

Abstract
In response to iron deplete aerobic conditions, bacteria often secrete low molecular weight, high-affinity iron(III)-complexing ligands, siderophores, to solubilize and sequester iron(III). Many marine siderophores are amphiphilic and are produced in suites, wherein each member within a particular suite has the same iron(III)-binding polar head group which is appended by one or two fatty acids of varying length, degree of unsaturation and hydroxylation, establishing the suite composition. We report herein the isolation and structural characterization of a suite of siderophores from marine bacterial isolate Vibrio sp. Nt1. Based on structural analysis, this suite of siderophores, the moanachelins, is amphiphilic and composed of two N-acetyl, N-hydroxy D-ornithines, one N-acetyl, N-hydroxy L-ornithine and either a glycine or an L-alanine, appended with various saturated and unsaturated fatty acid tails. The variation in the small side-chain amino acid is the first occurrence of variation in the peptidic head group structure of a set of siderophores produced by a single bacterium.
Keywords: Marine siderophores, Amphiphilic siderophores, peptide-based, amphibactins, moanachelins
Introduction
Siderophores are an important group of high affinity iron-coordinating molecules which are synthesized by bacteria under iron-limited conditions to facilitate the uptake of iron(III). Siderophores are composed of a variety of different building blocks, including, for example, amino acids, diamines, citric acid, succinic acid and dihydroxybenzoic acid. Siderophores coordinate iron(III) through hydroxamic acid, catechol or α-hydroxy carboxylic acid ligation, with exceptional affinity. A bacterium can produce either a single siderophore, such as desferrioxamine G produced by Vibrio sp. BLI-41 [1], multiple siderophores such as petrobactin and bacillibactin both produced by various Bacillus species [2], or an entire suite of structurally related amphiphilic siderophores, where the difference lies in the nature of the fatty acid appendage [3–9].
A particularly interesting class of siderophores is comprised of suites of amphiphilic peptide-based siderophores. This class of siderophores is produced by bacteria found in both terrestrial (e.g., mycobactins, serobactins, etc.) and marine (e.g., aquachelins, marinobactins, amphibactins, etc.) environments (Figure 1). Amphiphilic peptide-based siderophores have been isolated from pure bacterial cultures, and have been identified as products from nutrient enriched seawater incubations [10, 11]. Specifically, siderophore-like chelates with the same exact mass, as well as the appropriate mass fragmentation pattern of various amphibactin siderophores were identified [11]. While several of the amphibactins identified in seawater incubation experiments (m/z 832 and 858) were previously characterized [6], the two uncharacterized amphibactins in the previous work (m/z 804 and 832) [11] have now also been isolated and characterized [9].
Figure 1.

Select peptide-based siderophore suites. Terrestrial: mycobactins [8, 12, 13] and serobactins [14]. Marine: aquachelins [7, 9], marinobactins [7], and amphibactins [6, 9].
The biosynthetic pathways of peptidic marine siderophores have not been investigated, however the mycobactins, which are also produced as a suite of siderophores with varying fatty acid tails, are produced by a nonribosomal peptide synthetase (NRPS)-dependent biosynthetic pathway [8, 15, 16]. The NRPS adenylation domain (A-domain) plays an important role in amino acid substrate recognition and determining the order and identity of the amino acids incorporated into the peptide product. The selectivity of the A-domain is conferred by an ~10 amino acid sequence binding site [17–20]. In general, there is strict amino acid substrate specificity observed for NRPS A-domains. However, promiscuity in this domain in terms of the identity of the adenylated amino acid, with downstream acceptance, can lead to the production of a set of closely related molecules [21–23].
The NRPS-dependent biosynthetic pathways of a number of siderophores have been well studied, including enterobactin [24–26], pyochelin [27], yersiniabactin [28], vibriobactin [29], mycobactins [15], and pyoverdine [30, 31]. The amphiphilic peptidic marine siderophores are likely also assembled by NRPS multienzyme systems due to the inclusion of unique features such as D-amino acids, N-terminally attached fatty acid chains, and heterocyclic elements [23, 32]. Recent investigations of marine metagenomes indicate marine prokaryotes may use NRPSs in the biosynthesis of some siderophores [33].
We report herein the isolation and structural characterization of the moanachelins, a new suite of amphiphilic siderophores produced by Vibrio sp. Nt1 isolated from oligotrophic open ocean water. The moanachelins are the first suite of siderophores which exhibit variation not only in their fatty acid appendages, but also in the amino acid composition of the peptidic head group structure.
Materials and Methods
Bacterial strain
Vibrio sp. Nt1 was isolated from an oligotrophic surface seawater sample collected on July 15, 2009 at 20.30020 °N, −153.42077 °W while aboard the R/V Kilo Moana northeast of Hawaii [GenBank ID: JQ996144]. Bacterial isolation was performed as previously described [9]. The bacterium was identified by sequencing of the 16S small subunit rRNA gene as described in Gauglitz et al. [3]. Vibrio sp. HC0601C5 was also grown for isolation of amphibactin D (Figure 1, C14:0) for structural comparison using previously published methods [9].
Siderophore isolation
For siderophore production, Vibrio sp. Nt1 was cultured in 2 L natural seawater medium (NSW) in 4 L acid-washed Erlenmeyer flasks. NSW medium consists of 1 g ammonium chloride, 2 g bacto-casamino acids, and 0.1 g glycerol phosphate per liter of aged natural seawater. Cultures were grown on a rotary shaker (180 rpm) at room temperature until late exponential phase (about 14–18 hours) before harvesting. Siderophores were isolated from the culture supernatant using the methods described in Gauglitz et al. [3]. The siderophores were purified by reversed-phase high-performance liquid chromatography (RP-HPLC) on a preparative C4 column (250 mm length × 20 mm diameter, Higgins) with either a gradient from 100 % solvent A (0.05 % trifluoroacetic acid (TFA) in doubly deionized water (ddH2O, Barnstead Nanopure II)) to 100 % solvent B (0.05 % TFA in 80 % HPLC grade methanol (Fisher), 20 % ddH2O) over 37 minutes, holding at 100 % solvent B for an additional 10 minutes or a gradient from 50 % solvent A and 50 % solvent B to 100 % solvent B over 15 minutes, holding at 100 % solvent B for an additional 10 minutes, using a dual-pump Waters HPLC system. The eluent was continuously monitored at 215 nm and peaks were collected by hand and stored on dry ice. If necessary, fractions were ultrapurified on the same preparative C4 column using the same program as before. Purified siderophores were lyophilized and stored at −20 °C.
Structure determination
The masses of the siderophores and the siderophore fragments were determined by electrospray ionization mass spectrometry (ESI-MS) and tandem mass spectrometry on a Micromass Q-TOF2 (Waters Corp.).
The amino acid composition of the isolated siderophores was determined using the chiral derivatizing agent 1-fluoro-2-4-dinitrophenyl-5-L-alanine amide (FDAA; Marfey’s reagent) after hydrolysis of the siderophores with 55% hydroiodic acid (Spectrum Labs) at 110°C for 24hrs [34]. The hydrolyzed and derivatized amino acids were identified by co-injection with derivatized authentic amino acid standards and by comparison of the retention times when resolved by RP-HPLC. Solvents used for HPLC separation were 99.99% ddH2O (Barnstead Nanopure II) with 0.01% TFA and 100% HPLC grade acetonitrile (Fisher).
The fatty acids were identified by Gas Chromatography-Mass Spectrometry (Varian, Saturn 2100T GC-MS). Siderophores were incubated with 3N methanolic hydrochloride (Sigma) at 110°C for 3hrs to hydrolyze the amide bonds and generate the methyl esters of the fatty acid appendages. The methyl esters analyzed by GC-MS were identified by comparison to authentic fatty acid methyl ester standards (methyl dodecanoate, methyl tetradecanoate and methyl cis-9-hexadecenoate; Supelco) and by co-injection.
UV-Vis spectroscopy of the ferric moanachelin gly-D and ferric amphibactin D complexes was performed on a Cary 300 Bio UV-Visible Spectrophotometer (Varian). 1H, 13C, heteronuclear single quantum correlation spectroscopy (HSQC), correlation spectroscopy (COSY), and heteronuclear multiple bond correlation (HMBC) nuclear magnetic resonance (NMR) spectra of moanachelin gly-D and amphibactin D in d6-dimethyl sulfoxide (d6-DMSO, 99.9 % Cambridge Isotopes, Inc.) were recorded on a Bruker 800 MHz nuclear magnetic resonance (NMR) spectrometer. In addition, total correlation spectroscopy (TOCSY) was also performed for amphibactin D.
Results
A new suite of amphiphilic siderophores, the moanachelins, was isolated from the open ocean bacterium Vibrio sp. Nt1. The moanachelins are a group of closely related structures with variation not only in the fatty acid appendage of the siderophore, but also in the amino acid composition of the iron-coordinating peptidic head group. To differentiate between the two head groups, we refer to the siderophores which contain glycine as the moanachelins gly-B through gly-E, and to those which contain alanine, as the moanachelins ala-B and ala-D. The different members of the moanachelin suite of siderophores are designated as moanachelin gly or ala based on the unique amino acid present, i.e., alanine or glycine, and are further differentiated by the length of the fatty acid tail. Moanachelin gly-D and ala-D both have a C14:0 fatty acid, as does amphibactin D. During separation and isolation from the culture supernatant, the moanachelins behave similarly to the amphibactins, which have been isolated recently from the supernatant of Vibrio sp. HC0601C5 bacterial cultures [9] as well as other Vibrio sp. [6], and which contain serine in place of alanine or glycine, present in the moanachelins. The moanachelins elute as a cluster of peaks around 80 % methanol, indicating their relative hydrophobicity (Figure 2).
Figure 2.
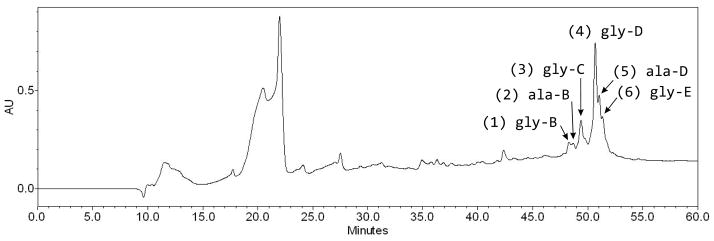
RP-HPLC chromatogram for total XAD extract of a Vibrio sp. Nt1 culture using a preparative Higgins C4 column. Sample is monitored at 215 nm. Siderophores elute between 47 and 53 minutes. Peak numbers 1–6 are noted.
A minimum of six moanachelins are produced. The [M+H]+ molecular ions of the moanachelins fall within the range of 774 m/z to 828 m/z. Starting with the most hydrophilic siderophore, moanachelin gly-B, the dominant siderophore identified in peak 1 (Figure 2), has a mass-over-charge ratio of 774.4. The dominant siderophores identified in peaks 2 and 3 are moanachelin ala-B (788.4 m/z) and moanachelin gly-C (800.4 m/z), respectively. The most abundant siderophore and the dominant siderophore in peak 4 is moanachelin gly-D (802.4 m/z). The later part of peak 4 has two small shoulder peaks, peaks 5 and 6, containing moanachelin ala-D and moanachelin gly-E (816.5 and 828.5 m/z, respectively). All of these peaks had positive responses to the liquid CAS test, indicating their ability to coordinate iron(III) [35].
The fragmentation patterns seen in the tandem mass spectra (Figure 3, Figure 4) and summarized in Table 1 indicate that the suite of siderophores can be broken up into two groups, depending on the identity of the second amino acid from the carboxy-terminus, and that they form a suite of siderophores with variations in the fatty acid tails (Figure 5). Tandem mass spectrometry reveals the majority of the ‘y’ (‘y’= [y+2H]+) and b-fragments expected for the moanachelins [36]. The ‘y’-fragments 191, 248, and 420 m/z are consistently observed for the moanachelins containing glycine, while the ‘y’-fragments 191, 262 and 434 m/z are observed for the moanachelins containing alanine (Table 1, Figures 3–4 and Figures S1–S4). The identical ‘y’-fragments, but different b-fragments, for each moanachelin (for both those containing a glycine or alanine residue) indicate that the suite contains members with distinct fatty acid appendages. For instance, the ‘y’-fragments 191, 248, 420 m/z, depicted in Figure 6 are observed in the tandem mass spectrum of moanachelin gly-D (Figure 3). In addition to moanachelins gly-B, gly-C, gly-D, gly-E and ala-B and ala-D, additional small quantities of other members of the suite were also detected based on their fragmentation patterns, including 732 m/z (initially named moanachelin gly-A), 786 m/z and 842 m/z. However, sufficient quantities were not obtained to verify their structures.
Figure 3.

Electrospray ionization-tandem mass spectrum of moanachelin gly-D (C14:0).
Figure 4.

Electrospray ionization-tandem mass spectrum of moanachelin ala-D (C14:0).
Table 1.
The b- and ‘y’-fragment values (m/z) observed by tandem mass spectrometry. ‘y’= [y+2H]+
| Moanachelin | ||||||
|---|---|---|---|---|---|---|
| gly-B (774 m/z) C12:0 |
Ala-B (788 m/z) C12:0 |
gly-C (800 m/z) C14:1 |
gly-D (802 m/z) C14:0 |
ala-D (816 m/z) C14:0 |
gly-E (828 m/z) C16:1 |
|
| B | ||||||
| B1 | 355 | 355 | 381 | 383 | 383 | 409 |
| B2 | 527 | 527 | 553 | 555 | 555 | 581 |
| B3 | 584 | 598 | 610 | 612 | 626 | 638 |
|
| ||||||
| ’y’ | ||||||
| Y1 | 191 | 191 | 191 | 191 | 191 | 191 |
| Y2 | 248 | 262 | 248 | 248 | 262 | 248 |
| Y3 | 420 | 434 | 420 | 420 | 434 | 420 |
Figure 5.

Suite of moanachelins. The position of the double bond in moanachelin gly-C was not determined.
Figure 6.
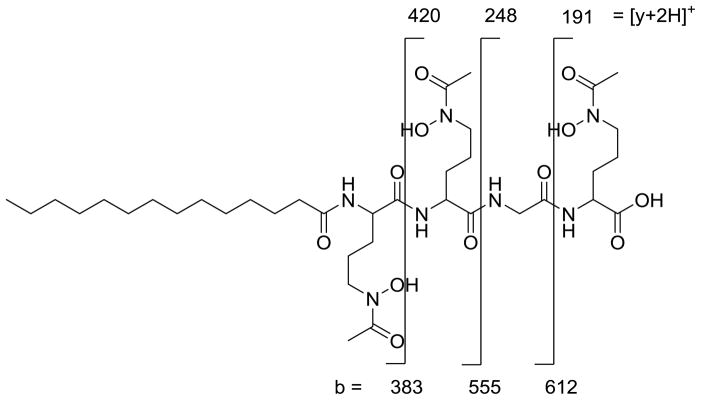
‘y’- and b- fragment analysis of the glycine-containing moanachelin gly-D (C14:0). ‘y’=[y+2H]+
Chiral amino acid analysis of members of the moanachelin suite confirmed the presence of two unique combinations of amino acids. The amino acids were identified by co-injection with authentic amino acid standards and by comparison of the retention times of the amino acid standards. Moanachelins gly-B, gly-C, gly-D and gly-E are composed of two modified D-ornithines, one modified L-ornithine and one glycine (Figure 5). Moanachelins ala-B and ala-D are composed of two modified D-ornithines, one modified L-ornithine and one L-alanine (Figure 5). The specific placement of the D- and L-ornithines was not determined.
The suite of moanachelin siderophores was hydrolyzed and derivatized with methanolic hydrochloride, extracted into hexanes and analyzed by GC-MS to determine the identity of the fatty acids (Figure 5). With the exception of moanachelin gly-C, for which the position of the double bond was not determined, the identification was confirmed by co-injection with fatty acid methyl ester standards (Supelco). The NMR analysis of moanachelin gly-D is consistent with this fatty acid tail analysis (Table 2).
Table 2.
Comparison of 1H and 13C chemical shifts of head group atoms for amphibactin D (C14:0) and moanachelin gly-D (C14:0)1.
| Position | Amphibactin D Head Group | Position | Moanachelin gly-D Head Group | ||
|---|---|---|---|---|---|
| 13C (ppm) | 1H (ppm) [m] | 13C (ppm) | 1H (ppm) [m] | ||
| N-Ac, N-OH Ornithine | N-Ac, N-OH Ornithine | ||||
| C1 | 173.2 | — | C1 | 173.3 | — |
| C2 | 51.41 | 4.13 | C2 | 51.45 | 4.15 |
| C3 | 28.38 | 1.67 | C3 | 28.36 | 1.55 |
| C4 | 22.9 | 1.54, 1.48 [d] | C4 | 23.09 | 1.54, 1.47 [d] |
| C5 | 46.5 | 3.45 | C5 | 46.3 | 3.44 |
| C6 | 170.2 | — | C6 | 170.2 | — |
| C7 | 20.07 | 1.95 | C7 | 20.1 | 1.96 |
| N1 | — | 7.78 | N1 | — | 7.98 |
|
| |||||
| Serine | Glycine | ||||
| C8 | 169.5 | — | C8 | 168.6 | — |
| C9 | 55.4 | 4.26 | C9 | 41.37 | 3.75, 3.73; 3.68, 3.66 [dd] |
| C10 | 61.68 | 3.55, 3.59 [d] | — | — | — |
| N2 | — | 7.83 | N2 | — | 8.1 |
|
| |||||
| N-Ac, N-OH Ornithine | N-Ac, N-OH Ornithine | ||||
| C11 | 171.43 | — | C10 | 171.6 | — |
| C12 | 52.5 | 4.2 | C11 | 52.16 | 4.19 |
| C13 | 28.9 | 1.46 | C12 | 29.0 | 1.46 |
| C14 | 22.9 | 1.54, 1.48 [d] | C13 | 22.89 | 1.54, 1.47 [d] |
| C15 | 46.5 | 3.47, 3.42 [d] | C14 | 46.31 | 3.47, 3.49 [d] |
| C16 | 170.2 | — | C15 | 170.2 | — |
| C17 | 20.07 | 1.95 | C16 | 20.1 | 1.96 |
| N3 | — | 8 | N3 | — | 8.11 |
|
| |||||
| N-Ac, N-OH Ornithine | N-Ac, N-OH Ornithine | ||||
| C18 | 172.89 | — | C17 | 172.0 | — |
| C19 | 52.1 | 4.23 | C18 | 52.28 | 4.23 |
| C20 | 28.8 | 1.68 | C19 | 29.12 | 1.46 |
| C21 | 22.9 | 1.54, 1.48 [d] | C20 | 23.0 | 1.52, 1.47 [d] |
| C22 | 46 | 3.44 | C21 | 46.31 | 3.47, 3.49 [d] |
| C23 | 170.3 | — | C22 | 170.2 | — |
| C24 | 20.07 | 1.95 | C23 | 20.1 | 1.96 |
| N4 | — | 8.17 | N4 | — | 7.93 |
See numbering in Figure 7.
Isolation procedures and structural analysis of the moanachelins indicate that they are structurally similar to the amphibactins, differing in the second amino acid from the carboxy-terminus. Like the moanachelins, the amphibactins are produced as a suite of siderophores with a conserved head group composed of two N-acetyl (Ac), N-hydroxy (OH) D-ornithines, one N-Ac, N-OH L-ornithine, one L-serine and a fatty acid appendage ranging from C12 to C18 [6, 9]. NMR analyses of the amphibactins and moanachelins in d6-DMSO (D, 99.9%) are presented here to confirm the structure and connectivity of these molecules.
A side-by-side comparison of the head group chemical shift values from the 1H and 13C NMR spectra of moanachelin gly-D and amphibactin D is presented in Table 2. Both siderophores are composed of a four amino acid head group with a tetradecanoic acid tail. Tandem mass spectral data as well as determination of the amino acid composition indicate that the structures of the two siderophores differ only in the second amino acid from the carboxy-terminus. Where the amphibactins have a serine residue [6], the moanachelins have either a glycine residue or an alanine residue. Comprehensive 1D and 2D NMR spectra allowed for the first full structural characterization of the moanachelins (moanachelin gly-D; C14:0) and the amphibactins (amphibactin D; C14:0) (Figures S7–S11 and S12–S17). The numbering for chemical shift assignments for moanachelin gly-D is given in Figure 7. The chemical shifts of d6-DMSO are noted at δH 2.49 in the 1H NMR and δC 39.5 in the 13C NMR spectrum, and were used as internal standards.
Figure 7.

1H and 13C number assignment for moanachelin gly-D. The chemical structure of amphibactin D differs at the C9 alpha carbon, where a serine group is present in contrast to the glycine depicted for moanachelin gly-D. Therefore, the numbering is the same for C1 to C8 and is shifted by 1 after C9 for amphibactin D due to the beta carbon of serine (see Table 2).
The 1H NMR spectrum of moanachelin gly-D reveals the presence of four amide protons at δH 7.93, δH 7.98, δH 8.1 and δH 8.11, three N-Ac, N-OH ornithine Cα protons at δH 4.15, δH 4.19, δH 4.23, and the glycine Cα protons at δH 3.73, δH 3.75 and δH 3.66, δH 3.68. The triplet at δH 0.84 corresponds to the methyl protons of the terminal carbon, while the aliphatic protons are located between δH 1 and 2. The 13C spectrum reveals the presence of four unique carbonyl signals at δC 168.6, δC 171.6, δC 172.0, δC 173.3, and one signal at δC 170.2 representing the carbonyl of the three modified ornithine side chains (C6, C15, C22). The carbon chemical shifts at C6, C15, and C22 are likely identical because of the locally identical surroundings of the N-acetylated N-hydroxylated ornithine moiety. The γ-carbons of the modified ornithine residues are found at approximately δC 46.3.
The 1H-1H COSY and 1H-13C HMBC reveal an alkyl group starting with the triplet methyl signal, H37, through to the methylene resonance H25. The number of carbon correlations to the alkyl chain seen in the 1H-13C HSQC and 13C NMR spectra confirm the fatty acid analysis that the alkyl group is a tetradecanoic acid. Through examination of the 1D and 2D NMR data the connectivity of the molecule has been established (Table S1). Similar analysis for amphibactin D reveals a remarkably related structure (Table S2).
The key difference between the amphibactin and moanachelin spectra lies with the Cα chemical shift of the serine present in amphibactin D versus the glycine present in moanachelin gly-D. The Cα carbon chemical shift for serine is δC 55.4, while it is δC 41.37 for glycine. The Cα for serine has only one proton, whereas glycine has two. In addition to a difference in the chemical shift of the serine Cα proton and carbon (C9 for amphibactin D and moanachelin gly-D), there is the additional signal of the serine beta carbon and protons (present in amphibactin D, C10, at δC 61.68 and δH 3.55, 3.59) (Table 2). All further spectral values are very similar.
These data, in combination with the tandem mass spectral data, and amino acid analysis, clearly demonstrate that the moanachelins are composed of two N-Ac, N-OH D-ornithines, one N-Ac, N-OH L-ornithine, and either a glycine or L-alanine subunit, followed by a fatty acid appendage ranging from C12 to C16 linked by an amide bond. In addition, the UV visible spectrum of iron(III)-moanachelin-gly D, as well as that of iron(III)-amphibactin D, reveal broad absorption maxima at 415 nm, characteristic of the hydroxamate to iron(III) charge-transfer band (Figures S5 and S6).
Discussion
We present here the structures of the moanachelins, a suite of amphiphilic marine siderophores produced by an open ocean Vibrio species. This suite of siderophores has varying fatty acid tail lengths and is composed of two N-Ac, N-OH D-ornithines, one N-Ac, N-OH L-ornithine and one variable amino acid, which form a peptidic head group that binds iron(III). The moanachelins are subcategorized depending on whether they contain glycine or alanine into the moanachelins gly-B through gly-E and moanachelins ala-B and ala-D. The existence of variation in the peptidic head group structure of a suite of amphiphilic siderophores produced by a single bacterium is unique to date.
While the moanachelins have either glycine or L-alanine at the same position in the peptide, no amino acid variation has been observed in the L-serine containing amphibactins (see Figure 1 and 5). The exclusive presence of L-serine in the amphibactins could be due to the highly conserved structure for the adenylation domain binding site specificity pocket for L-serine, which would be present in the NRPS system [19]. The glycine and alanine binding sites on the other hand have not been as well characterized and modeling studies indicate that they group into the same ‘small cluster’ [20]. This could indicate that they have similar binding pockets and may substitute for one another more readily or that there may be many degenerate solutions for the glycine and alanine binding pockets and multiple conformations of the binding pocket may recognize these small amino acids.
The occurrence of bacterial peptidic secondary metabolites which naturally incorporate different but structurally similar amino acids is rare. One instance is the production of a pyoverdine siderophore with two structural variants, whose amino acid structure differs in the incorporation of alanine or glycine [30]. However, the substitution of glycine was restricted to a specific alanine residue in the siderophore and the glycine-containing siderophore was a minor portion of the total siderophore production and could not be independently isolated. In the case of the moanachelins, the alanine and glycine-containing siderophores also had very similar physical properties. Flexibility in the NRPS system may result in the production of the two variants. Thus, it is plausible that either two separate biosynthetic pathways are present or that, in the case of a NRPS system, promiscuity of the adenylation-domain is accommodated in downstream domains, allowing either alanine or glycine to be incorporated into the siderophore.
Marine microbes have been shown to modify the hydrophilicity of their amphiphilic siderophores by varying the length, degree of unsaturation and hydroxylation of their fatty acid appendages. The large suites of amphiphilic siderophores that have been discovered in the past decade indicate that there is broad selectivity for the N-terminal fatty acid chain in the biosynthetic pathway, and it remains to be seen if these suites confer a specific advantage in an aqueous environment or if it is metabolically more favorable for the bacterium to incorporate a range of fatty acid tails. These linear peptide siderophores are produced by γ-proteobacteria isolated from the marine environment and are produced by a number of different genera. The continued detection of new siderophores with common structural features, isolated from numerous ocean environments, points toward an evolutionary lineage to siderophore production.
Supplementary Material
Acknowledgments
We thank the officers, crew, and scientific party aboard the R/V Kilo Moana during the 2009 POOB cruise. A.B. gratefully acknowledges NIH GM38130 and NSF CHE-1059067. J.M.G. is supported by a National Science Foundation Graduate Research Fellowship (2007052970). Any opinions, findings, conclusions or recommendations expressed in this publication are those of the author(s) and do not necessarily reflect the views of the National Science Foundation.
References
- 1.Martinez JS, Haygood MG, Butler A. Limnol Oceanogr. 2001;46:420–424. [Google Scholar]
- 2.Wilson M, Abergel R, Arceneaux J, Raymond K, Byers B. Biometals. 2010;23:129–134. doi: 10.1007/s10534-009-9272-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gauglitz JM, Zhou H, Butler A. J Inorg Biochem. 2012;107:90–95. doi: 10.1016/j.jinorgbio.2011.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Homann VV, Sandy M, Tincu J, Templeton A, Tebo BM, Butler A. J Nat Prod. 2009;72:884–888. doi: 10.1021/np800640h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Martin JD, Ito Y, Homann VV, Haygood MG, Butler A. J Biol Inorg Chem. 2006;11:633–641. doi: 10.1007/s00775-006-0112-y. [DOI] [PubMed] [Google Scholar]
- 6.Martinez JS, Carter-Franklin JN, Mann EL, Martin JD, Haygood MG, Butler A. Proc Natl Acad Sci USA. 2003;100:3754–3759. doi: 10.1073/pnas.0637444100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Martinez JS, Zhang GP, Holt PD, Jung HT, Carrano CJ, Haygood MG, Butler A. Science. 2000;287:1245–1247. doi: 10.1126/science.287.5456.1245. [DOI] [PubMed] [Google Scholar]
- 8.Snow G, White A. Biochem J. 1969;115:1031–1045. doi: 10.1042/bj1151031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vraspir JM, Holt PD, Butler A. BioMetals. 2011;24:85–92. doi: 10.1007/s10534-010-9378-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mawji E, Gledhill M, Milton JA, Zubkov MV, Thompson A, Wolff GA, Achterberg EP. Mar Chem. 2011;124:90–99. [Google Scholar]
- 11.Gledhill M, McCormack P, Ussher S, Achterberg EP, Mantoura RFC, Worsfold PJ. Mar Chem. 2004;88:75–83. [Google Scholar]
- 12.Ratledge C. Tuberculosis. 2004;84:110–130. doi: 10.1016/j.tube.2003.08.012. [DOI] [PubMed] [Google Scholar]
- 13.De Voss J, Rutter K, Schroeder B, Barry C. J Bacteriol. 1999;181:4443–4451. doi: 10.1128/jb.181.15.4443-4451.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rosconi F, Davyt D, Martinez V, Martinez M, Abin-Carriquiry JA, Zane H, Butler A, de Souza EM, Fabiano E. Environ Microbiol. 2012 doi: 10.1111/1462-2920.12075. In Press. [DOI] [PubMed] [Google Scholar]
- 15.Quadri L, Sello J, Keating T, Weinreb P, Walsh CT. Chem Biol. 1998;5:631–645. doi: 10.1016/s1074-5521(98)90291-5. [DOI] [PubMed] [Google Scholar]
- 16.Vergne A, Walz A, Miller M. Nat Prod Rep. 2000;17:99–116. doi: 10.1039/a809397k. [DOI] [PubMed] [Google Scholar]
- 17.Eppelmann K, Stachelhaus T, Marahiel M. Biochemistry. 2002;41:9718–9726. doi: 10.1021/bi0259406. [DOI] [PubMed] [Google Scholar]
- 18.Stachelhaus T, Mootz H, Marahiel M. Chem Biol. 1999;6:493–505. doi: 10.1016/S1074-5521(99)80082-9. [DOI] [PubMed] [Google Scholar]
- 19.Challis GL, Ravel J, Townsend C. Chem Biol. 2000;7:211–224. doi: 10.1016/s1074-5521(00)00091-0. [DOI] [PubMed] [Google Scholar]
- 20.Rausch C, Weber T, Kohlbacher O, Wohlleben W, Huson D. Nucleic Acids Res. 2005;33:5799–5808. doi: 10.1093/nar/gki885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Qiao K, Zhou H, Xu W, Zhang W, Garg N, Tang Y. Org Lett. 2011;13:1758–1761. doi: 10.1021/ol200288w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Crawford JM, Portmann C, Kontnik R, Walsh CT, Clardy J. Org Lett. 2011;13:5144–5147. doi: 10.1021/ol2020237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marahiel M, Essen L-O. Methods Enzymol. 2009;458:337–351. doi: 10.1016/S0076-6879(09)04813-7. [DOI] [PubMed] [Google Scholar]
- 24.Gehring AM, Mori I, Walsh CT. Biochemistry. 1998;37:2648–2659. doi: 10.1021/bi9726584. [DOI] [PubMed] [Google Scholar]
- 25.Rusnak F, Faraci WS, Walsh CT. Biochemistry. 1989;28:6827–6835. doi: 10.1021/bi00443a008. [DOI] [PubMed] [Google Scholar]
- 26.Shaw-Reid CA, Kelleher NL, Losey AM, Gehring C, Berg CT, Walsh HC. Chem Biol. 1999;6:385–400. doi: 10.1016/S1074-5521(99)80050-7. [DOI] [PubMed] [Google Scholar]
- 27.Quadri LEN, Keating TA, Patel HM, Walsh CT. Biochemistry. 1999;38:14941–14954. doi: 10.1021/bi991787c. [DOI] [PubMed] [Google Scholar]
- 28.Keating TA, Suo Z, Ehmann DE, Walsh CT. Biochemistry. 2000;39:2297–2306. doi: 10.1021/bi992341z. [DOI] [PubMed] [Google Scholar]
- 29.Keating TA, Marshall CG, Walsh CT. Biochemistry. 2000;39:15522–15530. doi: 10.1021/bi0016523. [DOI] [PubMed] [Google Scholar]
- 30.Barelmann I, Fernandez DU, Budzikiewicz H, Meyer J-M. Biometals. 2003;16:263–270. doi: 10.1023/a:1020615830765. [DOI] [PubMed] [Google Scholar]
- 31.Schalk IJ, Guillon L. Environ Microbiol. 2012 doi: 10.1111/1462-2920.12013. [DOI] [PubMed] [Google Scholar]
- 32.Sieber S, Marahiel M. Chem Rev. 2005;105:715–738. doi: 10.1021/cr0301191. [DOI] [PubMed] [Google Scholar]
- 33.Hopkinson BM, Barbeau KA. Environmental Microbiology. 2012;14:114–128. doi: 10.1111/j.1462-2920.2011.02539.x. [DOI] [PubMed] [Google Scholar]
- 34.B’Hymer C, Montes-Bayon M, Caruso JA. J Sep Sci. 2003;26:7–19. [Google Scholar]
- 35.Schwyn B, Neilands J. Anal Biochem. 1987;160:47–56. doi: 10.1016/0003-2697(87)90612-9. [DOI] [PubMed] [Google Scholar]
- 36.Roepstorff P, Fohlman J. Biomed Mass Spectrom. 1984;11:601. doi: 10.1002/bms.1200111109. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.