

Abstract
Our laboratory has investigated whether and how 17β-estradiol (E2) protects the brain against neurodegeneration associated with cerebrovascular stroke. We have discovered that low, physiological concentrations of E2, which are strikingly similar to low-basal circulating levels found in cycling mice, dramatically protect the brain against stroke injury, and consequently revealed multiple signaling pathways and key genes that mediate protective action of E2. Here we will review the discoveries comprising our current understanding of neuroprotective actions of estrogens against ischemic stroke. These findings may carry far reaching implications for improving the quality of life in aging populations.
Keywords: 17β-Estradiol, Estrogen receptor, Estrogen therapy, Ischemia, Middle cerebral artery occlusion, Inflammation, Neurogenesis, Stroke, Women’s health initiative
1. Introduction
Menopause marks the end of ovarian hormone production and the loss of reproductive function in women. Over the past century the age of menopause has remained fixed at age 51, while the average life expectancy has increased to over 80 years. Thus, today many women spend over three decades of their lives in a hypoestrogenic state. Several studies have suggested that this hypoestrogenic state may make women more vulnerable to a variety of age- and hormone-related diseases. Women are protected from stroke and other cardiovascular diseases until they reach menopause [6,49,62,92,93,108]. The lack of ovarian hormone production has been offered as an explanation for the fact that postmenopausal women are at higher risk for stroke compared to their male counterparts and account for approximately 61.0% of all stroke-related deaths (6–8; www.americanheart.org/statistics). Each year approximately 40,000 more women than men are affected by stroke [12]. This gender difference is related to a combination of both the longer life expectancy of women and the protective roles of estrogens, since the incidence of stroke in women dramatically increases after menopause and the risk continues to rise with age [53]. Over the past two decades, numerous observational and retrospective studies demonstrated that estrogen therapy (ET) given to postmenopausal women provides benefits against cardio- and cerebrovascular diseases [6,49,62,92,93,108].
Despite these studies, recent clinical trials including the women’s health initiative (WHI) reported negative impact of ET and spurred reconsideration of its use around the world. However, a careful review of the literature suggests that data from the WHI and other clinical trials can only be interpreted within the narrow context of their study designs [92]. In addition, some studies in animal models also suggest that estrogens are not universally protective and can be deleterious under some circumstances [47]. Therefore, it becomes even more important to continue to perform studies using different appropriate animal models to improve our understanding of the wide spectrum of estrogen’s actions, and to probe the mechanisms that underlie these seemingly divergent effects.
2. Discrepancies over the efficacy of estrogen therapy against stroke
Contrary to numerous studies that demonstrate beneficial actions of estrogen, the WHI reported that ET under certain circumstances increases the risk for stroke, and it does not exhibit any beneficial effects on long-term stroke outcomes [12,60,77,99]. The WHI consisted of a randomized, placebo-controlled clinical trial primarily designed to test whether ET is protective against coronary heart disease in postmenopausal women. In 2004, the WHI was terminated due to an increased risk of stroke in women receiving treatment [53]. It is important to remember that, in the WHI, the mean age of the subjects was 63 years, and the vast majority of subjects were on average 12 years postmenopause prior to the initiation of any hormone treatment [49,53]. In striking contrast, observational studies that previously reported cardio- and cerebrovascular benefits of ET examined women averaging 51 years of age, many of whom initiated hormone therapy in their menopausal transition [12,71,104].
To decipher the circumstances under which ET provides benefits against cerebral stroke, we attempted to mimic the delayed initiation of ET used in the WHI. We investigated whether a prolonged period of hypoestrogenicity disrupts the ability of 17β-estradiol (E2) to protect the brain against stroke injury [51]. We demonstrated that E2 exerts profound neuroprotective action only when administered immediately upon ovariectomy, but not when administered after 10 weeks of hypoestrogenicity. Therefore, our findings show that an extended period of hypoestrogenicity, similar to that experienced by the women included in the WHI, prevents E2 from protecting the brain against ischemia.
The secondary analysis of the WHI also suggests that the discrepancies between previous observational studies and recent clinical trials over the efficacy of ET may have resulted from the differences in the timing of hormone administration relative to the perimenopausal transition [33]. This recent re-evaluation of the WHI reported differential effects of ET on the risk of cardiovascular diseases based on the age and years since menopause. In this report, the use of postmenopausal conjugated equine estrogens (CEE) was associated with a reduced risk for coronary heart disease (a hazard ratio of 0.63 [95% confidence interval], 0.36–1.09) in a group of women aged 50–59 years [33]. In addition, the study reported a hazard ratio of 0.89 ([95% CI], 0.47–1.69) for stroke incidence in the CEE trial within the same age group, further supporting the concept that the timing of initiation of hormone therapy influences its efficacy against stroke injury [33]. Together, our findings, in combination with the results of the WHI and its recent re-evaluation, emphasize the tremendous importance of strengthening the collaboration between basic science and clinical researchers to take the maximum advantage of empirical and mechanism-based information and approaches to gain deeper understanding of the diverse mechanisms of E2’s protective actions.
3. Neuroprotective actions of estrogen against stroke
3.1. Low, physiological levels of E2 exert profound neuroprotective action
Our laboratory has used an animal model of stroke to decipher under what circumstances E2 protects against neuronal death and to uncover its mechanisms of action. We have shown that low, physiological levels of E2, which are strikingly similar to low-basal circulating levels found in cycling mice, exert profound neuroprotective actions in a stroke model in which the middle cerebral artery is permanently occluded (Fig. 1; [65,73,78,107]). Permanent blockage of the middle cerebral artery at its base leads to a severe metabolic impairment in the cerebral cortex and striatum, causing many neurons in these regions to die by necrosis within hours following injury. On the other hand, regions that surround the ischemic core (ischemic penumbra) undergo moderate metabolic impairment and are potentially salvageable by effective therapeutic agents. We have previously demonstrated that E2 exerts powerful neuroprotective action in ischemic penumbra where E2 protects neurons from delayed programmed cell death or apoptosis [65,73,78].
Fig. 1.
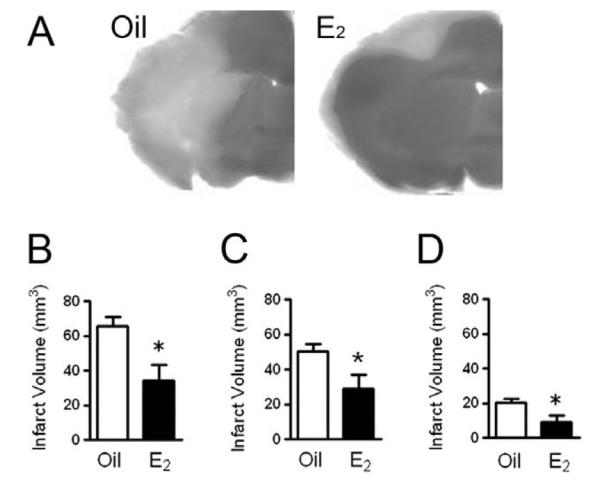
Low, physiological levels of E2 exert neuroprotection. E2 protects against neuronal death in a stroke model in which the middle cerebral artery is permanently occluded. A: C57BL/6 J mice were ovariectomized and immediately implanted with capsules containing either oil or E2 for 1 week. Subsequently, animals underwent experimental ischemia by middle cerebral artery occlusion and were killed 24 h after the onset of injury. Infarct volumes were measured using TTC staining in oil- or E2-treated mice. B–D: E2 treatment significantly reduced the total infarct volume (B, P < 0.02) and the extent of injury in both the cortex (C, P < 0.05) and striatum (D, P < 0.03). Data represent the mean ± SEM of 8–11 animals per group. Figure reprinted with permission from [85].
We have also demonstrated that E2 effectively reduces the infarct volume in both young and middle-aged animals compared to oil-treated counterparts, suggesting that a constellation of factors responsible for mediating E2’s protective actions is preserved during the initial stages of aging [11]. Further, we have found that, at low physiological doses (25 pg/ml serum [28]), E2 must be administered prior to the onset of injury, since acute administration of E2 at the time of injury does not reduce the extent of infarction [26]. This finding contrasts with the discoveries of Toung et al. [91] which show that supraphysiological concentrations of E2 administered immediately before the onset of ischemic injury exert neuroprotection. In addition, pharmacological levels of E2 also effectively protected against ischemic brain injury when administered as late as 6 h after the onset of injury [111,113]. The finding that acute, postischemic administration of E2 at supraphysiological doses exerts protective actions against ischemic injury may have strong clinical implications, especially if non-reproductive estrogen-like compounds can be designed, i.e. selective estrogen receptor modulators (SERMs), and used as the treatment as opposed to the preventive method. Together, our findings clearly demonstrate that administration of even basal levels of E2 profoundly protects the brain against stroke-like injury, and that the mechanisms by which estrogens achieve their protective actions are diverse and complex [10,58,66,71,98].
4. ERα mediates estrogen’s neuroprotective action
To date, two forms of nuclear estrogen receptor (ER) have been cloned. Since the discovery of the second form of ER (ERβ) in 1996, researchers have investigated the similarities and differences between the two forms of ER. Although ERα and ERβ share homologies in their structures and exhibit somewhat overlapping distributions throughout the body and brain, they exhibit very important differences in their functions. Classically, both ERα and ERβ are considered as transcription factors, though second-messenger activated pathways may also be important mechanisms that mediate the protective actions of the hormone. Our work has concentrated on whether classical ER-mediated gene regulation plays important roles to promote both trophic and protective actions in the ischemic brain.
Importantly, expression of ERα transiently increases in the developing cortex, and subsequently declines and remains at very low levels throughout adulthood [65,73,78]. A transient upregulation of ERα during neonatal development mediates E2’s pivotal neurotrophic actions in neurite growth, synaptogenesis and neuronal structural organization of cortical neurons. Recently, we found that ischemic injury also upregulates ERα expression in the cortex of ovariectomized animals without influencing ERβ expression [84]. Consequently, we hypothesized that the dramatic re-expression of ERα after stroke injury mediates E2’s profound neuroprotection against ischemia [27,28]. Furthermore, with the use of ERα (ERα −/−) and ERβ (ERβ −/−) knockout mice, we have discovered that the presence of ERα, but not ERβ, is a prerequisite for the ability of E2 to exert protective action against ischemic injury [28]; knocking out ERα blocks E2’s ability to reduce the extent of infarction. In marked contrast, E2 continued to exert powerful protective action against injury-induced neuronal death in ERβ-null mice. Taken together, the injured brain seems to provide signals conveying the need for the reappearance of ERα, which may mediate the ability of E2 to protect against neuronal apoptosis and possibly reinitiate differentiation of the injured brain.
5. Mechanisms of estrogen’s neuroprotective action
5.1. In vivo results
Using our experimental paradigm of stroke injury and low physiological levels of E2 replacement, our laboratory has investigated potential mechanisms that underlie E2’s neuroprotective action. To date, we have identified multiple downstream targets of E2 action in neuroprotection. E2 modulates the expression of arrays of genes in ischemic brains, including those that influence the balance between cell death and cell survival. For instance, we reported that low physiological levels of E2 prevent injury-induced downregulation of Bcl-2 without corresponding changes in the expression of other members of Bcl-2 family genes [27]. Because Bcl-2 is a protooncogene known to promote cell survival in a variety of tissues including the brain, we hypothesize that E2 decreases the extent of neuronal death by preventing downregulation of Bcl-2 in response to ischemic injury. We also examined the effects of stroke injury and E2 on immediate early genes (IEGs), since (a) they are induced in response to various forms of brain injury, (b) their expression is associated with injury-induced programmed cell death, and (c) steroids modulate their expression under a variety of experimental circumstances [27,67]. We have shown that the levels of IEGs including c-fos, fosB, c-jun, and junB increase following ischemic injury, and that E2 specifically attenuates injury-induced upregulation of c-fos mRNA and protein [67]. These findings strongly suggest that the ability of E2 to protect the brain against the late stages of neuronal death involves attenuation of c-fos upregulation. We then tested the hypothesis that E2 protects the brain against stroke injury by attenuating neuronal apoptosis. Our data confirmed that E2 attenuates markers of apoptosis including caspase-3 activation and injury-induced DNA fragmentation [68]. These events contribute to reduced neuronal apoptosis, as indicated by a decrease in the number of TUNEL-positive cells in the cerebral cortex of E2-treated ovariectomized animals compared to vehicle-treated counterparts (Fig. 2).
Fig. 2.

E2 attenuates markers of apoptosis. A, Photographs showing TUNEL-positive cells in the ischemic cortex from oil- and E2-treated animals at 4, 8, and 24 h after stroke injury. B, E2 significantly attenuates the number of TUNEL-positive cells during early (#P < 0.05) and late (*P < 0.05) phases of ischemic injury compared to oil-treated animals. Data represent the mean ± SEM of 8–10 animals per group. Figure reprinted with permission from [68].
5.2. In vitro results
To further explore the molecular and cellular mechanisms mediating protective actions of E2, we have implemented organotypic cerebral cortical explant cultures [106]. Explants were exposed to potassium cyanide/2-deoxyglucose (KCN/2-DG) to model metabolic inhibition observed during ischemic injury. This in vitro model induced by metabolic inhibition exhibits considerable parallelisms with our in vivo model of brain ischemic injury. Using this method, we found that E2 reduces neuronal death following injury in vitro, and that, as our in vivo model, this protection is mediated by estrogen receptors because (a) protection required pretreatment of hormone, (b) 17α-estradiol, the natural optical isomer of E2, which is generally considered to be less active due to its lower binding affinity to estrogen receptors, afforded no protection, and (c) protective action of E2 was blocked by the estrogen receptor antagonist ICI 182,780. We subsequently showed that protective action of E2 in vitro also involves attenuation of apoptosis [107]. E2 pretreatment significantly reduced the number of cells undergoing apoptotic cell death as indicated by nuclear condensation and TUNEL staining. To begin to decipher cell signaling pathways through which E2 prevents apoptosis, we then analyzed the levels of Akt kinase activation. We found that E2 enhances activation of Akt kinase, which is an important mediator of cell survival signaling pathways. These findings complement the work of several laboratories, whose findings have demonstrated the importance of several second messenger pathways in mediating the protective effects of estrogens [11]. Taken together, our findings clearly demonstrate that low physiological levels of E2 exhibit powerful neuroprotective actions both in vivo and in vitro, and that these protective actions are mediated by estrogen receptors and involve attenuation of neuronal apoptosis in response to ischemic brain injury.
6. Anti-inflammatory actions of estrogens
Recent studies describing the seemingly contradictory actions of estrogens in stroke injury have led us to reevaluate the circumstances under which E2 provides benefits against cerebral stroke and decipher its mechanisms of action. One prominent feature that follows stroke injury is massive inflammatory responses. Evidence now suggests that postischemic inflammation strongly contributes to the extent of brain injury, and E2 may protect the ischemic brain by exerting antiinflammatory actions [18,52,96]. Thus, we tested the hypothesis that an extended period of hypoestrogenicity both prevents E2 from protecting the brain against ischemia, and simultaneously suppresses its antiinflammatory actions. We found that E2 exerts profound neuroprotective action when administered immediately upon ovariectomy, but not when administered after 10 weeks of hypoestrogenicity.
6.1. Estrogens exhibit anti-inflammatory activity in the brain
Consistently, E2 treatment given immediately at the time of ovariectomy attenuated central and peripheral production of proinflammatory cytokines after ischemic stroke. In contrast, E2 did not suppress production of proinflammatory molecules when it was administered after 10 weeks postovariectomy [84]. These results demonstrate that a prolonged period of hypoestrogenicity disrupts both neuroprotective and antiinflammatory actions of E2. Our findings may help to explain the results of the WHI that reported no beneficial effect of ET against stroke because the majority of the subjects initiated ET after an extended period of hypoestrogenicity.
Over the past decade we have begun to appreciate that E2 modulates the brain’s immune response. Many neurological disorders such as Alzheimer’s disease (AD), Parkinson’s disease (PD), multiple sclerosis and cerebrovascular stroke all exhibit inflammation throughout the central nervous system and in the periphery. In vivo and in vitro models of brain injury and neurodegenerative diseases have provided substantial evidence that physiological levels of E2 suppress inflammation through ERα and ERβ [5,43,95–97]. These studies demonstrate the tremendous importance of understanding the crosstalk between the nervous, endocrine, and immune systems to fully appreciate the protective role of E2 during neurological diseases and injury [14,96].
Studies have shown that physiological levels of E2 suppress both microglial activation and iNOS-mediated immune response. iNOS is the time-limiting enzyme that produces nitric oxide (NO) as part of the innate immune response, and is tightly regulated after brain insults and injury [20]. Upregulation of iNOS following neurodegenerative insults results in increased production of NO and reactive oxygen species, which in turn disrupt cellular homeostasis and promote neuronal death. Using both rodent and human microglial cell lines, nanomolar E2 concentrations were shown to suppress microglial activation, iNOS expression as well as the production of several proinflammatory cytokines [10,25,39,87,97], and other proinflammatory mediators including matrix metalloproteinase 9 (MMP9), prostaglandin E2 (PGE2) and cyclooxygenase 2 (COX-2) (reviewed in [76,96]). Astrocytes also mediate the antiinflammatory properties of E2 following stimulation with lipopolysaccharide (LPS) [40], β-amyloid [24], and interleukin-1β (IL-1β) [43].
The anti-inflammatory actions of E2 are mediated through both ERα and ERβ, and they are expressed throughout various brain regions and in nearly all brain cell types including microglia and astrocytes (reviewed in [23,56,90,96]). Studies have shown that the ER antagonist ICI 182,780 blocks E2-induced downregulation of NO production [10,97]. This pharmacological data was subsequently supported by in vivo studies demonstrating that ERα plays a primary role as a regulator of E2’s anti-inflammatory actions in the brain. ERα mediates protective actions of E2 against LPS-induced microglial activation in the cortex (frontal, rhinal, and piriform cortex), subcortex (striatum, amydala, thalamus, and medial forebrain bundle), and hippocampus [94,95,97]. These studies indicate that ERα suppresses LPS-mediated signaling by inhibiting nuclear factor (NF)-κB, a transcription factor that regulates expression of many proinflammatory molecules including iNOS [98]. Although precise molecular mechanisms underlying ERβ’s anti-inflammatory actions are still unclear, studies demonstrate that ERβ plays an anti-inflammatory role in the BV2 murine cell line [5], young rat astrocyte cultures [43], and in primary neonatal mouse [8] and rat cultures [55]. The G-protein coupled membrane ER, GPR30, is also expressed in the brain but whether it plays a role in the anti-inflammatory actions of E2 is unclear [7,69].
6.2. Estrogens protect neurovascular unit during stroke
The neurovascular unit represents a conceptual framework described by the local cell to cell interactions among neuronal, glial, and vascular compartments, which all together work to maintain brain homeostasis [22]. Stroke and other neurological disorders such as AD, PD, and amyotrophic lateral sclerosis all disrupt the neurovascular unit [32]. Studies show that E2 may directly or indirectly regulate central inflammatory response by interacting with various components of the neurovascular unit including endothelial cells, smooth muscle cells, astrocytes, neurons, microglia, as well as circulating blood components such as platelets and leukocytes [13,112]. Thus, the efficacy of E2 as a therapeutic agent during focal cerebral ischemia is dependent upon its complex interactions with the entire neurovascular unit.
The anti-inflammatory properties of E2 in the cerebral circulation influence the incidence, outcome, and severity of injury in stroke and in other cerebrovascular disorders. Overall, vascular inflammatory responses are typically inhibited by E2 and enhanced by progesterone and testosterone [29]. One mechanism through which E2 protects the ischemic brain may involve increasing cerebral blood flow by enhancing the activity of endothelial nitric oxide synthase (eNOS). Within the vasculature, E2 suppresses COX-2 and interleukin 1-β (IL-1β), attenuates smooth muscle migration mechanisms, enhances endothelial cell survival through decreased apoptosis, suppresses leukocyte adhesion, and inhibits expression of adhesion molecules (VCAM-1, ICAM-1, and E-selectin) in cerebral endothelial cells [29,52]. E2 also influences the function of cerebrovascular mitochondria by improving the efficiency of electron transport and reducing oxidative stress by stabilizing ATP production [75,81]. Both ERs are expressed throughout the cerebrovasculature. ERα is expressed in smooth muscle cells [80], while both ERα and ERβ are present in the endothelial cell layer of cerebral arteries [19,80]. ERα [80] and ERβ are also present in mitochondria, with a growing body of evidence suggesting an emerging role for ERβ as a mitochondrial transcription factor [75,80]. In addition to classical ER signaling pathways, many of the neuroprotective effects of E2 in the endothelium and nearby glial cells may be attributed to membrane-bound ERα and/or ERβ in the membrane caveolae [15,29,46,100]. Interactions between ERα and the scaffolding protein, caveolin-1, have been shown to activate the phosphatidylinositol 3-kinase (PI3-K)/Akt pathway to enhance the expression of eNOS and inhibit apoptosis [2,46,79].
6.3. Estrogens suppress proinflammatory mediators during stroke
Acute inflammation mediates both toxic and reparative mechanisms during stroke, but injury promotes an excess of toxic inflammatory mechanisms. To date, evidence from various brain injury paradigms has provided a basis for evaluating the hypothesis that physiological concentrations of E2 possess anti-inflammatory properties that protect against delayed neuronal death during focal cerebral ischemia [42,96,109].
The primary source of NO that contributes to ischemia-induced apoptotic cell death is iNOS, which can be produced by several components of the neurovascular unit including neurons, microglia, astrocytes and endothelial cells [21,57]. Knockout mouse models for NOS2 [34,35,58] as well as wild-type (WT) C5/BL/6 J mice treated with inhibitors of iNOS demonstrate that NOS2 is a deleterious gene in models of both permanent and transient MCAO [35,82], but the role of E2 in iNOS knockout mice is not clear [44,63]. To gain a better understanding of the interactions between E2 and iNOS during stroke, we tested whether E2 protects the brain by suppressing iNOS expression in WT and iNOS knockout (iNOS −/−) female mice following permanent MCAO [9]. We quantified the extent of ischemic injury in the cortex, striatum, and hippocampus of injured mice and found that female iNOS−/− mice are protected during MCAO, but are not protected further in the presence of E2 (Fig. 3). We also found that E2 attenuated NOS2 mRNA, the gene product that codes for iNOS protein, in the cortex of both sham and injured WT mice (Fig. 3). These results demonstrate that one mechanism by which E2 exerts neuroprotection is by downregulation of iNOS, a critical gene in the stroke-associated inflammatory response. Thus, our findings suggest that E2 treatment is efficacious only when iNOS protein retains its functionality. A clearer understanding of the relationship between E2 and iNOS will provide greater insights into the molecular mechanisms underlying reported gender differences in stroke.
Fig. 3.

E2 and iNOS display complementary neuroprotective interactions during MCAO. A, B: E2 reduces infarct volume in the cortex and striatum of WT mice ( *, P < 0.05), but does not further suppress infarct size in iNOS−/− mice. iNOS−/− oil-treated mice were also protected during stroke compared to WT oil-treated mice (#, P < 0.05); n = 8–14 mice/treatment/genotype. C, D: NOS2 gene expression was significantly higher in WT oil-treated mice than in WT E-treated mice in the cortex and striatum (*P < 0.05). NOS2 gene expression was measured in 1-mm tissue micropunches adjacent to the tissue infarct in cortex and striatum of WT mice using qRT-PCR. n = 4–5 mouse cortex or striatum samples/hormone treatment. All values represent means ± SEM. Figure reprinted with permission from [9].
Increased cytokine production in the brain and periphery is part of the inflammatory response associated with cerebral ischemia [61,70]. In brain injury paradigms, E2 generally suppresses proinflammatory cytokines, and enhances the production anti-inflammatory cytokine responses [17,72]. Studies in human populations demonstrate a strong correlation between increased levels of serum or plasma pro-inflammatory cytokines and increased stroke severity, while increased levels of anti-inflammatory cytokines are associated with a better outcome after stroke [1,30,50,64,105]. However, there are virtually no clinical studies that address whether E2 decreases cytokine production following stroke. Therefore, we measured levels of central and peripheral cytokines in mice subjected to permanent MCAO. We found that E2 decreased the production of IL-6 on the ipsilateral side of brain injury and in serum. E2 also decreased the production of the monocyte chemoattractant protein-1 (MCP-1), while enhancing the production of vascular endothelial growth factor (VEGF), an important growth factor that promotes the integrity of the BBB [84].
To determine whether E2’s interactions with iNOS can also differentially suppress cytokine production, we measured peripheral cytokines in WT and iNOS−/− mice 24 h following permanent MCAO. We found that E2 and the absence of iNOS suppressed production of the chemokine macrophage inflammatory protein 1-α (MIP1α/CCL3) in WT mice, but E2 did not suppress MIP1α/CCL3 levels further in the iNOS−/− mouse (Fig. 4). Although precise roles of MIP1α/CCL3 during cerebral stroke are unclear, MIP1α/CCL3 serves to regulate microglia/macrophage functionality and regulates recruitment of polymorphonuclear cells [48]. MIPα/CCL3 serum levels are increased in human stroke patients [114] and its mRNA and protein levels are also increased in animal models of cerebral ischemia and hypoxia/ischemia [16,101]. This finding demonstrates that MIP1α/CCL3, similar to E2, serves a regulatory role only when iNOS is present, and provides further insight into the complex relationship between E2 and iNOS that contributes to the gender-based pathophysiology of stroke. Collectively, the findings from our studies are consistent with the aforementioned clinical studies and suggest another potential therapeutic target for ET against stroke-related brain injury.
Fig. 4.

E2 suppresses peripheral cytokines following MCAO. MIP1α /CCL3 was significantly suppressed by E2-treatment after MCAO injury in WT mice (*, P < 0.05), while E2 provided no further suppression in iNOS−/− mice compared to oil-treated controls. As with infarct volume, an effect of genotype was observed in iNOS−/− oil-treated mice which were also protected during stroke compared to WT oil-treated mice (#, P < 0.05, n = 8–12 mice/genotype/treatment). Values represent mean ± SEM.
7. Neurotrophic actions of estrogens
During brain development, E2 plays crucial roles in neuronal differentiation, neurite outgrowth, synaptogenesis and neuronal structural organization in the cerebral cortex and hippocampus. Consistently, expression of ERα transiently increases in these regions during fetal development, which in turn mediates neurotrophic actions of E2 [78]. Previously, we have demonstrated that expression of ERα highly increases in the ischemic cortex following stroke injury [27,28]. The dramatic re-expression of ERα after injury may thus represent a recapitulation of its expression during neonatal development when E2 plays pivotal trophic roles, allowing E2 to act once again as a trophic factor to promote repair and remodeling of the ischemic brain.
7.1. E2 stimulates glutamatergic synapse formation in the developing hippocampus through ERα
We recently demonstrated that E2 mediates subcellular changes in synaptic protein distribution to induce new synapses via an estrogen receptor (ER)-mediated process [37]. Synaptic protein distribution and size were identified with antibodies to the presynaptic vesicular glutamate transporter protein (vGlut1) and postsynaptic NMDA receptor (NR1 subunit). We have observed that E2 treatment (10 nM) for 48 h increases synapse density, as detected by NR1 and vGlut1 colocalization along dendrites of hippocampal neurons cultured in steroid-stripped media (Fig. 5). To test which ER subtype plays an important functional role in E2-induced synaptogenesis, we subsequently used ER antagonist [7α,17ß-[9[(4,4,5,5,5-pentafluoropentyl) sulfinyl]nonyl]estra-1,3,5(10)-triene-3,17-diol (ICI 182,780)] and the ER-α- and ER-β-specific agonists [1,3,5-tris(4-hydroxyphenyl)-4-propyl-1H-pyrazole (PPT) and 2,3-bis(4-hydroxyphenyl) propionitrile (DPN), respectively] [37]. As expected, ICI 182,780 blocked an increase in synapse density. We also found that treatment with PPT, but not DPN, induced significant increase in synapse density that mimicked the effect of E2 [37]. Together, the results of these studies demonstrate that E2 stimulates glutamatergic synapse formation in the developing hippocampus through an ERα-dependent mechanism. These findings carry profound implications regarding the potential of estrogen to influence learning, memory, and possibly hormone-modulated neurodegeneration.
Fig. 5.

E2 increases synaptic density and size. A–F, Double-label ICC for NR1 (green) and vGlut1 (red); yellow in the superimposed images indicates colocalization. G–H, E2 treatment significantly increased the density of colocalized NR1 and vGlut1 clusters and cluster size (48 h: P < 0.0005; 6 d: P < 0.003; Student’s t test). All values are mean ± SEM. Scale bars = 40 μm. Figure reprinted with permission from [37].
8. Neurogenic actions of estrogens following stroke injury
In addition to its profound neuroprotective and neurotrophic actions against ischemia, E2 also exerts critical neurogenic actions following stroke injury. Recently our laboratory demonstrated that E2 stimulates generation of newborn neurons in the subventricular zone (SVZ), one of two major neurogenic regions of the adult brain, under ischemic conditions.
8.1. E2 enhances neurogenesis following ischemic stroke
One of the most remarkable discoveries in modern neuroscience is that the adult brain continues to generate new neurons under both normal and neurodegenerative conditions. Studies have shown that adult neurogenesis is restricted to two major forebrain regions; the dentate gyrus of the hippocampus and the subventricular zone (SVZ) lining the lateral ventricle [3,88]. Under normal conditions, neural stem cells born in the SVZ differentiate into neuroblasts and migrate through the rostral migratory stream to the olfactory bulb, where they become functional interneurons [45]. However, several pathological processes induced by cortical aspiration, traumatic brain injury, and cerebral ischemia redirect the SVZ neuroblasts from their normal migratory route towards the sites of injury. Specifically, studies showed that a proportion of newborn neurons of the SVZ migrate towards the ischemic regions including the cerebral cortex and striatum [31,41,66]. The ability of newborn neurons of the SVZ to migrate to the ischemic regions to potentially replace damaged neurons points to the tremendous potential of the SVZ as a target for neuronal replacement therapy against stroke. However, recent reports indicate that the number of neurons that migrate to the injured regions is insufficient to assist proper recovery of the injured brain [4,89]. Therefore, we investigated whether E2 stimulates generation of newborn neurons in the SVZ under ischemic conditions. We have found that low, physiological levels of E2 replacement that produce approximately 25 pg/ml in serum [28], the same dose of E2 that sufficiently and profoundly protect neurons against ischemia, increases the number of newborn neurons in the SVZ after stroke injury (Fig. 6).
Fig. 6.

E2 significantly increases the number of BrdU+/Dcx+ newborn neurons. A, B: Confocal micrographs of BrdU+ cells (green) double-labeled with early neuronal marker doublecortin (Dcx, red) in the ipsilateral SVZ of oil- (A) vs. E2- (B) treated mice at 96 h after MCAO injury. C: Higher magnification of panel B to demonstrate colocalization of BrdU+/Dcx+. Arrows indicate representative double-labeled cells. D: E2 significantly increased the number of BrdU+/Dcx+ newborn neurons (*P = 0.0008, n = 6–7). E, F: BrdU+ cells (green) did not co-label with markers for mature neuron (NeuN, red; E) or astrocyte (GFAP, red; F). CC, corpus callosum; STR, striatum. Values represent mean ± SEM. Figure reprinted with permission from [85].
Fig. 6 shows confocal micrographs of BrdU+ cells (proliferating cellular marker, shown in green) double-stained with immature neuronal marker doublecortin (Dcx) in the SVZ of oil- (A) and E2-(B) treated animals following ischemic stroke, and illustrates that E2 significantly increases the number of BrdU+/Dcx+ newborn neurons (C). Notably, these newborn neurons have not differentiated into mature neurons at this time point (D). In addition, the proliferative action of E2 was confined to neuronal precursors and did not stimulate proliferation of astrocytes (E) or microglia (data not shown [85]).
8.2. E2 enhances rate of newborn neuron proliferation
Our study clearly demonstrates that low, physiological levels of E2 enhance neurogenesis in the SVZ following cerebral stroke, and has led to several fundamental discoveries regarding the circumstances and mechanisms of E2 actions on proliferating cells of the SVZ. First, E2 given to ovariectomized mice increases the number of proliferating cells in the SVZ after stroke injury without influencing the rate of apoptotic cell death [85]. The number of newborn neurons of the SVZ is determined by the equilibrium between proliferation and cell death. Previously, we have shown that E2 decreases the extent of ischemic injury by suppressing neuronal apoptosis in the cortex. Thus, we explored whether E2 continued to exert anti-apoptotic actions in the SVZ following stroke injury. We found that E2 does not influence the number of TUNEL-positive cells in the SVZ at 96 h after the onset of MCAO, a time point at which we observed neurogenic actions of E2 [85]. This finding indicates that an increase in the number of newborn neurons following E2 administration is not due to enhanced cell survival, but instead as a result of E2 stimulation on the expansion of neuronal populations by enhancing the rate of proliferation [85,86].
8.3. Low physiological doses of E2 are sufficient to enhance neurogenesis after ischemia but not in the absence of injury
In addition, we found that neurogenic actions of E2 is confined to the dorsal region of the SVZ, the site from which neuroblasts normally migrate to the olfactory bulb alongside the rostral migratory stream, and is absent from the ventral portion. Moreover, this low dose of E2 does not exert parallel actions in the absence of injury when uncontrolled proliferation may not be desirable. It is important to emphasize that we have discovered a dose of E2 that does not enhance neurogenesis in the SVZ in the absence of injury when uncontrolled proliferation may be undesirable. The enhanced neurogenic response following E2 treatment is desirable in the face of injury and subsequent neuronal loss, since it may enable the brain to facilitate the replacement of injured neurons and recover function. Furthermore, E2 exclusively increases the number of newborn cells that are destined to become mature neurons and does not influence gliosis (Fig. 6).
8.4. Neurogenic action of E2 requires both ERα and ERβ
We have begun to decipher some of the essential mechanistic elements that contribute to E2’s ability to influence neurogenesis in the face of a neurodegenerative stimulus. Previously we discovered that the presence of ERα, but not ERβ, is essential to attenuate neuronal cell death and the evolution of infarct volume after MCAO-induced injury [28], since knocking out ERα blocks E2’s ability to reduce the extent of infarction. On the other hand, E2 continued to exert powerful protective action against injury-induced neuronal death in ERβ-null mice.
In marked contrast, we demonstrated unequivocally that both ERα and ERβ play essential functional roles, and that the presence of both receptor forms is the prerequisite for E2 to enhance neurogenesis in the SVZ in an animal model of stroke [85]. The selective ablation of one form of ER completely prevents E2’s ability to modulate the number of immature neurons in the SVZ, an observation not expected if each receptor form plays an identical role [85]. Thus, the results of our study suggest that each receptor subtype exerts differential functional roles in modulating neurogenesis in the ischemic brain. ERα is known to exert proliferative actions in numerous tissue types including the breast, whereas ERβ plays a critical role in the organization of cortical neurons by influencing the number of cells in the SVZ during development [102]. Although precise roles for each ER form are yet to be determined, our study clearly demonstrates that the presence of both receptors is important in expansion of neuronal populations in the SVZ after ischemic injury [85].
8.5. Mechanisms of estrogen’s neurogenic action
Ischemic stroke elicits a cascade of inflammatory reactions that strongly contribute to the extent of ischemic brain injury. A hallmark of inflammatory responses induced by ischemic injury is the activation of microglia. The consequences of microglial activation can be deleterious or beneficial, depending on the state of activation as well as temporal and spatial pattern of its activation. Within hours following injury, microglia become activated and secrete a group of proteins called proinflammatory cytokines and chemokines including interleukin-6 (IL-6) that play detrimental roles in the pathophysiology of stroke. Cytokines are secreted proteins that mediate communications between immune cells and target tissues. Chemokines represent a subgroup of cytokines that facilitate the movement of microglia, monocytes and other infiltrating leukocytes. Fig. 7 shows micrographs of mouse brain sections stained with a marker for activated microglia (lectin), and demonstrates that E2 attenuates ischemia-induced microglial activation (A, B). Consistently, a cytokine multiplex proteomic array revealed that (a) ischemic injury increased the expression of a key proinflammatory cytokine IL-6 on the injured side of the brain compared to the uninjured side, and (b) E2 significantly attenuated ischemia-induced production of IL-6.
Fig. 7.

E2 attenuates ischemia-induced neuroinflammation. A, B: photomicrographs of mouse brain sections stained with a marker for activated microglia (lectin). Mice were ovariectomized and treated with oil or E2 for 1 week prior to MCAO-induced ischemic injury, and collected at 24 h after injury. C: Ischemic injury increased the expression of IL-6 (*, P < 0.0001; oil-treated mice; *, P < 0.005; E2-treated mice) on the injured (ipsilateral) side of the brain compared to the contralateral side. E2 treatment attenuated ischemia-induced production of IL-6 (#P = 0.0271) on the ipsilateral side of the ischemic brain (n = 5–6 per experimental group). All values represent mean ± SEM. Figure reprinted with permission from [84].
In addition to reported deleterious roles in the pathophysiology of stroke, IL-6 also impairs adult neurogenesis, and a non-steroidal antiinflammatory drug indomethacin restores neurogenesis by inhibiting microglial activation and IL-6 production [36,54]. Furthermore, IL-6 has been shown to promote astrocytic differentiation of neural progenitors in the SVZ [59]. Together, proinflammatory cytokines such as IL-6 may hinder the ischemic brain from undergoing repair and remodeling, by impairing generation of new neurons capable of migrating to injured regions to potentially replace damaged neurons. Consistently, we have shown that ischemic stroke significantly attenuates cell proliferation in the SVZ, and that indomethacin significantly enhances neurogenesis in the SVZ following stroke injury ([85], unpublished data). Taken together, our studies suggest that E2 increases the number of newborn neurons in the ischemic brain by attenuating ischemiainduced inflammatory responses.
8.6. E2 exerts neurorestorative actions in the ischemic brain
Under normal physiological conditions, the SVZ-derived neuroblasts migrate to the olfactory bulb where they differentiate into functional olfactory interneurons [45]. However, various neurodegenerative conditions including cerebral ischemia redirect the migration pattern of neuroblasts from their normal route towards the sites of injury including the cortex and striatum [4,31,38,66,83,89,115]. It has been well established that neuroblasts in the SVZ migrate toward the boundary zone of the ischemic lesion in rodent models of cerebral ischemia [4,31,38,115]. Once at the ischemic boundary, these cells start to exhibit multiple processes and ramification [4,38,115]. Recently, Yamashita et al. [110] have demonstrated that a proportion of the SVZ-derived neuroblasts differentiates into mature neurons and starts to express NeuN at the sites of injury, where they begin to incorporate into the existing neuronal circuit by forming synapses with neighboring cells. Thus, the SVZ is an important endogenous source of neuronal precursors that have the potential to migrate to the ischemic regions to replace damaged neurons.
However, even though the SVZ is a promising therapeutic target for neuronal replacement therapy, the numbers of newborn neurons that actually migrate to the sites of injury are believed to be insufficient for the functional recovery of the ischemic brain [4,89]. Therefore, the identification of an endogenous factor such as E2 that stimulates neurogenesis in the face of brain insults, as well as understanding the underlying mechanisms of its action may lead to the development of neuronal restorative therapies against neurodegenerative diseases and injury. Importantly, recent pharmacological therapies designed to enhance endogenous neurogenesis lead to functional recovery after stroke injury [74,103].
8.7. E2 facilitates migration of newborn neurons
Fig. 8 shows confocal fluorescent microphotographs of Dcx-positive newborn neurons on the ipsilateral hemisphere of oil- (A) vs. E2- (B) treated animals at 2 weeks after stroke injury, and demonstrates that E2 increases the number of newborn neurons in the ipsilateral cortex compared to oil-treated animals, suggesting that E2 facilitates migration of newborn neurons toward the ischemic regions. Whether E2 subsequently promotes differentiation and survival of the SVZ-derived neuroblasts at the site of ischemic injury, leading to the structural and functional recoveries of the brain is yet to be determined. Taken together, our findings lead to the conclusion that E2 protects the brain by suppressing neuronal apoptosis during the initial 24 h, and enhances neurogenesis within the first 96 h after ischemic stroke. It is clear that the timing of initiation of ET is critical. Finally, it is also clear that E2 acts, in part, by suppressing the inflammatory response. A better understanding of E2’s protective actions following stroke injury may lead to more effective therapies that can augment the ability of the SVZ to generate new neurons in the face of injury, and subsequently enhance migration, survival and maturation of newborn neurons.
Fig. 8.

E2 facilitates migration of newborn neurons. A, B: Confocal photomicrographs of Dcx+ cells migrating toward the ischemic boundary on the ipsilateral hemisphere from oil- (A) and E2- (B) treated mice at 2 weeks after the onset of MCAO-induced injury.
9. Summary
Here, we summarized recent studies that have contributed to our current understanding of the complex mechanisms that estrogens use to exert beneficial actions following stroke injury. We have shown that E2 dramatically protects the brain against stroke injury, and revealed multiple signaling pathways and key genes that mediate the effect of E2. In addition, E2 enhances production of adult-born neurons in the ischemic brain. This discovery may carry profound clinical implications, since these newborn neurons can migrate to the ischemic regions to potentially replace damaged neurons [31,41,66]. It is important to emphasize that doses of E2 that we used throughout our studies are low, physiological levels of E2, which are strikingly similar to low-basal circulating levels found in cycling mice. Low, physiological concentrations of E2 are sufficient to exert dramatic protection against stroke injury, simultaneously enhance neurogenesis only in the presence of injury, and do not exert any effects in the absence of injury when uncontrolled proliferation may be undesirable.
In light of recent clinical trials that reported negative impact of estrogen therapy and spurred reconsideration of its use around the world, it becomes even more important to continue to perform studies with the use of appropriate animal models to improve our understanding of the wide spectrum of estrogen’s protective action, and to probe the mechanisms that underlie its beneficial actions.
10. Concluding remarks
Over the past 20 years it has become clear that estrogens play critical non-reproductive functions. This family of hormones plays important and complex roles in maintaining normal brain function and in responding to injury and neurodegeneration. Our goal is to understand how estrogens protect the brain against neurodegeneration associated with cerebrovascular stroke.
Although we have primarily focused this chapter on the work performed in our laboratory over the past decade, we have attempted to put our studies in the context of the work of many other laboratories. It is our hope that investigations will continue to decipher the many seemingly contradictory actions of estrogens and the myriad of mechanisms by which they work.
Acknowledgments
This work was supported by National Institutes of Health Grant AG17164 and 02224 (P.M.W.), American Heart Association Postdoctoral Fellowship 0920086G (S.S.) and a Ruth L. Kirschstein NRSA Postdoctoral Fellowship AG 27614 (C.M.B.).
References
- [1].Acalovschi D, Wiest T, Hartmann M, Farahmi M, Mansmann U, Auffarth GU, Grau AJ, Green FR, Grond-Ginsbach C, Schwaniger M. Multiple levels of regulation of the interleukin-6 system in stroke. Stroke. 2003;34:1864–1870. doi: 10.1161/01.STR.0000079815.38626.44. [DOI] [PubMed] [Google Scholar]
- [2].Alvarez RJ, Gips SJ, Moldovan N, Wilhide CC, Milliken EE, Hoang AT, Hruban RH, Silverman HS, Dang CV, Goldschmidt-Clermont PJ. 17[beta]-Estradiol inhibits apoptosis of endothelial cells. Biochem. Biophys. Res. Commun. 1997;237:372–381. doi: 10.1006/bbrc.1997.7085. [DOI] [PubMed] [Google Scholar]
- [3].Alvarez-Buylla A, Garcia-Verdugo JM. Neurogenesis in adult subventricular zone. J. Neurosci. 2002;22:629–634. doi: 10.1523/JNEUROSCI.22-03-00629.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Arvidsson A, Collin T, Krik D, Kokaia Z, Lindvall O. Neuronal replacement from endogenous precursors in the adult brain after stroke. Nat. Med. 2002;8:963–970. doi: 10.1038/nm747. [DOI] [PubMed] [Google Scholar]
- [5].Baker AE, Brautigam VM, Watters JJ. Estrogen modulates microglial inflammatory mediator production via interactions with estrogen receptor beta. Endocrinology. 2004;145:5021–5032. doi: 10.1210/en.2004-0619. [DOI] [PubMed] [Google Scholar]
- [6].Behl C. Oestrogen as a neuroprotective hormone. Nat. Rev. Neurosci. 2002;3:433–442. doi: 10.1038/nrn846. [DOI] [PubMed] [Google Scholar]
- [7].Brailoiu E, Dun SL, Brailoiu GC, Mizuo K, Sklar LA, Oprea TI, Prossnitz ER, Dun NJ. Distribution and characterization of estrogen receptor G protein-coupled receptor 30 in the rat central nervous system. J. Endocrinol. 2007;193:311–321. doi: 10.1677/JOE-07-0017. [DOI] [PubMed] [Google Scholar]
- [8].Brown CM, Choi E, Xu Q, Vitek MP, Colton CA. The APOE4 genotype alters the response of microglia and macrophages to 17beta-estradiol. Neurobiol. Aging. 2007 doi: 10.1016/j.neurobiolaging.2007.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Brown CM, Cruz C.D. Dela, Yang E, Wise PM. Inducible nitric oxide synthase and estradiol exhibit complementary neuroprotective roles after ischemic brain injury. Exp. Neurol. 2008;210:782–787. doi: 10.1016/j.expneurol.2007.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Bruce-Keller AJ, Keeling JL, Keller JN, Huang FF, Camondola S, Mattson MP. Antiinflammatory effects of estrogen on microglial activation. Endocrinology. 2000;141:3646–3656. doi: 10.1210/endo.141.10.7693. [DOI] [PubMed] [Google Scholar]
- [11].Bryant D, Shedahl L, Marriott L, Shapiro R, Dorsa D. Multiple pathways transmit neuroprotective effects of gonadal steroids. Endocrine. 2006;29:199–207. doi: 10.1385/ENDO:29:2:199. [DOI] [PubMed] [Google Scholar]
- [12].Bushnell CD. Hormone replacement therapy and stroke: the current state of knowledge and directions for future research. Semin. Neurol. 2006;26:123–130. doi: 10.1055/s-2006-933316. [DOI] [PubMed] [Google Scholar]
- [13].Bushnell CD, Hurn P, Colton C, Miller VM, del Zoppo G, Elkind MSV, Stern B, Herrington D, Ford-Lynch G, Gorelick P, James A, Brown CM, Choi E, Bray P, Newby LK, Goldstein LB, Simpkins J. Advancing the Study of Stroke in women: summary and recommendations for future research from an NINDS-sponsored multidisciplinary working group. Stroke. 2006;37:2387–2399. doi: 10.1161/01.STR.0000236053.37695.15. [DOI] [PubMed] [Google Scholar]
- [14].Butts CL, Sternberg EM. Neuroendocrine factors alter host defense by modulating immune function. Cell Immunol. 2008;252:7–15. doi: 10.1016/j.cellimm.2007.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Chambliss KL, Yuhanna IS, Anderson RGW, Mendelsohn ME, Shaul PW. ER{beta} has nongenomic action in caveolae. Mol. Endocrinol. 2002;16:938–946. doi: 10.1210/mend.16.5.0827. [DOI] [PubMed] [Google Scholar]
- [16].Cowell RM, Xu H, Galasso JM, Silverstein FS. Hypoxic–ischemic injury induces macrophage inflammatory protein-1{alpha} expression in immature rat brain. Stroke. 2002;33:795–801. doi: 10.1161/hs0302.103740. [DOI] [PubMed] [Google Scholar]
- [17].Czlonkowska A, Ciesielska A, Gromadzka G, Kurkowska-Jastrzebska I. Estrogen and cytokines production – the possible cause of gender differences in neurological diseases. Curr. Pharm. Des. 2005;11:1017–1030. doi: 10.2174/1381612053381693. [DOI] [PubMed] [Google Scholar]
- [18].Czlonkowska A, Ciesielska A, Gromadzka G, Kurkowska-Jastrzebska I. Gender differences in neurological disease. Endocrine. 2006;29:243–256. doi: 10.1385/ENDO:29:2:243. [DOI] [PubMed] [Google Scholar]
- [19].Dan P, Cheung J, Scriven D, Moore E. Epitope-dependent localization of estrogen receptor-alpha, but not -beta, in en face arterial endothelium. Am. J. Physiol. Heart Circ. Physiol. 2003;284:H1295–306. doi: 10.1152/ajpheart.00781.2002. [DOI] [PubMed] [Google Scholar]
- [20].Danton GH, Dietrich WD. Inflammatory mechanisms after ischemia and stroke. J. Neuropathol. Exp. Neurol. 2003;62:127–136. doi: 10.1093/jnen/62.2.127. [DOI] [PubMed] [Google Scholar]
- [21].del Zoppo G, Ginis I, Hallenbeck JM, Iadecola C, Wang X, Feurstein GZ. Inflammation and stroke: putative role for cytokines, adhesion molecules and iNOS in brain response to ischemia. Brain Pathol. 2000;10:95–112. doi: 10.1111/j.1750-3639.2000.tb00247.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].del Zoppo GJ. Stroke and neurovascular protection. N. Engl. J. Med. 2006;354:553–555. doi: 10.1056/NEJMp058312. [DOI] [PubMed] [Google Scholar]
- [23].Dhandapani KM, Brann DW. Role of astrocytes in estrogen-mediated neuroprotection. Exp. Gerontol. 2007;42:70–75. doi: 10.1016/j.exger.2006.06.032. [DOI] [PubMed] [Google Scholar]
- [24].Dodel R, Du Y, Bales KR, Gao F, Paul S. Sodium salicylate and 17beta-estradiol attenuate nuclear transcription factor NF-κB translocation in cultured rat astroglial cultures following exposure to amyloid beta 1–40 and lipopolysaccharide. J. Neurochem. 1999;73:1453–1460. doi: 10.1046/j.1471-4159.1999.0731453.x. [DOI] [PubMed] [Google Scholar]
- [25].Drew PD, Chavis JA. Female sex steroids: effects upon microglial cell activation. J. Neuroimmunol. 2000;111:77–85. doi: 10.1016/s0165-5728(00)00386-6. [DOI] [PubMed] [Google Scholar]
- [26].Dubal DB, Kashon M, Pettigrew L, Ren J, Finklestein S, Rau SW, Wise PM. Estradiol protects against ischemic injury. J. Cereb. Blood Flow Metab. 1998;18:1253–1258. doi: 10.1097/00004647-199811000-00012. [DOI] [PubMed] [Google Scholar]
- [27].Dubal DB, Shughrue PJ, Wilson ME, Merchenthaler I, Wise PM. Estradiol modulates bcl-2 in cerebral ischemia: a potential role for estrogen receptors. J. Neurosci. 1999;19:6385–6393. doi: 10.1523/JNEUROSCI.19-15-06385.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Dubal DB, Zhu H, Yu J, Rau SW, Shughrue PJ, Merchenthaler I, Kindy MS, Wise PM. Estrogen receptor alpha, not beta is a critical link in estradiol-mediated protection against brain injury. Proc. Natl. Acad. Sci. USA. 2001;98:1952–1957. doi: 10.1073/pnas.041483198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Duckles SP, Krause DN. Cerebrovascular effects of oestrogen: multiplicity of action. Clin. Exp. Pharm. Physiol. 2007;34:801–808. doi: 10.1111/j.1440-1681.2007.04683.x. [DOI] [PubMed] [Google Scholar]
- [30].Fassbender K, Rossol S, Kammer T, Daffertshofer M, Wirth S, Dollman M, Hennerici M. Proinflammatory cytokines in serum of patients with acute cerebral ischemia: kinetics of secretion and relation to the extent of brain damage and outcome of disease. J. Neurol. Sci. 1994;122:135–139. doi: 10.1016/0022-510x(94)90289-5. [DOI] [PubMed] [Google Scholar]
- [31].Goings GE, Sahni V, Szele FG. Migration patterns of subventricular zone cells in adult mice change after cerebral cortex injury. Brain Res. 2004;996:213–226. doi: 10.1016/j.brainres.2003.10.034. [DOI] [PubMed] [Google Scholar]
- [32].Guo S, Lo EH. Dysfunctional cell–cell signaling in the neurovascular unit as a paradigm for central nervous system disease. Stroke. 2008 doi: 10.1161/STROKEAHA.108.534388. STROKEAHA.108.534388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Harman SM, Naftolin F, Brinton EA, Judelson DR. Is the estrogen controversy over? Deconstructing the women’s health initiative study: a critical evaluation of the evidence. Ann. NY Acad. Sci. 2005;1052:43–56. doi: 10.1196/annals.1347.004. [DOI] [PubMed] [Google Scholar]
- [34].Iadecola C, Zhang F, Casey R, Nagayama M, Ross ME. Delayed reduction of ischemic brain injury and neurological deficits in mice lacking the inducible nitric oxide synthase gene. J. Neurosci. 1997;17:9157–9164. doi: 10.1523/JNEUROSCI.17-23-09157.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Iadecola C, Zhang F, Xu S, Casey R, Ross ME. Inducible nitric oxide synthase gene expression in brain following cerebral ischemia. J. Cereb. Blood Flow Metab. 1995;15:378–384. doi: 10.1038/jcbfm.1995.47. [DOI] [PubMed] [Google Scholar]
- [36].Iosif RE, Ekdahl CT, Ahlenius H, Pronk CJH, Bonde S, Kokaia Z, Jacobsen S-EW, Lindvall O. Tumor necrosis factor receptor 1 is a negative regulator of progenitor proliferation in adult hippocampal neurogenesis. J. Neurosci. 2006;26:9703–9712. doi: 10.1523/JNEUROSCI.2723-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Jelks KB, Wylie R, Floyd CL, McAllister AK, Wise P. Estradiol targets synaptic proteins to induce glutamatergic synapse formation in cultured hippocampal neurons: critical role of estrogen receptor-alpha. J. Neurosci. 2007;27:6903–6913. doi: 10.1523/JNEUROSCI.0909-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Jin K, Sun Y, Xie L, Peel A, Mao XO, Batteur S, Greenberg DA. Directed migration of neuronal precursors into the ischemic cerebral cortex and striatum. Mol. Cell Neurosci. 2003;24:171–189. doi: 10.1016/s1044-7431(03)00159-3. [DOI] [PubMed] [Google Scholar]
- [39].Johnson AB, Sohrabji F. Estrogen’s effects on central and circulating immune cells vary with reproductive age. Neurobiol. Aging. 2005;26:1365–1374. doi: 10.1016/j.neurobiolaging.2004.12.006. [DOI] [PubMed] [Google Scholar]
- [40].Kipp M, Karakaya S, Johann S, Kampmann E, Mey J, Beyer C. Oestrogen and progesterone reduce lipopolysaccharide-induced expression of tumour necrosis factor-alpha and interleukin-18 in midbrain astrocytes. J. Neuroendocrinol. 2007;19:819–822. doi: 10.1111/j.1365-2826.2007.01588.x. [DOI] [PubMed] [Google Scholar]
- [41].Komitova M, Perfilieva E, Mattsson B, Eriksson PS, Johansson BB. Effects of cortical ischemia and postischemic environmental enrichment on hippocampal cell genesis and differentiation in the adult rat. J. Cereb. Blood Flow Metab. 2002;22:852–860. doi: 10.1097/00004647-200207000-00010. [DOI] [PubMed] [Google Scholar]
- [42].Lee SJ, McEwen BS. Neurotrophic and neuroprotective actions of estrogens and their therapeutic implications. Annu. Rev. Pharmacol. Toxicol. 2001;41:569–591. doi: 10.1146/annurev.pharmtox.41.1.569. [DOI] [PubMed] [Google Scholar]
- [43].Lewis DK, Johnson AB, Stohlgren S, Harms A, Sohrabji F. Effects of estrogen receptor agonists on regulation of the inflammatory response in astrocytes from young adult and middle-aged female rats. J. Neuroimmunol. 2008;195:47–59. doi: 10.1016/j.jneuroim.2008.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Loihl AK, Asensio V, Campbell IL, Murphy S. Expression of nitric oxide synthase (NOS)-2 following permanent focual ischemia and the role of nitric oxide in infarct generation in male, female, and NOS-2 gene-deficient mice. Brain Res. 1999;830:155–164. doi: 10.1016/s0006-8993(99)01388-8. [DOI] [PubMed] [Google Scholar]
- [45].Lois C, Alvarez-Buylla A. Long-distance neuronal migration in the adult mammalian brain. Science. 1994;264:1145–1148. doi: 10.1126/science.8178174. [DOI] [PubMed] [Google Scholar]
- [46].Luoma JI, Boulware MI, Mermelstein PG. Caveolin proteins and estrogen signaling in the brain. Mol. Cell Endocrinol. 2008;290:8–13. doi: 10.1016/j.mce.2008.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Macrae IM, Carswell HV. Oestrogen and stroke: the potential for harm as well as benefit. Biochem. Soc. Trans. 2006;34:1362–1365. doi: 10.1042/BST0341362. [DOI] [PubMed] [Google Scholar]
- [48].Maurer M, von Stebut E. Macrophage inflammatory protein-1. Int. J. Biochem. Cell Biol. 2004;36:1882–1886. doi: 10.1016/j.biocel.2003.10.019. [DOI] [PubMed] [Google Scholar]
- [49].McCullough LD, Hurn PD. Estrogen and ischemic neuroprotection: an integrated view. Trends Endocrinol. Metab. 2003;14:228–235. doi: 10.1016/s1043-2760(03)00076-6. [DOI] [PubMed] [Google Scholar]
- [50].Mergenthaler P, Dirnagl U, Meisel A. Pathophysiology of stroke: lessons from animal models. Metab. Brain Dis. 2004;19:151–167. doi: 10.1023/b:mebr.0000043966.46964.e6. [DOI] [PubMed] [Google Scholar]
- [51].Miller VM, Clarkson TB, Harman SM, Brinton EA, Cedars M, Lobo R, Manson JE, Merriam GR, Naftolin F, Santoro N. Women, hormones, and clinical trials: a beginning, not an end. J. Appl. Physiol. 2005;99:381–383. doi: 10.1152/japplphysiol.00248.2005. [DOI] [PubMed] [Google Scholar]
- [52].Miller VM, Duckles SP. Vascular actions of estrogens: functional implications. Pharmacol. Rev. 2008;60:210–241. doi: 10.1124/pr.107.08002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Mitka M. Studies explore stroke’s gender gap. JAMA. 2006;295:1755–1756. doi: 10.1001/jama.295.15.1755. [DOI] [PubMed] [Google Scholar]
- [54].Monje ML, Toda H, Palmer TD. Inflammatory blockade restores adult hippocampal neurogenesis. Science. 2003;302:1760–1765. doi: 10.1126/science.1088417. [DOI] [PubMed] [Google Scholar]
- [55].Mor G, Nilsen J, Horvath T, Bechmann I, Brown S, Garcia-Segura LM, Naftolin F. Estrogen and microglia: a regulatory system that affects the brain. J. Neurobiol. 1999;40:484–496. doi: 10.1002/(sici)1097-4695(19990915)40:4<484::aid-neu6>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- [56].Morissette M, Le Saux M, D’Astous M, Jourdain S, Al Sweidi S, Morin N, Estrada-Camarena E, Mendez P, Garcia-Segura LM, Di Paolo T. Contribution of estrogen receptors alpha and beta to the effects of estradiol in the brain. J. Steroid Biochem. Mol. Biol. 2008;108:327–338. doi: 10.1016/j.jsbmb.2007.09.011. [DOI] [PubMed] [Google Scholar]
- [57].Murphy S. Production of nitric oxide by glial cells: regulation and potential roles in the CNS. Glia. 2000;29:1–13. doi: 10.1002/(sici)1098-1136(20000101)29:1<1::aid-glia1>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- [58].Nagayama M, Aber T, Nagayama T, Ross ME, Iadecola C. Age-dependent increase in ischemic brain injury in wild-type mice and in mice lacking the inducible nitric oxide synthase gene. J. Cereb. Blood Flow Metab. 1999;19:661–666. doi: 10.1097/00004647-199906000-00009. [DOI] [PubMed] [Google Scholar]
- [59].Nakanishi M, Niidome T, Matsuda S, Akaike A, Kihara T, Sugimoto H. Microglia-derived interleukin-6 and leukaemia inhibitory factor promote astrocytic differentiation of neural stem/progenitor cells. Eur. J. Neurosci. 2007;25:649–658. doi: 10.1111/j.1460-9568.2007.05309.x. [DOI] [PubMed] [Google Scholar]
- [60].Nordell VL, Scarborough MM, Buchanan AK, Sohrabji F. Differential effects of estrogen in the injured forebrain of young adult and reproductive senescent animals. Neurobiol. Aging. 2003;24:733–743. doi: 10.1016/s0197-4580(02)00193-8. [DOI] [PubMed] [Google Scholar]
- [61].Offner H, Subramanian S, Parker SM, Afentoulis ME, Vandenbark AA, Hurn PD. Experimental stroke induces massive, rapid activation of the peripheral immune system. J. Cereb. Blood Flow Metab. 2005;26:654–665. doi: 10.1038/sj.jcbfm.9600217. [DOI] [PubMed] [Google Scholar]
- [62].Paganini-Hill A. Hormone replacement therapy and stroke: risk, protection or no effect? Maturitas. 2001;38:243–261. doi: 10.1016/s0378-5122(01)00167-0. [DOI] [PubMed] [Google Scholar]
- [63].Park E-M, Cho S, Frys KA, Glickstein SB, Zhou P, Anrather J, Ross ME, Iadecola C. Inducible nitric oxide synthase contributes to gender differences in ischemic brain injury. J. Cereb. Blood Flow Metab. 2006;26:392–401. doi: 10.1038/sj.jcbfm.9600194. [DOI] [PubMed] [Google Scholar]
- [64].Perini F, Morra M, Alecci M, Galloni E, Marchi M, Toso V. Temporal profile of serum anti-inflammatory and pro-inflammatory interleukins in acute ischemic stroke patients. Neurol. Sci. 2001;22:289–296. doi: 10.1007/s10072-001-8170-y. [DOI] [PubMed] [Google Scholar]
- [65].Prewitt AK, Wilson ME. Changes in estrogen receptor-alpha mRNA in the mouse cortex during development. Brain Res. 2007;1134:62–69. doi: 10.1016/j.brainres.2006.11.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Ramaswamy S, Goings GE, Soderstrom KE, Szele FG, Kozlowski DA. Cellular proliferation and migration following a controlled cortical impact in the mouse. Brain Res. 2005;1053:38–53. doi: 10.1016/j.brainres.2005.06.042. [DOI] [PubMed] [Google Scholar]
- [67].Rau S, Dubal D, Bottner M, Wise P. Estradiol differentially regulates c-Fos after focal cerebral ischemia. J. Neurosci. 2003;23:10487–10494. doi: 10.1523/JNEUROSCI.23-33-10487.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Rau SW, Dubal DB, Bottner M, Gerhold LM, Wise PM. Estradiol attenuates programmed cell death after stroke-like injury. J. Neurosci. 2003;23:11420–11426. doi: 10.1523/JNEUROSCI.23-36-11420.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Raz L, Khan MM, Mahesh VB, Vadlamudi RK, Brann DW. Rapid estrogen signaling in the brain. Neurosignals. 2008;16:140–153. doi: 10.1159/000111559. [DOI] [PubMed] [Google Scholar]
- [70].Rodriguez-Yanez M, Castillo J. Role of inflammatory markers in brain ischemia. Curr. Opin. Neurol. 2008;21:353–357. doi: 10.1097/WCO.0b013e3282ffafbf. [DOI] [PubMed] [Google Scholar]
- [71].Rossouw JE, Anderson GL, Prentice RL, LaCroix AZ, Kooperberg C, Stefanick ML, Jackson RD, Beresford SA, Howard BV, Johnson KC, Kotchen JM, Ockene J. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results from the women’s health initiative randomized controlled trial. JAMA. 2002;288:321–333. doi: 10.1001/jama.288.3.321. [DOI] [PubMed] [Google Scholar]
- [72].Salem ML. Estrogen, a double-edged sword: modulation of the TH1- and TH2-mediated inflammations by differential regulation of TH1/TH2 cytokine production. Curr. Drug Targets – Inflamm. Allergy. 2004;3:97–104. doi: 10.2174/1568010043483944. [DOI] [PubMed] [Google Scholar]
- [73].Shughrue P, Stumpf W, Maclusky N, Zielinski N, Hochberg R. Developmental changes in estrogen receptors in mouse cerebral cortex between birth and postweaning: studied by autoradiography with 11 beta-methoxy-16 alpha-[125I]iodoestradiol. Endocrinology. 1990;126:1112–1124. doi: 10.1210/endo-126-2-1112. [DOI] [PubMed] [Google Scholar]
- [74].Shyu W-C, Lin S-Z, Yang H-I, Tzeng Y-S, Pang C-Y, Yen P-S, Li H. Functional recovery of stroke rats induced by granulocyte colony-stimulating factor-stimulated stem cells. Circulation. 2004;110:1847–1854. doi: 10.1161/01.CIR.0000142616.07367.66. [DOI] [PubMed] [Google Scholar]
- [75].Simpkins JW, Yang S-H, Sarkar SN, Pearce V. Estrogen actions on mitochondria–physiological and pathological implications. Mol. Cell Endocrinol. 2008;290:51–59. doi: 10.1016/j.mce.2008.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Sohrabji F. Guarding the blood–brain barrier: a role for estrogen in the etiology of neurodegenerative disease. Gene Expr. 2007;13:311–319. doi: 10.3727/000000006781510723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Sohrabji F, Bake S. Age-related changes in neuroprotection: is estrogen pro-inflammatory for the reproductive senescent brain? Endocrine. 2006;29:191–197. doi: 10.1385/ENDO:29:2:191. [DOI] [PubMed] [Google Scholar]
- [78].Solum DT, Handa RJ. Localization of estrogen receptor alpha (ER[alpha]) in pyramidal neurons of the developing rat hippocampus. Dev. Brain Res. 2001;128:165–175. doi: 10.1016/s0165-3806(01)00171-7. [DOI] [PubMed] [Google Scholar]
- [79].Stirone C, Boroujerdi A, Duckles SP, Krause DN. Estrogen receptor activation of phosphoinositide-3 kinase, akt, and nitric oxide signaling in cerebral blood vessels: rapid and long-term effects. Mol. Pharmacol. 2005;67:105–113. doi: 10.1124/mol.104.004465. [DOI] [PubMed] [Google Scholar]
- [80].Stirone C, Duckles SP, Krause DN. Multiple forms of estrogen receptor-alpha in cerebral blood vessels: regulation by estrogen. Am. J. Physiol. Endocrinol. Metab. 2003;284:E184–192. doi: 10.1152/ajpendo.00165.2002. [DOI] [PubMed] [Google Scholar]
- [81].Stirone C, Duckles SP, Krause DN, Procaccio V. Estrogen increases mitochondrial efficiency and reduces oxidative stress in cerebral blood vessels. Mol. Pharmacol. 2005;68:959–965. doi: 10.1124/mol.105.014662. [DOI] [PubMed] [Google Scholar]
- [82].Sugimoto K, Iadecola C. Effects of aminoguanidine on cerebral ischemia in mice: comparison between mice with and without inducible nitric oxide synthase gene. Neurosci. Lett. 2002;331:25–28. doi: 10.1016/s0304-3940(02)00834-0. [DOI] [PubMed] [Google Scholar]
- [83].Sundholm-Peters NL, Yang HK, Goings GE, Walker AS, Szele FG. Subventricular zone neuroblasts emigrate toward cortical lesions. J. Neuropathol. Exp. Neurol. 2005;64:1089–1100. doi: 10.1097/01.jnen.0000190066.13312.8f. [DOI] [PubMed] [Google Scholar]
- [84].Suzuki S, Brown CM, Dela Cruz CD, Yang E, Bridwell DA, Wise PM. Timing of estrogen therapy after ovariectomy dictates the efficacy of its neuroprotective and antiinflammatory actions. Proc. Natl. Acad. Sci. USA. 2007;104:6013–6018. doi: 10.1073/pnas.0610394104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Suzuki S, Gerhold LM, Bottner M, Rau SW, Cruz C. Dela, Yang E, Zhu H, Yu J, Cashion AB, Kindy MS, Merchenthaler I, Gage FH, Wise PM. Estradiol enhances neurogenesis following ischemic stroke through estrogen receptors alpha and beta. J. Comput. Neurol. 2007;500:1064–1075. doi: 10.1002/cne.21240. [DOI] [PubMed] [Google Scholar]
- [86].Smith MT, Pencea V, Wang Z, Luskin MB, Insel TR. Increased number of BrdU-labeled neurons in the rostral migratory stream of the estrous prairie vole. Horm. Behav. 2001;39:11–21. doi: 10.1006/hbeh.2000.1630. [DOI] [PubMed] [Google Scholar]
- [87].Tapia-Gonzalez S, Carrero P, Pernia O, Garcia-Segura LM, Diz-Chaves Y. Selective oestrogen receptor (ER) modulators reduce microglia reactivity in vivo after peripheral inflammation: potential role of microglial ERs. J. Endocrinol. 2008;198:219–230. doi: 10.1677/JOE-07-0294. [DOI] [PubMed] [Google Scholar]
- [88].Taupin P, Gage FH. Adult neurogenesis and neural stem cells of the central nervous system in mammals. J. Neurosci. Res. 2002;69:745–749. doi: 10.1002/jnr.10378. [DOI] [PubMed] [Google Scholar]
- [89].Tonchev AB, Yamashima T, Sawamoto K, Okano H. Enhanced proliferation of progenitor cells in the subventricular zone and limited neuronal production in the striatum and neocortex of adult macaque monkeys after global cerebral ischemia. J. Neurosci. Res. 2005;81:776–788. doi: 10.1002/jnr.20604. [DOI] [PubMed] [Google Scholar]
- [90].Toran-Allerand CD. Minireview: a plethora of estrogen receptors in the brain: where will it end? Endocrinology. 2004;145:1069–1074. doi: 10.1210/en.2003-1462. [DOI] [PubMed] [Google Scholar]
- [91].Toung TJK, Traystman RJ, Hurn PD. Estrogen-mediated neuroprotection after experimental stroke in male rats. Stroke. 1998;29:1666–1670. doi: 10.1161/01.str.29.8.1666. [DOI] [PubMed] [Google Scholar]
- [92].Turgeon JL, Carr MC, Maki PM, Mendelsohn ME, Wise PM. Complex actions of sex steroids in adipose tissue, the cardiovascular system, and brain: insights from basic science and clinical studies. Endocrinol. Rev. 2006;27:575–605. doi: 10.1210/er.2005-0020. [DOI] [PubMed] [Google Scholar]
- [93].Turgeon JL, McDonnell DP, Martin KA, Wise PM. Hormone therapy: physiological complexity belies therapeutic simplicity. Science. 2004;304:1269–1273. doi: 10.1126/science.1096725. [DOI] [PubMed] [Google Scholar]
- [94].Vegeto E, Belcredito S, Etteri S, Ghisletti S, Brusadelli A, Meda C, Krust A, Dupont S, Ciana P, Chambon P, Maggi A. Estrogen receptor-a mediates the brain antiinflammatory activity of estradiol. Proc. Natl. Acad. Sci. USA. 2003;100:9614–9619. doi: 10.1073/pnas.1531957100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Vegeto E, Belcredito S, Ghisletti S, Meda C, Etteri S, Maggi A. The endogenous estrogen status regulates microglia reactivity in animal models of neuroinflammation. Endocrinology. 2006;147:2263–2272. doi: 10.1210/en.2005-1330. [DOI] [PubMed] [Google Scholar]
- [96].Vegeto E, Benedusi V, Maggi A. Estrogen anti-inflammatory activity in brain: a therapeutic opportunity for menopause and neurodegenerative diseases. Front. Neuroendocrinol. 2008;29:507–519. doi: 10.1016/j.yfrne.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Vegeto E, Bonincontro C, Pollio G, Sala A, Viappiani S, Nardi F, Brusadelli A, Vivani B, Ciana P, Maggi A. Estrogen prevents the lipopolysaccharide-induced inflammatory response in microglia. J. Neurosci. 2001;21:1809–1818. doi: 10.1523/JNEUROSCI.21-06-01809.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Vegeto E, Ghisletti S, Meda C, Etteri S, Belcredito S, Maggi A. Regulation of the lipopolysaccharide signal transduction pathway by 17[beta]-estradiol in macrophage cells. J. Steroid Biochem. Mol. Biol. 2004;91:59–66. doi: 10.1016/j.jsbmb.2004.02.004. [DOI] [PubMed] [Google Scholar]
- [99].Viscoli CM, Brass LM, Kernan WN, Sarrel PM, Suissa S, Horwitz RI. A clinical trial of estrogen-replacement therapy after ischemic stroke. N. Engl. J. Med. 2001;345:1243–1249. doi: 10.1056/NEJMoa010534. [DOI] [PubMed] [Google Scholar]
- [100].Viviani B, Corsini E, Binaglia M, Lucchi L, Galli CL, Marinovich M. The anti-inflammatory activity of estrogen in glial cells is regulated by the PKC-anchoring protein RACK-1. J. Neurochem. 2002;83:1180–1187. doi: 10.1046/j.1471-4159.2002.01235.x. [DOI] [PubMed] [Google Scholar]
- [101].Wang HK, Park UJ, Kim SY, Lee JH, Kim SU, Gwag BJ, Lee YB. Free radical production in CA1 neurons induces MIP-1{alpha} expression, microglia recruitment, and delayed neuronal death after transient forebrain ischemia. J. Neurosci. 2008;28:1721–1727. doi: 10.1523/JNEUROSCI.4973-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Wang L, Andersson S, Warner M, Gustafsson J. Estrogen receptor (ER) beta knockout mice reveal a role for ER beta in migration of cortical neurons in the developing brain. Proc. Natl. Acad. Sci. USA. 2003;100:703–708. doi: 10.1073/pnas.242735799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Wang L, Zhang Z, Wang Y, Zhang R, Chopp M. Treatment of stroke with erythropoietin enhances neurogenesis and angiogenesis and improves neurological function in rats. Stroke. 2004;35:1732–1737. doi: 10.1161/01.STR.0000132196.49028.a4. [DOI] [PubMed] [Google Scholar]
- [104].Wassertheil-Smoller S, Hendrix SL, Limacher M, Heiss G, Kooperberg C, Baird A, Kotchen T, Curb JD, Black H, Rossuw JE, Aragaki A, Safford M, Stein E, Laowattana S, Mysiw WJ. Effect of estrogen plus progestin on stroke in postmenopausal women. JAMA. 2003;289:2673–2684. doi: 10.1001/jama.289.20.2673. [DOI] [PubMed] [Google Scholar]
- [105].Welsh P, Lowe GDO, Chalmers J, Campbell DJ, Rumley A, Neal BC, MacMahon SW, Woodward M. Associations of proinflammatory cytokines with the risk of recurrent stroke. Stroke. 2008;39:2226–2230. doi: 10.1161/STROKEAHA.107.504498. [DOI] [PubMed] [Google Scholar]
- [106].Wilson ME, Dubal DB, Wise PM. Estradiol protects against injury-induced cell death in cortical explant cultures: a role for estrogen receptors. Brain Res. 2000;873:235–242. doi: 10.1016/s0006-8993(00)02479-3. [DOI] [PubMed] [Google Scholar]
- [107].Wilson ME, Liu Y, Wise PM. Estradiol enhances Akt activation in cortical explant cultures following neuronal injury. Mol. Brain Res. 2002;102:48–54. doi: 10.1016/s0169-328x(02)00181-x. [DOI] [PubMed] [Google Scholar]
- [108].Wise PM, Dubal DB, Rau SW, Brown CM, Suzuki S. Are estrogens protective or risk factors in brain injury and neurodegeneration? Reevaluation after the women’s health initiative. Endocrinol. Rev. 2005;26:308–312. doi: 10.1210/er.2004-0014. [DOI] [PubMed] [Google Scholar]
- [109].Wise PM, Dubal DB, Wilson ME, Rau SW, Liu Y. Estrogens: trophic and protective factors in the adult brain. Front. Neuroendocrinol. 2001;22:33–66. doi: 10.1006/frne.2000.0207. [DOI] [PubMed] [Google Scholar]
- [110].Yamashita T, Ninomiya M, Hernandez Acosta P, Garcia-Verdugo JM, Sunabori T, Sakaguchi M, Adachi K, Kojima T, Hirota Y, Kawase T, Araki N, Abe K, Okano H, Sawamoto K. Subventricular zone-derived neuroblasts migrate and differentiate into mature neurons in the post-stroke adult striatum. J. Neurosci. 2006;26:6627–6636. doi: 10.1523/JNEUROSCI.0149-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Yang S-H, Liu R, Wu SS, Simpkins JW. The use of estrogens and related compounds in the treatment of damage from cerebral ischemia. Ann. NY Acad. Sci. 2003;1007:101–107. doi: 10.1196/annals.1286.010. [DOI] [PubMed] [Google Scholar]
- [112].Yang S-H, Ran L, Perez EJ, Xiaofei W, Simpkins JW. Estrogens as protectants of the neurovascular unit against ischemic stroke. Curr. Drug. Targets – CNS Neurol. Disord. 2005;4:169–177. doi: 10.2174/1568007053544174. [DOI] [PubMed] [Google Scholar]
- [113].Yang S-H, Shi J, Day AL, Simpkins JW, Robinson SE. Estradiol exerts neuroprotective effects when administered after ischemic insult. Stroke. 2000;31:745–750. doi: 10.1161/01.str.31.3.745. [DOI] [PubMed] [Google Scholar]
- [114].Zaremba J, Ilkowski J, Losy J. Serial measurements of the chemokines CCL2, CCL3, and CCL5 in serum of patients with acute ischaemic stroke. Folia Neuropathol. 2006;44:282–289. [PubMed] [Google Scholar]
- [115].Zhang RL, Zhang Z, Wang L, Wang Y, Gousev A, Zhang L, Ho K, Morshead C, Chopp M. Activated neural stem cells contribute to stroke-induced neurogenesis and neuroblast migration toward the infarct boundary in adult rats. J. Cereb. Blood Flow Metab. 2004;24:441–448. doi: 10.1097/00004647-200404000-00009. [DOI] [PubMed] [Google Scholar]