

Abstract
Background
Mutations in STAT1 cause a broad spectrum of disease, ranging from severe viral and bacterial infections (amorphic alleles), to mild disseminated mycobacterial disease (hypomorphic alleles), to chronic mucocutaneous candidiasis (hypermorphic alleles). The hypermorphic mutations are also associated with arterial aneurysms, autoimmunity and squamous cell cancers.
Objective
To investigate the role of STAT1 gain of function mutations in phenotypes other than CMC.
Methods
We initially screened patients with chronic mucocutaneous candidiasis and autoimmunity for STAT1 mutations. We functionally characterized mutations in vitro and studied immune profiles and regulatory T cells. After our initial case identifications we explored two large cohorts of FOXP3WT IPEX-like patients for STAT1 mutations.
Results
We identified 5 children with polyendocrinopathy, enteropathy, and dermatitis, reminiscent of IPEX syndrome, all but one had a variety of mucosal and disseminated fungal infections. All patients lacked FOXP3 mutations but had uniallelic STAT1 mutations [c.629 G>T, p.R210I; c.1073 T>G, p.L358W, c.796G>A; p.V266I; c.1154C>T, T385M (2 patients)]. STAT1 phosphorylation in response to IFN-γ, IL-6 and IL-21 was increased and prolonged. CD4+ IL-17 producing T cells were diminished. All patients had a normal percentage of regulatory T cells in the CD4+ T cell compartment and their function was intact in the two patients tested. Patients with cells available for study had normal levels of IL-2-induced STAT5 phosphorylation..
Conclusions
Gain-of-function mutations in STAT1 can cause an IPEX-like syndrome with normal frequency and function of regulatory T cells.
Keywords: STAT1, IPEX, FOXP3, Treg, CMC, aneurysms
INTRODUCTION
Immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) is characterized by multi-organ autoimmunity (type I diabetes, thyroiditis, enteropathy, villus atrophy, dermatitis, and other autoimmune phenomena) and typically begins in infancy5–10. Classical IPEX is caused by mutations in FOXP3, the gene critical for production of regulatory T cells (Tregs, CD4+CD25+FOXP3+)5, 10. In the absence of Tregs, effector T cells are thought to be free to attack end organs, leading to a distinctive clinical presentation and pathology11.
A significant number of patients who have symptoms of IPEX have wild-type (WT) FOXP3 gene sequence and are therefore classified as IPEX-like. IL2RA (CD25) and STAT5B defects have been identified in a small number of patients with IPEX-like clinical phenotypes and regulatory T cell dysfunction. However, the molecular defects responsible for the remainder of patients with this clinical presentation are unknown.
We identified a small number of patients who had clinical features of IPEX, some of whom also had chronic mucocutaneous candidiasis (CMC) or other infections. Syndromes in which pronounced immune dysregulation is combined with specific infectious susceptibilities are few and variable. Although infections do occur in the IPEX syndrome, their etiology is difficult to assign, since most patients have poor skin and gut barrier function and are receiving aggressive immunosuppression8. CMC and sinopulmonary infections occur in about 16% of FOXP3 deficient IPEX patients8 (unpublished data, TR Torgerson). Sequencing of the FOXP3 gene was normal in our patients. IL-2 receptor alpha chain (CD25) deficiency is typically associated with low numbers of NK and T cells and while infections are observed in CD25 deficiency, CMC is not reported12. Patients with mutations in STAT5B often present with CMV infection, CMC, lung disease related to T cell dysfunction17.
Dominant gain-of-function mutations in STAT1 have recently been associated with chronic mucocutaneous candidiasis (CMC) 1–3,, disseminated coccidioidomycosis, and disseminated histoplasmosis 2–4. Interestingly, in one series they were also associated with an increased incidence of autoimmunity (19%), squamous cell cancer (9%), and cerebral aneurysms (4%)2. These mutations in the coiled-coil and DNA binding domains lead to impaired dephosphorylation of STAT1 after stimulation and reduced numbers of IL-17 producing Th17 cells2, 3.
In view of the recognized link of gain-of-function STAT1 mutations with CMC, disseminated fungal infections, thyroid autoimmunity, and aneurysm formation, we examined patients with FOXP3WT IPEX-like autoimmunity with and without CMC to determine whether gain-of-function STAT1 mutations may underlie IPEX-like disease.
METHODS
All patients or their guardians provided informed consent under approved protocols of the National Institute of Allergy and Infectious Diseases, Seattle Children’s Hospital, or Anna Meyer Children’s Hospital in Florence, Italy. Normal blood was obtained under approved protocols of these centers.
Cell lines
EBV-transformed B cell lines derived from patients and normal donors were maintained in RPMI 1640 with 20% fetal calf serum (FCS; Gibco BRL, Carlsbad, CA), 2mM L-glutamine, penicillin 100U/ml, 100μg/ml streptomycin (Gibco), at 37°C in a humidified 5% CO2 incubator. STAT1 deficient U3A cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM; Gibco) supplemented with 10% FCS, 2mM L-glutamine and antibiotics.
DNA sequencing
Genomic DNA (PureGene Gentra DNA isolation kit, QIAGEN) and total RNA (RNeasy isolation kit, QIAGEN) were extracted from EBV-transformed B cell lines or polymorphonuclear leukocytes. Primers for STAT1 and full-length cDNA were designed using Primer Select (DNAstar Lasergene). Genomic amplification was performed with Platinum PCR Supermix High Fidelity (Invitrogen). Samples were treated with ExoSAP (Affymetrix) and resulting product was used for sequencing with Big Dye Terminators v3.1 (Applied Biosystems, Foster City, CA), purified with Performa DTR short well plate kit (Edge Biosystems), run on an Applied Biosystems 3730XL sequencer and aligned to the consensus sequence NM_007315.3 using Sequencer software (Gene Codes).
Nuclear extracts and nuclear complex binding
Nuclear extracts from transfected U3A cells (Amaxa nucleofector, Lonza, Walkersville, MD), stimulated or not with IFN-γ (400 IU/ml) or IFN-α (IFN-α 2b, PBL Biomedical Laboratories, Piscataway, NJ) 1,000 IU/ml, were prepared using the Panomics kit (Panomics, Fremont, CA). For determination of DNA binding activity, an ELISA-like colorimetric assay (TRANSAM, Active Motif, Carlsbad, CA) using a plate coated with a STAT1 binding oligonucleotide derived from the GAS, was used. Absorbance was measured on a spectrophotometer at 450nm.
Reporter gene assay
STAT1 deficient U3A cells were co-transfected with wild type (WT) and/or mutant STAT1 expression constructs along with a plasmid containing tandem IFN-response elements driving a luciferase reporter gene (1μg; Panomics, Fremont, CA). Following overnight incubation, cells were stimulated or not with IFN-γ or IFN-α (1,000 IU/ml) for 6 h and resuspended in lysis buffer prior to a dual luciferase assay (Promega, Madison, WI). Experiments were done in triplicate and relative luciferase units were corrected for Renilla luciferase activity, which was co-transfected to control for transfection efficiency. Data are expressed as fold increase in response to interferon over the non-stimulated samples.
Flow cytometry
STAT1 and STAT5 activation
To assay STAT1 and STAT5 activation, EBV-B cells or PBMCs, when available, were stimulated either with IL-2, IFN-γ, IL-6 or IL-21 for 15 minutes, fixed with paraformaldehyde and permeabilized in methanol. Cells were stained with Alexa488 conjugated anti-phospho STAT1 (pY701) and PE-conjugated anti- phospho STAT5 (BD Biosciences). Levels of phosphorylation were assessed in the CD3+ lymphocyte gate (PBMCs) or live cell population (EBV-B cells).
STAT1 dephosphorylation
U3A cells were transiently transfected with plasmids encoding WT or mutant STAT1 using lipofectamine (Invitrogen). The following day, culture media were replaced and cells were treated or not with IFN-γ (400 IU/ml) for 30 min and incubation continued for the indicated periods of time, when cells were recovered, fixed and permeabilized in methanol. Cells were stained for total (Alexa 647 conjugated anti-STAT1) and phosphorylated tyrosine Y701 STAT1 (Alexa 488 conjugated anti-pSTAT1; BD Biosciences). For U3A cells, the levels of phosphorylation were assessed in the cells gated for the expression of total STAT1. Data were collected using a FACS Caliber (BD Biosciences). All flow data were analyzed using FLowJo. (Treestar, Ashland, OR).
Treg phenotyping and intracellular cytokine analysis
Staining for intracellular cytokines and FOXP3 was performed on fresh peripheral blood mononuclear cells (PBMCs) prepared by density centrifugation and resuspended in complete RPMI medium (106 cells/ml). For Treg phenotyping, the PBMCs were surface labeled for CD3, CD4 and CD8 followed by intracellular staining for FOXP3 (clone 259D) and HELIOS after fixation and permeabilization using the eBioscience FOXP3 staining kit (all antibodies from Biolegend). For intracellular cytokine analysis, the PBMCs were examined after stimulation with PMA 50ng/ml and ionomycin 1μM (Calbiochem) in the presence of brefeldin A (1μg/ml; Sigma) for 5 h at 37°C. The PBMCs were surface labeled for CD3, CD4 and CD8, followed by fixation and permeabilization, and then intracellular staining for IL-2, IFN-γ, IL-17A, TNF-α, IL-10 and FOXP3 (all antibodies from Biolegend). Data collection was done with BD LSR Fortessa and analyzed with FlowJo (Tree Stars, Inc).
Treg induction and functional analysis
PBMCs from healthy controls and patients were surface labeled for CD4, CD127, CD25, CD45RA and CD31. The PBMCs were sorted on BD Aria II for Tregs (CD4+CD127−CD25hi) and naïve T cells (CD4+CD127+CD25−CD45RA+CD31+). The naïve CD4+ T cells were stimulated in 24 well plates (Nunc) at 250,000 cells per well that had been coated with anti-CD3 (3μg/ml, UCHT1) and anti-CD28 (1μg/ml, CD28.2) antibodies (Biolegend). The cells were cultured in 200 U/ml IL-2 containing complete RPMI 1640 medium for 5 days in the presence or absence of TGF-β1 (5ng/ml), IFN-γ (20ng/ml) and/or IL-21 (20ng/ml) (Peprotech). FOXP3 and HELIOS expression were analyzed. For in vitro Treg suppression, the responders, Tregs and HLA-DR+ antigen presenting cells (APCs) were obtained from buffy coats of healthy donors. CD4+ cells were isolated from PBMCs using CD4 microbeads and AutoMACS (Miltenyi Biotec) as per manufacturer’s protocol. The CD4+ cells were labeled for CD4, CD127 and CD25 to sort for responders (CD4+CD127+CD2−) and Tregs (CD4+CD127−CD25hi). The HLA-DR+ APCs were isolated from the CD− PBMCs using HLA-DR microbeads and AutoMACS. The responders were labeled with carboxyfluorescein succinimidyl ester (CFSE) using the CellTrace CFSE Kit (Invitrogen). In 96 flat-bottom well plates (Nunc), 50,000 responders were stimulated with 25,000 non-irradiated HLA-DR+ APCs and 0.25μg/ml OKT3 alone or in the presence of decreasing numbers of Tregs or effector T cells (Teffs, CD4+CD127+CD2−). On day 4, the cells were labeled for surface CD4 and intracellular FOXP3 and analyzed for level of CFSE dilution within CD4+ cells.
Autoantibody detection
Plasma anti-cytokine autoantibodies were determined as described 18.
Statistics
Student’s t test was used for analysis of in vitro data where indicated (GraphPad Prism Software, San Diego, CA). The statistical significance level adopted was p < 0.05.
RESULTS
Patients
In the cohort of patients at the NIH, we identified 5 patients with IPEX-like features and CMC but with no specific molecular etiology. Sequencing of STAT1 identified heterozygous defects in 3 patients (patients 1, 2, and 3); two of whom (1 & 2) had already been tested for FOXP3 mutations in Seattle and found to have wild-type sequence. To determine whether STAT1 GOF mutations might be associated more broadly with an IPEX-like phenotype, two cohorts of IPEX-like patients were screened: 38 patients (four with CMC) from a cohort collected at Anna Meyer Children’s Hospital in Florence, Italy and 35 patients with an IPEX-like phenotype and normal regulatory T cell numbers (14 with CMC or candidemia) collected in Seattle (see Table 3 for patient characteristics). No cases were identified in the Italian cohort but two patients (4 and 5) were found to have heterozygous STAT1 mutations in the Seattle cohort. Subsequent screening of 2 additional Italian patients after submission of this manuscript identified one more patient with a STAT1 mutation. Cases are described in detail in the Online Supplement and summarized clinically in Table 1 and their laboratories in Table 2. Figure 1 shows elements of their phenotypes. Four of the five STAT1 patients had CMC (Patient 1; Figure 1A) but in one of these (Patient 5), the CMC was very mild, occurring only after the patient had been treated with antibiotics. Patients 2 and 3 had significant intracranial aneurysms like those described previously (Figure 1B)1.
Table 3.
Summary of Cohorts studied
| Number of patients screened | Number of STAT1 mutants | Number of patients with CMC | Enteropathy | Skin disease | Endocrinopathy | Autoimmune cytopenia | Recurrent infections other than CMC | |
|---|---|---|---|---|---|---|---|---|
| NIH | 5 | 3 | 5/5 | 3/5 | 2/5 | 5/5 | 2/5 | 5/5 |
| Seattle | 35 | 2** | 14/35 | 35/35 | 26/35 | 21/35 | 12/35 | 15/35 |
| Florence | 38 | 0 | 4/38 | 38/38 | 20/38 | 20/38 | 13/38 | 19/38 |
2 out of 3 NIH patients were also previously screened for FOXP3 mutation by the Seattle Group and they are not included in their cohort of 35.
Table 1.
Clinical characteristics
| PATIENT 1 | PATIENT 2 | PATIENT 3 | PATIENT 4 | PATIENT 5 | |
|---|---|---|---|---|---|
| AGE | 13 | 4 | 14 | 4 | 13 |
| GENDER | M | M | F | M | M |
| INITIAL PRESENTATION | INFANCY | INFANCY | INFANCY | INFANCY | TODDLER |
| Deceased | YES (invasive fungemia) | YES (intracranial bleed) | |||
| ETHNICITY | Hispanic | Caucasian | Korean | Caucasian | Caucasian |
| CMC | Oral, esophageal, fingernail; C. parapsilosis candidemia (catheter related) | Limited oral mucosal | Oral, esophageal, fingernail | None | Limited oral mucosal |
| SKIN and APPENDAGES | Generalized eczema with hyperkeratosis Acrodermatitis enteropathica |
Mild eczema Poor tooth enamelation |
Mild eczema | Staphylococcal superinfections of severe atopic dermatitis | Mild to moderate eczema |
| Sinopulmonary infections | Encapsulated bacteria; RSV bronchiolitis; Bronchiectasis with M. avium; Aspergillus pneumonia | Encapsulated bacteria; RSV bronchiolitis | Encapsulated bacteria; RSV bronchiolitis | Recurrent pneumonia; otitis, sinusitis, external otitis | Recurrent otitis |
| PULMONARY | Bronchiectasis Pulmonary hypertension |
Bronchiectasis | Bronchiectasis | Recurrent pneumonia | Recurrent croup |
| Blood-borne infections | MRSA; Disseminated aspergillosis (with immunosuppression) | MRSA | None | None |
C. parapsilosis candidemia (catheter related) Enterococcus (catheter related) |
| Herpes infections | HSV stomatitis | HSV skin infections, limited | Herpes zoster twice CNS VZV |
None | None |
| CNS | Normal vasculature by MRI | Bilateral basilar and carotid artery aneurysms hemiparesis and secondary hydrocephalus | Bilateral basilar and carotid artery aneurysms CVAs at 12 and 14yrs |
None | None |
| CARDIOVASCULAR | Persistent foramen ovale, hypertension, heart failure | None | Calcification of aorta, celiac and superior mesenteric arteries; renal artery stenosis | None | None |
| ENDOCRINE | Type I DM Multinodular goiter, anti-thyroid antibody + |
Type I DM, anti-GAD antibody + Hypothyroidism, anti-thyroid antibody+ |
Type I DM | None | Hypothyroidism, anti-thyroid antibodyneg; Hyperglycemia while on steroids; Growth hormone insufficiency |
| GROWTH AND DEVELOPMENT | Short stature Delayed puberty |
Short stature | Short stature Delayed puberty |
None | Short stature Delayed puberty Speech delay |
| GI | Hepatosplenomegaly Soft palate dysfunction Esophageal dysmotility Protein losing enteropathy Villus blunting |
Hepatosplenomegaly Lymphocytic and eosinophilic enteritis Villus blunting Recurrent intussusception Recurrent C. difficile colitis |
Hepatosplenomegaly | No organomegaly Protein losing enteropathy Lymphocytic and eosinophilic enteritis |
No organomegaly Chronic gastritis; Chronic lymphocytic enteritis. Villus atrophy |
| RENAL | Renovascular hypertension renal scarring (at autopsy) | None | Renovascular hypertension (renal artery stenosis) | None | None |
| HEMATOLOGY | Thrombocytopenia Hemolytic anemia |
None | None | Neutropenia | None |
| BONE | Generalized osteopenia and osteoporosis | Generalized osteopenia and osteoporosis Multiple bone fractures Dactylitis Osteomyelitis |
Generalized osteopenia | No osteoporosis | No osteoporosis L2 Hemi-vertebrae Spina Bifida occulta |
Table 2.
Laboratory Data
| IMMUNE CHARACTERISTICS OF PATIENTS
| |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Patient 1 | Patient 2 | Patient 3 | Patient 4 | Patient 5 | |||||||
|
| |||||||||||
| STAT1 mutation | c.629G>T, p.R210I Coiled-coil domain |
c.1073T>G, p.L358W DNA binding domain |
c.1154C>T, p.T385M DNA binding domain |
c.796G>A; p.V266I Coiled-coil domain |
c.1154C>T, p.T385M DNA binding domain |
||||||
|
| |||||||||||
| Lymphocyte Subsets | |||||||||||
| Normal (cells/ul) | 4 yrs | 13 yrs | 1 yr | 3.5 yrs | 4 yrs | 4 yrs | 13yrs | 3 months | 5 months | 4 yrs | 11 yrs |
| CD3+/CD4+ (359–1565) | 599 | 119 | 2,219 | 1057 | 761 | 700 | 301 | 3136 | 5427 | 622 | 276 |
| CD4+ Naïve (82–755) | N/A | 4 | N/A | N/A | N/A | N/A | 80 | 1975 | 4342 | 180 | 77 |
| CD3+/CD8+ (178–853) | 304 | 269 | 680 | 503 | 367 | 490 | 569 | 1372 | 1850 | 190 | 310 |
| CD3+/CD4−/CD8− (18–185) | 134 | 149 | 100 | 115 | 707 | 80 | 103 | 166 | 494 | 46 | 114 |
| CD20 + (59–329) | 1486 | ~1 | 2,040 | 707 | 745 | 330 | 55 | 4704 | 5181 | 358 | 425 |
| CD20+/IgM−/IgD−/CD27+ (5–46) | N/A | 0 | N/A | 0 | N/A | N/A | 0 | N/A | 311 | N/A | N/A |
| CD3-/CD16+ or CD56+ (126–729) | 81 | 130 | 277 | 568 | 137 | 50 | 45 | 588 | 740 | 39 | 46 |
|
| |||||||||||
|
FOXP3+ Tregs:
% of CD4+ T cells (Normal 4–8%) |
5.5% | 6.5% | 6.2% | 5.2% | 4.5% | ||||||
|
| |||||||||||
| Lymphocyte Proliferation* | PHA: Normal | PHA: Decreased (65% of normal) | PHA: Decreased (35% of normal) | CD3: Decreased | PHA: Decreased (69% of normal) | ||||||
| Mitogens | ConA: Normal | ConA: Decreased (46% of normal) | ConA: 54% Normal | CD3+IL2: Normal | PWM: Normal | ||||||
| PWM: Normal | PWM: Normal | CD3: Decreased (41% of normal) | |||||||||
| Recall Antigens | Tetanus: Normal | Tetanus: Normal | Tetanus: No response | ||||||||
| Candida: Normal | Candida: Decreased (80% of normal) | Candida: Decreased (25% of normal) | |||||||||
|
| |||||||||||
| Cytokine Profiles of stimulated PBMCs | PHA induced IL-10 production <15% of healthy control, LPS induced TNF α production increased by 25 fold | PHA induced IL-10 production ~ 32% of healthy control, LPS induced TNF α production increased by ~2 fold | PHA induced IL-10 production 3 % of healthy control, LPS induced TNF α production similar to control | N/A | N/A | ||||||
|
| |||||||||||
| Immunoglobulin levels (mg/dl) | 4 yrs (nl value) | 13 yrs (nl value) | 1 yr (nl value) | 3.5 yrs (nl value) | 4 yrs (nl value) | 14 yrs (nl value) | 1.5 yrs | 4 yrs (nl value) | 11 yrs (nl value) | ||
| IgG | 800 (386–1,470) | 271 | 1100 (246–904) | 1470 | 800 (386–1,470) | 1200 | 735 | 455 (444–1187) | 527 (695–1602) | ||
| IgA | 29 (29–256) | <10 | 178 (27–66) | 192 (27–246) | 29 (29–256) | 16 (52–319) | 49 | 99 (61–345) | 81 (51–223) | ||
| IgM | 166 (37–224) | <10 | 92 (40–143) | 84 (37–184) | 166 (37–224) | <21 (45–244) | 45 | 30 (48–226) | 50 (39–167) | ||
| IgE IU/ml | N/A | N/A | 186 (<90) | 81 (<90) | N/A | <1 | 57 | 24 (<193) | 5 (<397) | ||
|
| |||||||||||
| Immune globulin therapy | Begun at 11 years Rituximab at 12 years |
Begun at 3 years | Begun at 13 years | None | None | ||||||
|
| |||||||||||
| Anti-Pneumococcal antibody titers | Protective for 2/14 (at 1yr) | Protective for 7/14 (at 7 months) 0/14 (at 2 yrs) | Protective 1/14 (at 12 yrs) | Protective 7/7 (at 16 months) | Protective 0/14 (at 4 yrs) | ||||||
|
| |||||||||||
| HLA | DQB1 02, 06 | DQ 02, 02 | DQB1 03, 05:01 | N/A | N/A | ||||||
These studies were carried out at different centers with different normal value
Figure 1. Clinical Phenotypes.


(A): Patient 1, extensive candidiasis of nails, causing dystrophy and paronychia.
(B): Patient 3, Multiple intracranial aneurysms in the Circle of Willis, as detected by CT angiogram and MR angiogram.
(1C): Bilateral lower lobe bronchiectasis with endobronchial/peribronchial consolidation seen on chest CT.
(1D): multiple calcifications in descending aorta detected by CT of patient 3.
Immunophenotyping and immune function in patients
All patients had normal T cell numbers and subsets when tested under 5 years of age, but patients 1, 3, and 5 developed significant CD4+ T cell lymphopenia and patients 1 and 3 developed B cell lymphopenia by the early teen years, while CD8+ T cells and NK cells were preserved (Table 2). Based on a recent review of classical IPEX, patients usually have lymphocytosis but the percentage of CD3, CD4, CD8, CD16, CD19 remains unchanged, CD4/CD8 ratio is maintained or increased and the percentages of naive and memory T cells are mostly comparable to their age-matched controls 19. Class-switched memory B cells (Ig−/Ig−/CD20+/CD27+) were not present in patients 1, 2 or 3, although this was not evaluated at the earliest time points (Table 2). Patient 4 had markedly elevated class switched memory B cells at 16 months, but we do not know their trajectory. Patients 1, 2, and 3 received immune globulin replacement for poor vaccine responses. In addition, patients 1 and 3 received IVIG for immune modulation. Lymphocyte proliferation assays did not show significant or consistent abnormalities in the patients. In summary, CD4+ T cell lymphopenia, diminished class switched memory B cells and defective vaccine responses were common among the identified patients and appeared to progress with age (Table 2).
STAT1 mutations
Full length sequencing of STAT1 in genomic DNA identified heterozygous mutations in either the coiled-coil or DNA binding domains (Table 2). None of these mutations were previously reported among patients with disseminated mycobacterial infections or isolated CMC. However, the p.T385M mutation identified in patients 3 and 5 has also been observed in an adult patient with disseminated histoplasmosis but without autoimmunity3. In all cases, sequencing of full-length cDNA demonstrated equal representation of mutant and wild-type alleles, indicating that these mutations do not lead to cDNA instability. None of these mutations were found in dbSNP 132, or the 1000 Genomes Project.
Gain-of-function mutations in STAT1 lead to enhanced DNA binding and transactivation
Mutant STAT1 binding activity to a GAS oligonucleotide in response to IFN-γ was studied in EBV transformed B cells from all patient mutations (R201I, L358W, T385M, V266I). Following IFN-γ stimulation the mean fold increase over baseline in STAT1 binding to a GAS oligonucleotide was higher in all 4 mutant cell lines (mean: 2.84 ± 0.31 SD; range 2.46 to 3.16) compared to WT (p <0.05) (Supplemental Figure 1A). STAT1-deficient U3A cells transfected with mutant constructs showed similarly enhanced STAT1 activation as detected by luciferase activity in response to IFN-γ (mean fold induction 3.03 ± 0.26; range 2.72 – 3.35; p=0.04) (Supplemental Figure 1B). STAT1 mutant constructs also showed enhanced GAS-luciferase activity in response to IFN-α (3.67 ± 1.52). These activities were unaffected by co-transfection of mutant with WT STAT1 constructs, confirming the dominant gain-of-function aspect of these mutants.
IFN-γ induces prolonged STAT1 phosphorylation in gain of function mutants
Flow cytometric analysis of CD3+ T cells (Figure 2A) or EBV B cells (Figure not shown) from the patients confirmed IFN-γ-induced STAT1 hyperphosphorylation. Higher STAT1 phosphorylation in CD3+ T cells was also detected following stimulation with IL-6 and IL-21, cytokines that signal through STAT3 as well as STAT1.
Figure 2. STAT1 staining.
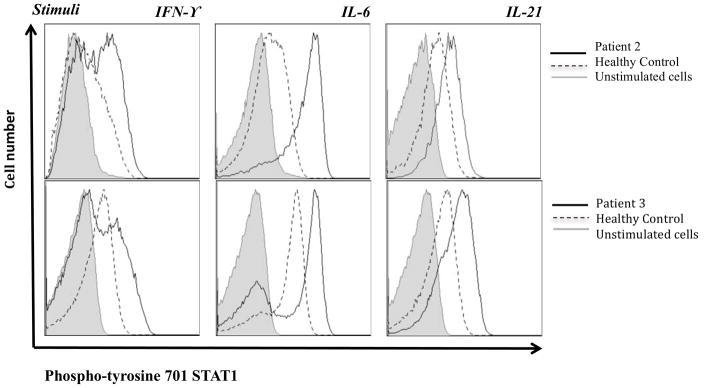

(A): Intracellular staining of phospho-tyrosine 701 STAT1 in CD3+ lymphocytes isolated from healthy controls and STAT1 gain of function mutants (patient 2 and 3) after stimulation with IFN-γ, IL-6, or IL-21 for 15 minutes.
(B): Kinetics of the STAT1 phosphorylation were studied by intracellular phospho-tyrosine 701 STAT1 staining in U3A cells transfected with mutant or WT constructs and stimulated with IFN-γ. Stimulation indices at 60 minutes following stimulation (SI: MFI of pSTAT1 at 60′ after stimulation /MFI pSTAT1 at rest) were higher for mutants (SI: 2.6 for R210I, 4.0 for L358W, and 4.4 for T385M), than for WT STAT1 (SI of 0.9, p <0.05).
STAT1 normally undergoes rapid phosphorylation followed by dephosphorylation (Figure 2B, top panel), whereas the gain-of-function mutants do not dephosphorylate normally. To quantify dephosphorylation of transfected U3A cells following stimulation, we calculated a median stimulation index [mean fluorescence index (MFI) of pSTAT1 after stimulation for 1 hour/MFI pSTAT1 at rest]. These values were 2.6 for R210I, 4.0 for L358W, and 4.4 for T385M, while cells expressing the WT STAT1 showed an SI of 0.9 (p <0.05) (Figure 2B). Data for V266I is not available.
Normal Tregs and diminished Th17 in gain of function mutants
The lack of functional FOXP3+ Treg cells is the cause of disease in classical IPEX syndrome. The percentage of CD25hiFOXP3+ cells present in the CD4+ T cell population and their level of FOXP3 expression were repeatedly within normal range in all patients (Table 2 and Figure 3A). While it has been shown in murine studies that Helios can be expressed in peripheral induced Tregs, the Helios+ Tregs represent a more stable and suppressive subset. 20, 21. Therefore we examined this subset and found that there were 84% and 64% of Tregs that expressed Helios in patients 2 and 3, respectively, comparable to controls (Figure 3A). In addition, their naïve CD4+ T cells were capable of being induced by TGFβ1 in vitro to express FOXP3 (Figure 3B). As previously reported, the vast majority of these iTregs failed to express HELIOS in vitro20. Interestingly, there was significant expression of FOXP3 in the absence of TGFβ1, unlike in healthy controls. While IFN-γ and IL-21 negatively affected TGFβ1 induction of FOXP3 in controls, only IL-21 had an inhibitory effect in these patients. Therefore, patient naïve CD4+ cells were capable of expressing FOXP3 in vitro, even in the presence of STAT1 signaling cytokines. Their suppressive function was not tested because human in vitro TGFβ1-induced iTregs are not suppressive22.
Figure 3. Treg subsets, FOXP3 expression, cytokine profiles and STAT5 signaling.




(A): Percentage and level of FOXP3 and HELIOS expression within CD3+CD4+ gated T cells from patients 2 and 3 compared to healthy controls.
(B): Induction of FOXP3 and absence of HELIOS expression in naïve T cells from healthy control and patients 2 and 3 on day 5 after anti-CD3/CD28 stimulation in the presence of IL-2 (NONE) only, + TGF-β1, + TGF-β1 and IFN-γ or + TGF-β1 and IL-21.
(C): Comparison of cytokine expression between FOXP3+ and FOXP−CD4+ T cells within fresh PBMCs of healthy controls and patients 2 and 3 stimulated with PMA/ionomycin.
(D): Normal IL-2 induced STAT5 phosphorylation in CD3+ lymphocytes of patient 3 (solid line histogram) compared to healthy control (dashed histogram) and unstimulated cells (shaded histogram). Patient 2 had similar expression profile (data not shown).
Another cardinal feature of Tregs is anergy or the absence of significant cytokine production. Examination of cytokine production within CD4+ T cells among patients and controls showed that the vast majority of Tregs lack significant expression of IL-2, IFN-γ and TNF-α as compared to FOXP− nonTregs. The predominant cytokine producers within the FOXP3+ population were localized to the HELIO−Treg subset (data not shown). Consistent with previous reports, there was significant diminution of IL-17+FOXP3−CD4+ cells (Th17) in patients 2 (0%) and 3 (0.3%), as compared to controls (1% and 3%) (Figure 3C)2, 3. There were low levels of IL10 signal predominately within the FOXP− nonTregs in both patients and controls.
Since Tregs are functionally dependent on IL-2 signaling through CD25 and STAT5 and since CD25 and STAT5b mutations are associated with autoimmune disease, we examined patient IL-2-induced STAT5 phosphorylation; it was normal in patients 2 and 3 (Figure 3D). Regardless of numbers of phenotypic Tregs, their ability to suppress T cell proliferation is the critical functional output. Despite patients 2 and 3 having IPEX-like phenotypes, their Tregs were fully capable of suppressing the proliferation of allogeneic responder cells (Figure 4). Unfortunately, we did not have sufficient cells from the patients to perform a comprehensive study of Treg suppressor function, particularly in the presence of STAT1 signaling cytokines.
Figure 4. Tregs suppressive function.


(A) : Responders only histogram represents CFSE dilution on day 4 within CD4+ T cells. The histograms in the two right columns demonstrate the level of proliferative suppression within CFSE+ cells in the presence of 1/2 and 1/4 the numbers of Tregs and Teffs relative to responders. Left FACS plot column shows the level of FOXP3 expression within CFS− Tregs/Teffs and CFSE+ responders on day 4. Patient 2 and control 1 are allogeneic and control 2 is autologous to responders and HLA-DR+ APCs.
(B) Suppressive function of Tregs from patient 3 and healthy controls. Patient 3 and control 1 are allogeneic and control 2 is autologous to responders and HLA-DR+ APCs.
Autoantibodies to IFN-α, IL-17 AND IL-22
In view of the autoimmunity characteristically seen in IPEX syndrome and the demonstrated role for anti-IL-17 and anti-IL-22 autoantibodies in CMC, we screened patient sera for autoantibodies to IFN-α, IL-17 and IL-22 as described18, but found none.
DISCUSSION
We describe an IPEX-like clinical disease in five patients with heterozygous gain of function mutations in STAT1. In the initial patients that we identified, CMC was prominent, and together with widespread autoimmunity suggested the underlying molecular defect. In the subsequent patients however, the IPEX-like symptoms were accompanied by no CMC (patient 4) or mild CMC that came to light only after detailed questioning and review of the clinical history. Between the 2 cohorts of IPEX-like patients screened for this study, STAT1 mutations may account for 4% of patients, but it is not clear whether pre-selecting patients with specific clinical or laboratory features may help stratify those most likely to have a STAT1 mutation.
The STAT1 mutations we identified led to prolonged phosphorylation of STAT1 with exaggerated signaling through direct STAT1 homodimeric (IFN-γ), direct heterodimeric (IFN-α) and indirect heterodimeric (IL-6, IL-21) pathways. While low IL-17 producing CD4+ T cell numbers may account for the CMC seen in these patients, they do not account for the extended phenotype of infection, enteropathy, aneurysms, and autoimmunity. Autoimmunity and enteropathy have not been significant aspects of patients with defects in IL-17Rα, IL-17F, or STAT3. However, increased autoimmunity is seen in IFN-α therapy, which may be a phenocopy of the increased IFN-α signaling seen in this condition. While most of our cases had varying degrees of CMC, four had significant bacterial infections and three had recurrent or severe viral infections as well including RSV, HSV, VZV. This broader spectrum of infection may reflect, at least in part, the progressive lymphopenia, loss of memory B cells and hypogammaglobulinemia that appears to emerge over time and may not have been fully developed in the younger children we identified. Respiratory Aspergillus and M. avium complex may were seen as pulmonary pathogens in the context of bronchiectasis, which may not reflect an innate immune defect. The disseminated Aspergillus in Patient 1 may reflect iatrogenic immunosuppression.
Three patients had insulin-dependent diabetes mellitus, suggestive of an autoimmune basis for their loss of insulin production. However, only one patient had demonstrated anti-GAD autoantibodies. Patient 1 had numerous autoimmune complications (type 1 DM, hypothyroidism, Evan’s syndrome), the management of which contributed to his fatal disseminated aspergillosis. Three patients had hypothyroidism and all patients had growth delay with or without growth hormone insufficiency. Other manifestations of autoimmunity seen in these patients were modest compared to those observed in classic IPEX caused by FOXP3 mutations. The metabolic bases of the osteopenia, growth failure and delayed puberty remain unclear, but were significant clinical manifestations that brought these children repeatedly to attention. In addition, we do not have an explanation for the hepatosplenomegaly seen in three patients.
Gastrointestinal manifestations and growth failure contributed to the initial diagnostic consideration of an IPEX-like syndrome. Villus blunting or atrophy was seen in three patients, and a mixed eosinophilic/lymphocytic infiltrate was detected in four patients. The structural and functional defects of palatal dysfunction, esophageal dysfunction and diaphragmatic hernia in Patient 1 were clinically significant and suspected in Patient 2. We follow another STAT1 hypermorphic mutation patient with midgut malrotation and CMC who does not have the IPEX-like phenotype (Holland SM, unpublished), suggesting that gut development may be influenced by hypermorphic STAT1 mutation.
While it is clear that these mutations lead to hyperactivation of STAT1 with enhanced and prolonged responses to IFN-γ and IFN-α, the precise mechanisms by which these mutations lead to autoimmunity, enteropathy, and infection susceptibility are unclear. It is clear, however, that there can be widely variable expression of this disease, since the mutation T385M caused an infantile-onset IPEX-like syndrome in this report, but also predisposed to disseminated histoplasmosis in a different patient described in a companion report 3. The mechanism for the intracellular infection susceptibility appears to be IFN tachyphylaxis, leading to impaired restimulation of the IFN-γ pathway3. Importantly, Treg frequency and function appeared to be intact, in accord with their normal FOXP3 expression. However, it should be noted that our interrogation of Treg function was limited to just the Tregs from these patients and in the absence of STAT1 signaling cytokines. It is conceivable that the Treg suppressor function could be abrogated in the presence of STAT1 signaling cytokines. Likewise, their responder cells or APCs could be more resistant to Treg-mediated suppression.
Although patients with CD25 or STAT5B deficiency can have enteropathy, eczema, type I diabetes and opportunistic infections, IL-2-mediated STAT5 phosphorylation was intact in these patients’ cells. Therefore, the mechanism of their IPEX-like syndrome is likely distinct from those previously identified.
Three patients had obvious cardiac or vascular defects, ranging from cerebral aneurysms, to premature atherosclerosis and renal artery stenosis, to patent foramen ovale, pulmonary hypertension and congestive heart failure. The recent report of a role for IL-17 blockade in development of aneurysms in a mouse model is provocative, since the development of IL-17 producing T cells is impaired in this disease 23.
Gain-of-function mutations in STAT1 lead to a hypermorphic phenotype in vitro, which is clearly capable of a range of manifestations and developmental effects. They regulate numerous immune and possibly extra-immunologic phenotypes, the most common, but not universal, of which remains CMC. These multiple anomalies suggest that STAT1 has a broader role in hematopoietic, gastrointestinal, autoimmune, arterial and cardiac biology than previously appreciated.
Key Message.
STAT1 gain of function mutations can cause a syndrome of enteropathy and endocrinopathy, with or without mucocutaneous candidiasis, that resembles IPEX. However, regulatory T cell function is preserved.
Acknowledgments
Declaration of all sources of funding: This research was supported by the Division of Intramural Research, NIAID, NIH and in part by the National Cancer Institute, National Institutes of Health (contract HHSN261200800001E), Bethesda, MD 20892. The views expressed in this article are those of the authors and do not reflect the official policy of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the US government.
Abbreviations
- STAT1
Signal transducer and activator of transcription 1
- FOXP3
Forkhead box P3
- IPEX
Immune Dysregulation, Polyendocrinopathy, Enteropathy, X-linked
- TREG
regulatory T cell
- CMC
chronic mucocutaneous candidiasis
- GAS
Gamma activating sequence
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Boisson-Dupuis S, Kong XF, Okada S, Cypowyj S, Puel A, Abel L, et al. Inborn errors of human STAT1: allelic heterogeneity governs the diversity of immunological and infectious phenotypes. Curr Opin Immunol. 2012;24:364–78. doi: 10.1016/j.coi.2012.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liu L, Okada S, Kong XF, Kreins AY, Cypowyj S, Abhyankar A, et al. Gain-of-function human STAT1 mutations impair IL-17 immunity and underlie chronic mucocutaneous candidiasis. The Journal of experimental medicine. 2011;208:1635–48. doi: 10.1084/jem.20110958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.van de Veerdonk FL, Plantinga TS, Hoischen A, Smeekens SP, Joosten LA, Gilissen C, et al. STAT1 mutations in autosomal dominant chronic mucocutaneous candidiasis. The New England journal of medicine. 2011;365:54–61. doi: 10.1056/NEJMoa1100102. [DOI] [PubMed] [Google Scholar]
- 4.Sampaio Eea. Human Autosomal Dominant STAT1 Mutations Are Associated With Disseminated Coccidioidomycosis And Histoplasmosis. Journal of Allergy and Clinical Immunology. 2012 accompanying submission. [Google Scholar]
- 5.Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, et al. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nature genetics. 2001;27:20–1. doi: 10.1038/83713. [DOI] [PubMed] [Google Scholar]
- 6.Bennett CL, Ochs HD. IPEX is a unique X-linked syndrome characterized by immune dysfunction, polyendocrinopathy, enteropathy, and a variety of autoimmune phenomena. Current opinion in pediatrics. 2001;13:533–8. doi: 10.1097/00008480-200112000-00007. [DOI] [PubMed] [Google Scholar]
- 7.Moraes-Vasconcelos D, Costa-Carvalho BT, Torgerson TR, Ochs HD. Primary immune deficiency disorders presenting as autoimmune diseases: IPEX and APECED. Journal of clinical immunology. 2008;28 (Suppl 1):S11–9. doi: 10.1007/s10875-008-9176-5. [DOI] [PubMed] [Google Scholar]
- 8.Ochs HD, Gambineri E, Torgerson TR. IPEX, FOXP3 and regulatory T-cells: a model for autoimmunity. Immunologic research. 2007;38:112–21. doi: 10.1007/s12026-007-0022-2. [DOI] [PubMed] [Google Scholar]
- 9.Torgerson TR, Ochs HD. Regulatory T cells in primary immunodeficiency diseases. Current opinion in allergy and clinical immunology. 2007;7:515–21. doi: 10.1097/ACI.0b013e3282f1a27a. [DOI] [PubMed] [Google Scholar]
- 10.Torgerson TR, Ochs HD. Immune dysregulation, polyendocrinopathy, enteropathy, X-linked: forkhead box protein 3 mutations and lack of regulatory T cells. The Journal of allergy and clinical immunology. 2007;120:744–50. doi: 10.1016/j.jaci.2007.08.044. quiz 51–2. [DOI] [PubMed] [Google Scholar]
- 11.Bacchetta R, Passerini L, Gambineri E, Dai M, Allan SE, Perroni L, et al. Defective regulatory and effector T cell functions in patients with FOXP3 mutations. The Journal of clinical investigation. 2006;116:1713–22. doi: 10.1172/JCI25112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Caudy AA, Reddy ST, Chatila T, Atkinson JP, Verbsky JW. CD25 deficiency causes an immune dysregulation, polyendocrinopathy, enteropathy, X-linked-like syndrome, and defective IL-10 expression from CD4 lymphocytes. The Journal of allergy and clinical immunology. 2007;119:482–7. doi: 10.1016/j.jaci.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 13.Sharfe N, Dadi HK, Shahar M, Roifman CM. Human immune disorder arising from mutation of the alpha chain of the interleukin-2 receptor. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:3168–71. doi: 10.1073/pnas.94.7.3168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cohen AC, Nadeau KC, Tu W, Hwa V, Dionis K, Bezrodnik L, et al. Cutting edge: Decreased accumulation and regulatory function of CD4+ CD25(high) T cells in human STAT5b deficiency. Journal of immunology. 2006;177:2770–4. doi: 10.4049/jimmunol.177.5.2770. [DOI] [PubMed] [Google Scholar]
- 15.Kofoed EM, Hwa V, Little B, Woods KA, Buckway CK, Tsubaki J, et al. Growth hormone insensitivity associated with a STAT5b mutation. The New England journal of medicine. 2003;349:1139–47. doi: 10.1056/NEJMoa022926. [DOI] [PubMed] [Google Scholar]
- 16.Bernasconi A, Marino R, Ribas A, Rossi J, Ciaccio M, Oleastro M, et al. Characterization of immunodeficiency in a patient with growth hormone insensitivity secondary to a novel STAT5b gene mutation. Pediatrics. 2006;118:e1584–92. doi: 10.1542/peds.2005-2882. [DOI] [PubMed] [Google Scholar]
- 17.Nadeau K, Hwa V, Rosenfeld RG. STAT5b deficiency: an unsuspected cause of growth failure, immunodeficiency, and severe pulmonary disease. The Journal of pediatrics. 2011;158:701–8. doi: 10.1016/j.jpeds.2010.12.042. [DOI] [PubMed] [Google Scholar]
- 18.Ding L, Mo A, Jutivorakool K, Pancholi M, Holland SM, Browne SK. Determination of Human Anticytokine Autoantibody Profiles Using a Particle-Based Approach. Journal of clinical immunology. 2011 doi: 10.1007/s10875-011-9621-8. [DOI] [PubMed] [Google Scholar]
- 19.Barzaghi F, Passerini L, Bacchetta R. Immune dysregulation, polyendocrinopathy, enteropathy, x-linked syndrome: a paradigm of immunodeficiency with autoimmunity. Front Immunol. 2012;3:211. doi: 10.3389/fimmu.2012.00211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thornton AM, Korty PE, Tran DQ, Wohlfert EA, Murray PE, Belkaid Y, et al. Expression of Helios, an Ikaros transcription factor family member, differentiates thymic-derived from peripherally induced Foxp3+ T regulatory cells. Journal of immunology. 2010;184:3433–41. doi: 10.4049/jimmunol.0904028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McClymont SA, Putnam AL, Lee MR, Esensten JH, Liu W, Hulme MA, et al. Plasticity of human regulatory T cells in healthy subjects and patients with type 1 diabetes. Journal of immunology. 2011;186:3918–26. doi: 10.4049/jimmunol.1003099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tran DQ, Ramsey H, Shevach EM. Induction of FOXP3 expression in naive human CD4+FOXP3 T cells by T-cell receptor stimulation is transforming growth factor-beta dependent but does not confer a regulatory phenotype. Blood. 2007;110:2983–90. doi: 10.1182/blood-2007-06-094656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Piggott K, Deng J, Warrington K, Younge B, Kubo JT, Desai M, et al. Blocking the NOTCH pathway inhibits vascular inflammation in large-vessel vasculitis. Circulation. 2011;123:309–18. doi: 10.1161/CIRCULATIONAHA.110.936203. [DOI] [PMC free article] [PubMed] [Google Scholar]