
- 9.Kozlov SV, Graham ME, Jakob B, Tobias F, Kijas AW, Tanuji M, et al. Autophosphorylation and ATM activation: additional sites add to the complexity. J Biol Chem. 2011;286:9107–19. doi: 10.1074/jbc.M110.204065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guo Z, Kozlov S, Lavin MF, Person MD, Paull TT. ATM activation by oxidative stress. Science. 2010;330:517–21. doi: 10.1126/science.1192912. [DOI] [PubMed] [Google Scholar]
- 11.Nam EA, Cortez D. ATR signalling: more than meeting at the fork. Biochem J. 2011;436:527–36. doi: 10.1042/BJ20102162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ball HL, Cortez D. ATRIP oligomerization is required for ATR-dependent checkpoint signaling. J Biol Chem. 2005;280:31390–6. doi: 10.1074/jbc.M504961200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nam EA, Zhao R, Glick GG, Bansbach CE, Friedman DB, Cortez D. Thr-1989 phosphorylation is a marker of active ataxia telangiectasia-mutated and Rad3-related (ATR) kinase. J Biol Chem. 2011;286:28707–14. doi: 10.1074/jbc.M111.248914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pawelczak KS, Bennett SM, Turchi JJ. Coordination of DNA-PK activation and nuclease processing of DNA termini in NHEJ. Antioxid Redox Signal. 2011;14:2531–43. doi: 10.1089/ars.2010.3368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dobbs TA, Palmer P, Maniou Z, Lomax ME, O’Neill P. Interplay of two major repair pathways in the processing of complex double-strand DNA breaks. DNA Repair (Amst) 2008;7:1372–83. doi: 10.1016/j.dnarep.2008.05.001. [DOI] [PubMed] [Google Scholar]
- 16.Singleton BK, Torres-Arzayus MI, Rottinghaus ST, Taccioli GE, Jeggo PA. The C terminus of Ku80 activates the DNA-dependent protein kinase catalytic subunit. Mol Cell Biol. 1999;19:3267–77. doi: 10.1128/mcb.19.5.3267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gell D, Jackson SP. Mapping of protein-protein interactions within the DNA-dependent protein kinase complex. Nucleic Acids Res. 1999;27:3494–502. doi: 10.1093/nar/27.17.3494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weterings E, Verkaik NS, Keijzers G, Florea BI, Wang SY, Ortega LG, et al. The Ku80 carboxy terminus stimulates joining and artemis-mediated processing of DNA ends. Mol Cell Biol. 2009;29:1134–42. doi: 10.1128/MCB.00971-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bennett SM, Woods DS, Pawelczak KS, Turchi JJ. Multiple protein-protein interactions within the DNA-PK complex are mediated by the C-terminus of Ku 80. Int J Biochem Mol Biol. 2012;3:36–45. [PMC free article] [PubMed] [Google Scholar]
- 20.Hammel M, Yu Y, Mahaney BL, Cai B, Ye R, Phipps BM, et al. Ku and DNA-dependent protein kinase dynamic conformations and assembly regulate DNA binding and the initial non-homologous end joining complex. J Biol Chem. 2010;285:1414–23. doi: 10.1074/jbc.M109.065615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pawelczak KS, Turchi JJ. A mechanism for DNA-PK activation requiring unique contributions from each strand of a DNA terminus and implications for microhomology-mediated nonhomologous DNA end joining. Nucleic Acids Res. 2008;36:4022–31. doi: 10.1093/nar/gkn344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Meek K, Douglas P, Cui X, Ding Q, Lees-Miller SP. trans Autophosphorylation at DNA-dependent protein kinase’s two major autophosphorylation site clusters facilitates end processing but not end joining. Mol Cell Biol. 2007;27:3881–90. doi: 10.1128/MCB.02366-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Neal JA, Dang V, Douglas P, Wold MS, Lees-Miller SP, Meek K. Inhibition of homologous recombination by DNA-dependent protein kinase requires kinase activity, is titratable, and is modulated by autophosphorylation. Mol Cell Biol. 2011;31:1719–33. doi: 10.1128/MCB.01298-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen BP, Uematsu N, Kobayashi J, Lerenthal Y, Krempler A, Yajima H, et al. Ataxia telangiectasia mutated (ATM) is essential for DNA-PKcs phosphorylations at the Thr-2609 cluster upon DNA double strand break. J Biol Chem. 2007;282:6582–7. doi: 10.1074/jbc.M611605200. [DOI] [PubMed] [Google Scholar]
- 25.Yajima H, Lee KJ, Chen BP. ATR-dependent phosphorylation of DNA-dependent protein kinase catalytic subunit in response to UV-induced replication stress. Mol Cell Biol. 2006;26:7520–8. doi: 10.1128/MCB.00048-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Peng Y, Woods RG, Beamish H, Ye R, Lees-Miller SP, Lavin MF, et al. Deficiency in the catalytic subunit of DNA-dependent protein kinase causes down-regulation of ATM. Cancer Res. 2005;65:1670–7. doi: 10.1158/0008-5472.CAN-04-3451. [DOI] [PubMed] [Google Scholar]
- 27.Quennet V, Beucher A, Barton O, Takeda S, Löbrich M. CtIP and MRN promote non-homologous end-joining of etoposide-induced DNA double-strand breaks in G1. Nucleic Acids Res. 2011;39:2144–52. doi: 10.1093/nar/gkq1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Langerak P, Mejia-Ramirez E, Limbo O, Russell P. Release of Ku and MRN from DNA ends by Mre11 nuclease activity and Ctp1 is required for homologous recombination repair of double-strand breaks. PLoS Genet. 2011;7:e1002271. doi: 10.1371/journal.pgen.1002271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sertic S, Pizzi S, Lazzaro F, Plevani P, Muzi-Falconi M. NER and DDR: classical music with new instruments. Cell Cycle. 2012;11:668–74. doi: 10.4161/cc.11.4.19117. [DOI] [PubMed] [Google Scholar]
- 30.Shuck SC, Short EA, Turchi JJ. Eukaryotic nucleotide excision repair: from understanding mechanisms to influencing biology. Cell Res. 2008;18:64–72. doi: 10.1038/cr.2008.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sertic S, Pizzi S, Cloney R, Lehmann AR, Marini F, Plevani P, et al. Human exonuclease 1 connects nucleotide excision repair (NER) processing with checkpoint activation in response to UV irradiation. Proc Natl Acad Sci U S A. 2011;108:13647–52. doi: 10.1073/pnas.1108547108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gibson BA, Kraus WL. New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nat Rev Mol Cell Biol. 2012;13:411–24. doi: 10.1038/nrm3376. [DOI] [PubMed] [Google Scholar]
- 33.Jackson SP. The DNA-damage response: new molecular insights and new approaches to cancer therapy. Biochem Soc Trans. 2009;37:483–94. doi: 10.1042/BST0370483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lukas J, Lukas C, Bartek J. More than just a focus: The chromatin response to DNA damage and its role in genome integrity maintenance. Nat Cell Biol. 2011;13:1161–9. doi: 10.1038/ncb2344. [DOI] [PubMed] [Google Scholar]
- 35.Cleaver JE. γH2Ax: biomarker of damage or functional participant in DNA repair “all that glitters is not gold!”. Photochem Photobiol. 2011;87:1230–9. doi: 10.1111/j.1751-1097.2011.00995.x. [DOI] [PubMed] [Google Scholar]
- 36.Riballo E, Kühne M, Rief N, Doherty A, Smith GCM, Recio MJ, et al. A pathway of double-strand break rejoining dependent upon ATM, Artemis, and proteins locating to gamma-H2AX foci. Mol Cell. 2004;16:715–24. doi: 10.1016/j.molcel.2004.10.029. [DOI] [PubMed] [Google Scholar]
- 37.Cook PJ, Ju BG, Telese F, Wang X, Glass CK, Rosenfeld MG. Tyrosine dephosphorylation of H2AX modulates apoptosis and survival decisions. Nature. 2009;458:591–6. doi: 10.1038/nature07849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jungmichel S, Stucki M. MDC1: The art of keeping things in focus. Chromosoma. 2010;119:337–49. doi: 10.1007/s00412-010-0266-9. [DOI] [PubMed] [Google Scholar]
- 39.Lowndes NF, Toh GW. DNA repair: the importance of phosphorylating histone H2AX. Curr Biol. 2005;15:R99–102. doi: 10.1016/j.cub.2005.01.029. [DOI] [PubMed] [Google Scholar]
- 40.Yoo HY, Kumagai A, Shevchenko A, Shevchenko A, Dunphy WG. Ataxia-telangiectasia mutated (ATM)-dependent activation of ATR occurs through phosphorylation of TopBP1 by ATM. J Biol Chem. 2007;282:17501–6. doi: 10.1074/jbc.M701770200. [DOI] [PubMed] [Google Scholar]
- 41.Shuck SC, Turchi JJ. Targeted inhibition of Replication Protein A reveals cytotoxic activity, synergy with chemotherapeutic DNA-damaging agents, and insight into cellular function. Cancer Res. 2010;70:3189–98. doi: 10.1158/0008-5472.CAN-09-3422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu S, Opiyo SO, Manthey K, Glanzer JG, Ashley AK, Amerin C, et al. Distinct roles for DNA-PK, ATM and ATR in RPA phosphorylation and checkpoint activation in response to replication stress. Nucleic Acids Res. 2012;40:10780–94. doi: 10.1093/nar/gks849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Serrano MA, Li Z, Dangeti M, Musich PR, Patrick S, Roginskaya M, et al. DNA-PK, ATM and ATR collaboratively regulate p53-RPA interaction to facilitate homologous recombination DNA repair. Oncogene. 2012 doi: 10.1038/onc.2012.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liaw H, Lee D, Myung K. DNA-PK-dependent RPA2 hyperphosphorylation facilitates DNA repair and suppresses sister chromatid exchange. PLoS One. 2011;6:e21424. doi: 10.1371/journal.pone.0021424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Feng J, Wakeman T, Yong S, Wu X, Kornbluth S, Wang XF. Protein phosphatase 2A-dependent dephosphorylation of replication protein A is required for the repair of DNA breaks induced by replication stress. Mol Cell Biol. 2009;29:5696–709. doi: 10.1128/MCB.00191-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Flynn RL, Zou L. ATR: a master conductor of cellular responses to DNA replication stress. Trends Biochem Sci. 2011;36:133–40. doi: 10.1016/j.tibs.2010.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang X, Zou L, Lu T, Bao S, Hurov KE, Hittelman WN, et al. Rad17 phosphorylation is required for claspin recruitment and Chk1 activation in response to replication stress. Mol Cell. 2006;23:331–41. doi: 10.1016/j.molcel.2006.06.022. [DOI] [PubMed] [Google Scholar]
- 48.Reinhardt HC, Yaffe MB. Kinases that control the cell cycle in response to DNA damage: Chk1, Chk2, and MK2. Curr Opin Cell Biol. 2009;21:245–55. doi: 10.1016/j.ceb.2009.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ahn JY, Schwarz JK, Piwnica-Worms H, Canman CE. Threonine 68 phosphorylation by ataxia telangiectasia mutated is required for efficient activation of Chk2 in response to ionizing radiation. Cancer Res. 2000;60:5934–6. [PubMed] [Google Scholar]
- 50.Ahn J, Urist M, Prives C. The Chk2 protein kinase. DNA Repair (Amst) 2004;3:1039–47. doi: 10.1016/j.dnarep.2004.03.033. [DOI] [PubMed] [Google Scholar]
- 51.Jin J, Ang XL, Ye X, Livingstone M, Harper JW. Differential roles for checkpoint kinases in DNA damage-dependent degradation of the Cdc25A protein phosphatase. J Biol Chem. 2008;283:19322–8. doi: 10.1074/jbc.M802474200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jin J, Shirogane T, Xu L, Nalepa G, Qin J, Elledge SJ, et al. SCFbeta-TRCP links Chk1 signaling to degradation of the Cdc25A protein phosphatase. Genes Dev. 2003;17:3062–74. doi: 10.1101/gad.1157503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ferguson AM, White LS, Donovan PJ, Piwnica-Worms H. Normal cell cycle and checkpoint responses in mice and cells lacking Cdc25B and Cdc25C protein phosphatases. Mol Cell Biol. 2005;25:2853–60. doi: 10.1128/MCB.25.7.2853-2860.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lopergolo A, Tavecchio M, Lisanti S, Ghosh JC, Dohi T, Faversani A, et al. Chk2 phosphorylation of survivin-DeltaEx3 contributes to a DNA damage-sensing checkpoint in cancer. Cancer Res. 2012;72:3251–9. doi: 10.1158/0008-5472.CAN-11-4035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ilves I, Tamberg N, Botchan MR. Checkpoint kinase 2 (Chk2) inhibits the activity of the Cdc45/MCM2-7/GINS (CMG) replicative helicase complex. Proc Natl Acad Sci U S A. 2012;109:13163–70. doi: 10.1073/pnas.1211525109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hu J, Sun L, Shen F, Chen Y, Hua Y, Liu Y, et al. The intra-S phase checkpoint targets Dna2 to prevent stalled replication forks from reversing. Cell. 2012;149:1221–32. doi: 10.1016/j.cell.2012.04.030. [DOI] [PubMed] [Google Scholar]
- 57.Keating MJ, Bach C, Yasothan U, Kirkpatrick P. Bendamustine. Nat Rev Drug Discov. 2008;7:473–4. doi: 10.1038/nrd2596. [DOI] [PubMed] [Google Scholar]
- 58.Leoni LM, Bailey B, Reifert J, Bendall HH, Zeller RW, Corbeil J, et al. Bendamustine (Treanda) displays a distinct pattern of cytotoxicity and unique mechanistic features compared with other alkylating agents. Clin Cancer Res. 2008;14:309–17. doi: 10.1158/1078-0432.CCR-07-1061. [DOI] [PubMed] [Google Scholar]
- 59.Hartmann M, Zimmer C. Investigation of cross-link formation in DNA by the alkylating cytostatic IMET 3106, 3393 and 3943. Biochim Biophys Acta. 1972;287:386–9. doi: 10.1016/0005-2787(72)90282-1. [DOI] [PubMed] [Google Scholar]
- 60.Strumberg D, Harstrick A, Doll K, Hoffmann B, Seeber S. Bendamustine hydrochloride activity against doxorubicin-resistant human breast carcinoma cell lines. Anticancer Drugs. 1996;7:415–21. doi: 10.1097/00001813-199606000-00007. [DOI] [PubMed] [Google Scholar]
- 61.Episkopou H, Kyrtopoulos SA, Sfikakis PP, Fousteri M, Dimopoulos MA, Mullenders LH, et al. Association between transcriptional activity, local chromatin structure, and the efficiencies of both subpathways of nucleotide excision repair of melphalan adducts. Cancer Res. 2009;69:4424–33. doi: 10.1158/0008-5472.CAN-08-3489. [DOI] [PubMed] [Google Scholar]
- 62.Gaul L, Mandl-Weber S, Baumann P, Emmerich B, Schmidmaier R. Bendamustine induces G2 cell cycle arrest and apoptosis in myeloma cells: the role of ATM-Chk2-Cdc25A and ATM-p53-p21-pathways. J Cancer Res Clin Oncol. 2008;134:245–53. doi: 10.1007/s00432-007-0278-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Landau HJ, McNeely SC, Nair JS, Comenzo RL, Asai T, Friedman H, et al. The checkpoint kinase inhibitor AZD7762 potentiates chemotherapy-induced apoptosis of p53-mutated multiple myeloma cells. Mol Cancer Ther. 2012;11:1781–8. doi: 10.1158/1535-7163.MCT-11-0949. [DOI] [PubMed] [Google Scholar]
- 64.Beeharry N, Rattner JB, Bellacosa A, Smith MR, Yen TJ. Dose dependent effects on cell cycle checkpoints and DNA repair by bendamustine. PLoS One. 2012;7:e40342. doi: 10.1371/journal.pone.0040342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jalal S, Earley JN, Turchi JJ. DNA repair: from genome maintenance to biomarker and therapeutic target. Clin Cancer Res. 2011;17:6973–84. doi: 10.1158/1078-0432.CCR-11-0761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cruet-Hennequart S, Glynn MT, Murillo LS, Coyne S, Carty MP. Enhanced DNA-PK-mediated RPA2 hyperphosphorylation in DNA polymerase eta-deficient human cells treated with cisplatin and oxaliplatin. DNA Repair (Amst) 2008;7:582–96. doi: 10.1016/j.dnarep.2007.12.012. [DOI] [PubMed] [Google Scholar]
- 67.Pabla N, Huang S, Mi QS, Daniel R, Dong Z. ATR-Chk2 signaling in p53 activation and DNA damage response during cisplatin-induced apoptosis. J Biol Chem. 2008;283:6572–83. doi: 10.1074/jbc.M707568200. [DOI] [PubMed] [Google Scholar]
- 68.Colton SL, Xu XS, Wang YA, Wang G. The involvement of ataxia-telangiectasia mutated protein activation in nucleotide excision repair-facilitated cell survival with cisplatin treatment. J Biol Chem. 2006;281:27117–25. doi: 10.1074/jbc.M602826200. [DOI] [PubMed] [Google Scholar]
- 69.Sangster-Guity N, Conrad BH, Papadopoulos N, Bunz F. ATR mediates cisplatin resistance in a p53 genotype-specific manner. Oncogene. 2011;30:2526–33. doi: 10.1038/onc.2010.624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lewis KA, Lilly KK, Reynolds EA, Sullivan WP, Kaufmann SH, Cliby WA. Ataxia telangiectasia and rad3-related kinase contributes to cell cycle arrest and survival after cisplatin but not oxaliplatin. Mol Cancer Ther. 2009;8:855–63. doi: 10.1158/1535-7163.MCT-08-1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Prevo R, Fokas E, Reaper PM, Charlton PA, Pollard JR, McKenna WG, et al. The novel ATR inhibitor VE-821 increases sensitivity of pancreatic cancer cells to radiation and chemotherapy. Cancer Biol Ther. 2012;13:1072–81. doi: 10.4161/cbt.21093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Peasland A, Wang LZ, Rowling E, Kyle S, Chen T, Hopkins A, et al. Identification and evaluation of a potent novel ATR inhibitor, NU6027, in breast and ovarian cancer cell lines. Br J Cancer. 2011;105:372–81. doi: 10.1038/bjc.2011.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhao H, Piwnica-Worms H. ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol Cell Biol. 2001;21:4129–39. doi: 10.1128/MCB.21.13.4129-4139.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Fuse E, Kuwabara T, Sparreboom A, Sausville EA, Figg WD. Review of UCN-01 development: a lesson in the importance of clinical pharmacology. J Clin Pharmacol. 2005;45:394–403. doi: 10.1177/0091270005274549. [DOI] [PubMed] [Google Scholar]
- 75.Wagner JM, Karnitz LM. Cisplatin-induced DNA damage activates replication checkpoint signaling components that differentially affect tumor cell survival. Mol Pharmacol. 2009;76:208–14. doi: 10.1124/mol.109.055178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Peters GJ, van der Wilt CL, van Moorsel CJ, Kroep JR, Bergman AM, Ackland SP. Basis for effective combination cancer chemotherapy with antimetabolites. Pharmacol Ther. 2000;87:227–53. doi: 10.1016/S0163-7258(00)00086-3. [DOI] [PubMed] [Google Scholar]
- 77.Karnitz LM, Flatten KS, Wagner JM, Loegering D, Hackbarth JS, Arlander SJ, et al. Gemcitabine-induced activation of checkpoint signaling pathways that affect tumor cell survival. Mol Pharmacol. 2005;68:1636–44. doi: 10.1124/mol.105.012716. [DOI] [PubMed] [Google Scholar]
- 78.Huelsenbeck SC, Schorr A, Roos WP, Huelsenbeck J, Henninger C, Kaina B, et al. Rac1 protein signaling is required for DNA damage response stimulated by topoisomerase II poisons. J Biol Chem. 2012;287:38590–9. doi: 10.1074/jbc.M112.377903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Flatten K, Dai NT, Vroman BT, Loegering D, Erlichman C, Karnitz LM, et al. The role of checkpoint kinase 1 in sensitivity to topoisomerase I poisons. J Biol Chem. 2005;280:14349–55. doi: 10.1074/jbc.M411890200. [DOI] [PubMed] [Google Scholar]
- 80.Kennedy DR, Beerman TA. The radiomimetic enediyne C-1027 induces unusual DNA damage responses to double-strand breaks. Biochemistry. 2006;45:3747–54. doi: 10.1021/bi052334c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kennedy DR, Ju J, Shen B, Beerman TA. Designer enediynes generate DNA breaks, interstrand cross-links, or both, with concomitant changes in the regulation of DNA damage responses. Proc Natl Acad Sci U S A. 2007;104:17632–7. doi: 10.1073/pnas.0708274104. [DOI] [PMC free article] [PubMed] [Google Scholar]