

Abstract
Nanoparticles have received enormous attention as a promising tool to enhance target-specific drug delivery and diagnosis. Various in vitro and in vivo techniques are used to characterize a new system and predict its clinical efficacy. These techniques enable efficient comparison across nanoparticles and facilitate a product optimization process. On the other hand, we recognize their limitations as a prediction tool, which owe to inadequate applications and overly simplified test conditions. This article provides a critical review of in vitro and in vivo techniques currently used for evaluation of nanoparticles and introduces emerging techniques and models that may be used complementarily.
Keywords: Nanomedicine, nanoparticles, particle characterization, in vitro, animal models
1. Introduction
The field of nanomedicine has seen significant progress in the past decades, both in design and the scope of applications. Various techniques are used to characterize nanoparticles (NPs) and predict their ultimate fates in human body. However, current technology is challenged in a sense that the characterization is often performed in a condition that does not reflect the complexity of physiological environment. Moreover, in vivo studies based on animal models largely remain a black box approach, where pharmacokinetics and biodistribution of NPs are driven by a series of biological events that are not readily predicted in vitro. In order to expedite the transition of a bench-top effort to a clinically effective product, it is imperative that investigators employ adequate methodologies to characterize nanomedicine, correlate their effects and biological consequences, and predict the therapeutic outcomes in clinical subjects in the early stage of product development. The purpose of this review is to overview current techniques used in NP evaluation from a critical perspective, discuss potential pitfalls and cautions, and introduce emerging technologies that deserve keen attention from the field of nanomedicine.
2. In vitro characterization of NPs
2.1. Physical properties
2.1.1. Particle size
Particle size is the most basic information of NPs, one of the main determinants of biodistribution and retention of the NPs in target tissues. Dynamic light scattering (DLS) is commonly used for the particle size determination. DLS measures Brownian motion of NPs in suspension and relates its velocity, known as translational diffusion coefficient, to the size of NPs according to the Stokes-Einstein equation.1 The particle size is defined as the size of a hypothetical hard sphere that diffuses in the same fashion as that of the NPs being measured. The result is reported as a mean particle size and homogeneity of size distribution. The latter is expressed as polydispersity index (PDI), a dimensionless parameter calculated from a cumulants analysis of the DLS-measured intensity autocorrelation function.2 A PDI value from 0.1 to 0.25 indicates a narrow size distribution, and a PDI value greater than 0.5 does a broad distribution.3 While DLS provides a simple and speedy estimate of the particle size, several studies suggest inherent limitations of DLS. For example, DLS is relatively poor at analyzing multimodal particle size distribution.3, 4 For example, when a mixture of 20 nm and 100 nm NPs is measured, the signal of smaller particle is lost because the intensity of a spherical particle with a radius r is proportional to r6; thus, the scattering intensity of small particles tends to be masked by that of larger particles.
Microscopy provides an accurate assessment of the size and shape of an NP; however, it often requires complicated sample preparation steps specific to microscope techniques,4 which can change samples and create artifacts (e.g., NP agglomeration during drying process for electron microscopy5). Moreover, due to the limited throughput, it is difficult to obtain particle size distribution.6 Another imaging-based method is the NP tracking analysis (NTA), a single particle tracking technique based on dark field or fluorescence microscopy and automatic imaging analysis.7 In this method, NP size is derived from the average displacement of NPs undergoing Brownian motion within a fixed time frame.7, 8 An advantage of this method is that it tracks individual NP and thus provides a high resolution for multimodal samples and aggregation.7 On the other hand, it requires a sample to be sufficiently diluted so that the observation fields may not be overly crowded.5 Alternatively, NP size may be estimated by disc centrifugation, which depends on sedimentation speed of NPs. Since NPs with a few % size differences settle at significantly different rates, the disc centrifugation method can resolve a very small size difference (as little as 2%).9 Moreover, this method can analyze a broad range of particle size, ranging from 5 nm to 75 μm. On the other hand, it takes longer than other methods, lasting 15 to 30 minutes, and requires that NPs be denser than the suspended fluid.9
Since these techniques rely on different physical principles and sample preparation, the results vary according to the employed methods.5 For example, electron microscopy, DLS, NTA, and disc centrifugation gave rise to highly variable results even for the well-defined, homogeneous NPs. Depending on the methods and the type of averages reported (intensity, number, or volume), silver NPs (70 nm) and gold NPs (15 nm) were measured as 40 nm–124 nm and 11 nm–52 nm, respectively.5 In addition to the underlying principles, it should be considered that sample status in each method is not the same. For example, the size of NPs measured in solution is generally much larger than the dried NPs because of the hydration layer.
The measured particle size can also be different depending on how the samples are prepared even in the same method. For example, in DLS measurement, it is critical to ensure that the NPs are well dispersed. NPs are typically dispersed via probe/bath sonication or vortex mixing. A high energy dispersion method can temporarily reduce agglomeration, but the NPs do not remain dispersed for a long time and agglomerate again.10 Therefore, it is often observed that the increased duration of sonication and/or higher energy sonication method ultimately promote agglomeration after initial dispersion due to the enhanced interaction of NPs with high surface energy.10–12 Ionic strength of the NP suspension is another important factor.11 When TiO2 NPs or quantum dots were analyzed, an increase in ionic strength from 0.001M to 0.1M resulted in 50-fold increase in the hydrodynamic diameter.11 This is because the increasing ions shield the electrical layer on NPs that has kept the NPs apart at a lower ionic strength. The pH of the NP suspension also plays a role in particle size measurement.11 When pH is distant from the isoelectric point of NPs, the electrostatic repulsive force is dominant over the van der Waals force, and NPs are well dispersed. On the other hand, the repulsive force decreases and the hydrodynamic size increases, when the pH is close to the isoelectric point and, thus, the NP surface is less charged. Due to the dependence on pH and ionic strength, the size distribution in a condition where the bioactivity of NPs is tested is quite different that measured in water. Murdock et al. measured sizes of various inorganic and organic NPs in water or cell culture medium (with or without serum) with DLS.10 In many cases, NPs aggregated to a greater extent in serum-free medium than in water.10 The presence of serum proteins attenuated the size increase, likely due to surface stabilization by the adsorbed proteins. Therefore, it is recommended that the conditions in which NP size is measured be recorded when DLS is used for size measurement.
Additional cautions are needed in measuring sizes of NPs with non-spherical shape. DLS assumes spherical shape for NPs; therefore, it is important to validate this assumption via microscopic examination. When the shape significantly deviates from a sphere, the DLS measurement may be less accurate; thus, image analysis must be accompanied.13 It is also noteworthy that the particle size can differ by a factor of 2 to 4 depending on the type of particle size distribution used in DLS (i.e. intensity, volume, and number-based); therefore, one should report the type in addition to the size.5
2.1.2. Surface charge
Surface charge, expressed as zeta potential, critically influences the interaction of an NP with the environment.3 There are two liquid layers surrounding an NP: strongly bound inner part (Stern layer) and weakly bound outer layer. Zeta potential is commonly measured by laser Doppler electrophoresis, which evaluates electrophoretic mobility of suspended NPs in the medium, thus measuring the potential at the boundary of the outer layer. Generally, particles with zeta potential more positive than +30 mV or more negative than −30 mV have colloidal stability maintained by electrostatic repulsion. One limitation is that in bimodal samples the zeta potential value of larger particles dominates the scattering signal of smaller particles, similar to DLS size measurements.10
The zeta potential measurement depends on the strength and valency of ions contained in the NP suspension. High ionic strength and high valency ions compress the electric double layer, resulting in reduction of the zeta potential.14, 15 The pH, the concentration of hydrogen ions in the medium, greatly influences the zeta potential as well. When the suspension is acidic, the NPs acquire more positive charge, and vice versa. Therefore, a zeta potential value without indication of solution pH is a virtually meaningless number.1 It is recommended that information of the NP suspension be precisely described in reporting the zeta potential, including the ionic strength, composition of the medium, and the pH.16, 17 For comparison of results across different studies, it is conceivable to normalize the zeta potential by pC (negative logarithm of concentration of counterion species).17
2.1.3. Drug release kinetics
When NPs are used for the delivery of a drug, the ability of NP to release the drug is evaluated over a period of time, since it ultimately determines the availability of a drug at target tissues, thereby the therapeutic outcomes.3 There are three possible mechanisms of drug release: desorption of the surface bound/adsorbed drug, diffusion from the polymer matrix, and release subsequent to polymer erosion. In the case of a matrix-type polymer NP, in which the drug is uniformly distributed in the matrix, the release occurs by diffusion and/or erosion of the matrix. If the diffusion occurs more rapidly than matrix degradation, diffusion is likely to be a main mechanism of drug release. Rapid initial burst release is attributed to the fraction of the drug adsorbed or weakly bound to the surface of the NPs.18
Drug release from NPs is studied in at least three ways: sampling and separation, dialysis membrane diffusion, and in situ analytical technique.19 In the sampling and separation technique, the released drug is separated from NPs by filtration, centrifugation, or centrifugal filtration, and quantified by various analytical methods. The NPs are supplemented with fresh release medium, resuspended, and incubated further for next sampling. While this method can be performed with a small amount of samples and simple analytical equipment, several shortcomings exist. Irrespective of the separation methods, the process is slow and inefficient, which make them less suitable for studying NPs that rapidly release a drug. Moreover, centrifugal force or shear stress during filtration required for NP separation, which becomes increasingly strong when NPs are relatively small, can alter the NPs and the release kinetics. Dialysis membrane diffusion depends on continuous diffusion across the dialysis membrane filtration. Advantages of this method are that the NPs are not subject to invasive separation processes and sampling is quick and simple. On the other hand, the dialysis membrane can attenuate the drug release as a diffusion barrier or an adsorptive surface; therefore, a control experiment with a free drug should be accompanied to account for the membrane effect. The dialysis membrane diffusion method typically employs a large volume of release medium. While the large volume helps maintain a sink condition for drug release, drug analysis may become difficult due to the low concentration. In situ analytical technique is useful for studying nanocrystals, which is made almost exclusively of a drug. This technique analyzes the properties of NPs in situ to determine the quantity of the released (dissolved) drug indirectly. Various analytical techniques, including electrochemical analysis, solution calorimetry, or turbidimetric method, and light scattering technique, are employed for this purpose.19 This technique does not need NP separation and enables real-time assessment of the release kinetics. However, it is limited in determining the integrity of the released drug.
2.2. Prediction of physical and chemical stability of NPs
Maintaining NP stability in the bloodstream is a crucial requirement for successful drug delivery to target tissues. The fate of NPs in vivo is in large part determined by its ability to maintain the size, to retain drug payload external to the target tissues, and to properly release drug to the cells. Ideally, a NP must remain stable (i.e., resist aggregation or degradation and retain drug) in the blood until it reaches the target sites. Instability of NPs results in altered biodistribution and premature drug release, thereby compromising the efficacy of the delivery system. Hence, evaluation of NP stability is an important aspect of NP characterization and an essential component to the success of the system. This section reviews commonly used techniques to investigate NP stability in various biologically relevant media in vitro (Fig. 1). Well-established studies of micelles or similar self-assembled NP systems are mainly used as examples. Although the NP stability needs to be ultimately measured in vivo, these techniques provide reasonable prediction of NP stability in a physiological environment.
Figure 1.

Techniques used for prediction of physical and chemical stability of various NPs. Techniques are listed in the order of required time and resources (top to bottom: least to most).
2.2.1. Determination of critical aggregation/micelle concentration
The critical association or aggregation concentration (CAC) or critical micelle concentration (CMC) can be used to evaluate the stability of self-assembled NP systems including polymeric or surfactant micelle systems. The CAC, or CMC, is defined as the concentration at which a self-assembled particle or micelle associates/dissociates. This value provides a quantitative measure of physical stability of NPs. A relatively low CAC/CMC indicates a more stable micelle system than one with a high CAC/CMC. In other words, NPs with a low CAC/CMC is more likely to resist dissociation upon dilution in the blood.
CMC can be measured using a variety of different detection methods, such as conductivity, chromatography, surface tension, fluorescent probes, and light scattering. When measuring CMC using surface tension, the CMC is defined as the concentration of a surfactant (i.e., an amphiphilic polymer) above which the surface tension becomes constant. At concentrations below CMC, a surfactant has not yet saturated the surface and lends itself to reduce surface tension of the solution. On the other hand, at concentrations above CMC, this saturation has occurred, and the excess surfactants form micelles and do not contribute to the surface tension change. Many studies have utilized this method to determine the CMC of micelles.20–23 Another commonly used method to measure CAC/CMC is to utilize fluorescence probes, such as pyrene, as an indicator of micelle dissociation. Pyrene is a hydrophobic aromatic hydrocarbon, which partitions in the hydrophobic domain of self-assembled NPs during assembly.24 When a NP dissociates, pyrene is exposed to water, where it shows a different fluorescence profile than when in the hydrophobic domain of the NP. Therefore, the CAC/CMC can be determined by monitoring the change in fluorescence profile of pyrene, defined as the concentration at which a drastic band shift is observed. The pyrene technique has widely been used as an indicator of relative micelle stability.25–29 In addition, light scattering is used to determine CAC/CMC. This technique measures the count rate (the intensity of scattered light in DLS), which is proportional to the number of NPs in solution when NP size is constant.30 The count rate is plotted with respect to NP concentration, and the CAC/CMC is defined as a concentration above which the count rate shows a linear increase with concentration of the components of NPs.30 The CAC/CMC measurement is a relatively simple and sensitive method of evaluating NP stability, but a disadvantage is that the application is limited to micelles and self-assembled NPs, whose formation is influenced by concentrations of the components.
2.2.2. Determination of low critical solution temperature
Temperature-sensitive micelle systems composed of a co-polymer of hydrophobic block and thermosensitive block can utilize lower critical solution temperature (LCST) as a measure of their stability. The LCST is defined as the temperature at which phase transition of a thermosensitive polymer occurs (from hydrophilic to hydrophobic with increase in temperature).31 This phase change provides the system with the ability to release a drug in response to an external thermal stimuli at a specific temperature via local and controlled hyperthermia in a specific region of the body.32 At temperatures below LCST, the polymer is amphiphilic and the drug remains encapsulated in micelles; however, at temperatures above LCST, the thermosensitive block becomes hydrophobic, destabilizing the micelle structure and releasing the drug.33 As an inherent property of a thermosensitive polymer, LCST can be utilized in comparing the stability of NPs based on such polymers. For example, LCST of poly(N-isopropylacrylamide-co-maleic anhydride) copolymer increased from 31.1 to 45°C as the content of maleic anhydride and molecular weight increased.34 Conversely, LCST of Pluronics and poly(N-isopropylacrylamide) decreased when mixed with saccharides.35 One may expect that NPs based on a polymer with a relatively high LCST will be more resistant to thermal dissociation.
2.2.3. Monitoring NP size or turbidity of NP suspension
Changes in NP size can be used to predict the stability of most NPs. Assuming that the assembled NPs form within a constant size range, deviations from the average NP size range can be interpreted as an indication of NP dissociation or instability in that particular environment or concentration. In one study, the stability of different NPs in 10% bovine serum albumin (BSA) and 10% human plasma solution was studied by monitoring their size change.36 Here, an increase in NP size provides evidence of protein adsorption, and therefore, potential instability in vivo. A similar study evaluated micellar NP stability by incubating the NPs with 5% BSA and measuring the size using DLS.37 Changes in turbidity of NP suspension may also be used as an indication of instability. For example, one study monitored the change in absorbance at 550 nm of NPs in a suspension and utilized the kinetics of absorbance decay (decomposition of NPs) as an indicator of NP stability.38 In another example, turbidity of a sulfonamide-containing hydrogel NP suspension was measured at different pHs to study the pH-sensitive aggregation behavior of the NPs.39 The NPs showed constant particle size and turbidity at pH 7 or higher but increased turbidity and size at pH below 7, indicative of NP aggregation due to hydrophobic interactions of the deionized polymer.39 These methods may be used for virtually any types of NP systems and performed with basic analytical equipment. However, they take into account any materials present in the medium; thus, it is difficult to monitor the stability of NPs in a complex fluid that contains additional components such as serum proteins.
2.2.4. Gel permeation chromatography
Gel permeation chromatography (GPC) can be used to determine the physical stability of self-assembled NPs. This technique separates self-assembled NPs from degraded NPs or their components. NP stability can be estimated based on the elution times, given that degrading NPs or their components are eluted later than intact NPs. Yokoyama et al. used GPC to study the formation of different polymeric micelles and their stability in aqueous media.40 In subsequent studies, GPC was used to study the stability of drug-loaded micellar structures of different compositions in the presence of serum41, 42 or purified serum albumins.42 This technique was also used to assess the formation and dissociation of an insulin-hydrophobized pullulan NP assemblies.43 The assemblies showed high colloidal stability in water and buffer, but insulin was released rapidly from the assemblies upon the addition of bovine serum albumin.43 Many other studies have also used GPC as a means to evaluate stability of self-assembled NPs in the presence of serum proteins.44–48 While this technique is straightforward, interactions between the column beads and NPs may affect the outcome of the analysis.
2.2.5. Forster Resonance Energy Transfer technique
Recently, Forster Resonance Energy Transfer (FRET) has been employed to study the stability of NPs at the molecular level. Cheng and colleagues encapsulated a FRET pair, consisting of hydrophobic fluorescent probes DiO (donor) and DiI (acceptor), in a polymeric micelle to study the stability of micelles.49 The FRET pair retained in the hydrophobic core of the micelles shows a FRET signal due to their proximity to each other, whereas the FRET signal disappears as the micelles dissociate and release the dyes. Using this phenomenon, micelle stability during cellular uptake has been studied.49 When FRET dye-loaded micelles were incubated with KB cells for 2 hours, a strong DiO signal was observed on the plasma membrane, indicating that the dyes were already released from the micelle core while passing the cell membrane.49 Following internalization, a FRET signal was partially restored, suggesting the two dyes were trafficked to and concentrated in the same endosomal vesicles.49 The FRET technique was used to study the stability of NPs in vivo as combined with intravital microscopy.50 Many other studies have utilized FRET in evaluating the stability of the NP systems in vitro or in vivo.25, 48, 50, 51 One challenge in FRET analysis is the need for technical adjustment to avoid optical artifacts that may interfere with FRET detection. For example, an acceptor dye can be excited directly with light that is supposed to excite the donor.52 Alternatively, fluorescence from the donor can leak into the detection channel of the acceptor fluorescence (bleed-through).52 For accurate assessment of FRET signals, several optical corrections need to be made to account for these issues. Furthermore, for NP systems that require covalent labeling of NP-dye, this conjugation may affect the formation or chemical conformation of the NPs, thus potentially changing its properties.
2.3. In vitro prediction of in vivo fates of NPs
Once NPs are introduced into the circulation, plasma proteins almost instantaneously adsorb to the surface. The protein corona changes the nature of the NPs and induces sequential immune responses, leading to uptake by phagocytes of the reticuloendothelial system (RES) and rapid clearance from the circulation.53, 54 Therefore, it is important that NPs resist the adsorption of plasma proteins and premature clearance by the immune system.55 Protein adsorption to the NP surface is mainly influenced by the hydrophobicity and surface among others.56 Typically, hydrophilic and electrically neutral NPs are less likely to engage with serum proteins; therefore, polyethylene glycol (PEG) is widely used to decorate the NP surface (‘PEGylate’) and prevent binding of plasma proteins to the NPs. The PEGylated stealth NPs thus acquire a longer half-life in circulation,55, 57, 58 which translates to greater accumulation in solid tumors via the enhanced permeability and retention (EPR) effect.59, 60 On the other hand, an accelerated blood clearance of PEGylated liposomes has been reported following the injection of the first dose in animals,61 mediated by the production of anti-PEG IgM.62 Clinical studies shows an occurrence of anti-PEG antibodies in human subjects following the treatment with PEGylated agents.63 Therefore, alternative stealth coatings are investigated to circumvent the immunogenicity of PEG, such as polysaccharides including dextran, heparin, and low molecular weight chitosan, or synthetic polymers.55, 64–66
2.3.1. Protein adsorption
Given the immunogenicity of an NP has only been detected in costly in vivo studies, there has been increasing interest in predicting the immunological responses to NPs in earlier phases of NP development, preferably in vitro. One of the basic techniques to study the stealth properties of NPs is to examine the extent of protein adsorption on NP surface, the first step of phagocytic removal of NPs.67–69 To assess the extent of protein adsorption, NPs with constant surface area are incubated in serum for a period of time and washed with water to remove proteins loosely adsorbed to the surface. The proteins bound onto NPs are desorbed with a surfactant like sodium dodecyl sulfate and subject to gel electrophoresis and/or quantitative protein assay. Attempts have been made to correlate protein adsorption to NP surface and in vivo fate of the NP.55 For example, polystyrene NPs coated with a series of amphiphilic polyethylene oxide-polypropylene oxide block copolymers showed much reduced protein adsorption in vitro and a prolonged circulation in rats.55 However, protein adsorption alone provides only a rough prediction of the potential immunogenicity, as it does not reflect the complex nature of subsequent immune reactions leading to elimination of NPs.
2.3.2. Phagocytic uptake
Another technique to predict the fate of NPs in blood is to measure the degree of phagocytic uptake of NPs by incubating fluorescently-labeled NPs with macrophages for a certain period of time and quantifying the amount of NPs taken up by the phagocytes.70–72 The size and surface properties are important determinants of phagocytosis.73 According to a study with microparticles, their shape at the initial contact with a phagocyte is shown to be critical in the phagocytosis process.74 The shape has similar importance in NPs.75, 76 For example, linear polymer micelles (filomicelles) had a longer circulation time than their spherical counterparts with similar chemistry due to their resistance to phagocytic uptake in flow.77 Similarly, PEGylated gold nanorods, as compared to spherical NPs, were taken up by macrophages to a lesser extent and showed a longer circulation time upon injection in mice.75 One limitation of the phagocytosis assay is that it is carried out in cell culture medium, which does not completely resemble the concentration and composition of proteins in blood; thus, the extent of protein adsorption and phagocytosis can be underestimated.
2.3.3. Complement activation
Adsorption of a group of soluble plasma proteins, also called as the complement system, on NP surface initiates a biochemical cascade leading to NP clearance from the circulation via complement receptor-mediated phagocytosis.54 The degree of complement system activation can be measured to predict the ability of NPs to evade or elicit the phagocytic clearance. As foreign particles trigger the system activation, one of the soluble protein components, C3, is cleaved into C3b and C3a. Therefore, the ratio of C3b to C3 is determined as a measure of the extent of complement activation by NPs, via crossed immunoelectrophoresis of serum solution incubated with NPs.78–81 The complement system activation assay has been used to analyze the effect of chain length, conformation, charge, and composition of a surface-decorating polymer on its stealth functions.73, 82–84 When chitosans with different chain lengths (8.8 to 80 kDa) were compared, complement activation increased with chain length and number of NH2 groups.85 Difference in the conformation of polymer chains on the NP surface can dramatically influence the ability to bind to plasma proteins and activate the complement system. For example, polyalkylcyanoacrylate (PACA) NPs with dextran coating prepared in two different methods had two types of dextran conformation and density, which had opposite complement activation effects.82 Dextran chains bound forming flexible “loops” on the NP surface had a strong complement activation effect, but dense “brush” like conformation showed resistance to protein adsorption.82 Similar observations were reported with poly(isobutylcyanoacrylate) (PIBCA) NPs coated with dextran or chitosan.78 Vauthier et al. reported that the complement activation effects of PIBCA NPs with different dextran coatings did not necessarily correlate with their albumin binding, indicating that each protein interacts with surface coatings uniquely according to its size, conformation, and flexibility.83
2.3.4. NCL protocols
The Nanotechnology Characterization Laboratory (NCL) has published a series of in vitro protocols specifically designed for the evaluation of nanomaterials compatibility with various biological environments and the immune system, which include tests of blood coagulation, complement activation, protein binding, platelets aggregation, and phagocytosis due to nanomaterials.86 Although no in vitro test may exactly mimic real physiological conditions in vivo, combinations of these approaches can help predict in vivo behaviors of NPs.
3. Cell-based evaluation of NPs
Once NPs are characterized with respect to their physical and chemical properties, their biological effects are tested in cell culture models prior to in vivo applications. This section describes widely-used cell models, their advantages and weaknesses.
3.1. Two-dimensional (2D) monolayer cell culture
In frequently used 2D cell culture, cells are grown as a monolayer on a plate or flask surface, which is treated via physical methods or adhesive biological materials to encourage cell attachment. The cells are bathed in culture medium supplemented with nutrients and grown at 37°C in a humidified environment that provides uniform exposure to oxygen (and carbon dioxide). These conditions provide minimum requirements for maintaining cell viability. As a consequence of convenience, the monolayer cell culture model is extremely beneficial for quick determination of cellular uptake and intracellular trafficking of NPs, bioactivity of drugs delivered as NPs, and toxicity of the vehicles. These studies are usually done with multiple established cell lines.
3.1.1. Cellular uptake
Confocal microscopy and flow cytometry are widely used to study the cellular uptake of NPs. These methods require that NPs be labeled with a fluorescent marker, which is done by physical entrapment or covalent conjugation. While the former has the advantage of simplicity, one should be aware that a lipophilic dye may leach out of NPs upon contact with amphiphilic or lipophilic components and misrepresent NPs.87 When the intention is to track a vehicle, it is desirable to label the component by covalent conjugation of a dye and confirm the stability of the conjugation in a solution similar to physiological fluid. Ideally, a drug and a vehicle should be separately labeled so that drug delivery attributable to the vehicle may be accurately evaluated.
Ekkapongpisit et al. studied the potential of silica (10 nm, no surface modification) and polystyrene NPs (30 nm, carboxyl surface modification) as theranostic agents in the treatment of ovarian cancer.88 Cellular uptake of the fluorescently labeled NPs were studied with OVCAR-3 and SKOV-3 ovarian cancer cell lines using fluorescence microscopy.88 Initially mesoporous silica NPs were associated along nuclei, with subsequent diffusion into the cytoplasm.88 Polystyrene NPs were observed as punctate signals restricted to cell peripheries, which disappeared in 120 min.88 At subtoxic levels of the NPs, mesoporous silica NPs showed faster cellular uptake and longer intracellular retention than polystyrene NPs.88
While confocal microscopy helps localize NPs within cells, quantitative analysis of NP uptake relies on flow cytometry. In flow cytometry, cells in suspension are passed through an interrogation point, where the cells are individually examined by a laser with respect to their optical or fluorescent properties.89 Quantitative information is acquired based on the number of fluorescent cells or an average fluorescence intensity of the cell population and used to determine the fraction of cells killed by therapeutic treatments or the amount of a fluorescent drug internalized by the cells. In one example, magnetic NPs (MNPs) were used as a drug carrier to tumor cells.90 Iron oxide (Fe3O4) was covered with carboxymethyl chitosan (CMCS),90 in which montmorillonite (MMT) was intercalated to enhance cellular uptake of the NPs.90, 91 A lipophilic fluorescent dye, coumarin-6, was covalently intercalated to MMT to label the CMCS/MMT-covered MNPs for a cellular uptake study.90, 92 Flow cytometry was used in quantifying the uptake of the coumarin-6 labeled CMCS/MMT-MNPs by HeLa cells.90 The researchers observed that cellular uptake of their delivery system increased with increasing MMT content.90 On the other hand, the amount of NPs associated with the cells did not increase in proportion to the NP concentration, indicating that the cellular uptake was a saturable process.90 Gratton et al. used flow cytometry to evaluate cellular uptake kinetics of PEG hydrogel particles with different sizes and shapes in HeLa cells, prepared with a lithographic fabrication (Particle Replication In Non-wetting Templates, PRINT) technique.93 This study found that submicron particles were taken up by HeLa cells to a greater extent than microparticles and the high aspect ratio (height: diameter = 3:1) particles showed a greater rate and extent of cellular uptake than the low aspect ratio (1:1) particles with a comparable volume.93
Alternatively, cellular uptake can be quantified by direct measurement of intracellular drug or dye contents. Here, cells are destroyed at the end of a treatment to release the internalized drug. For example, in the evaluation of doxorubicin (DOX)-loaded polymeric micelles, SiHa human cervical tumor cells were incubated with the micelles or a free drug for various time periods, washed, and extracted with dimethyl sulfoxide (DMSO).94 DOX content in the cell extract was determined according to the fluorescence intensity.94 This study found no difference between free DOX and DOX-loaded micelles in cellular uptake at each time point but significant difference in cytotoxicity (concentration for 50% cell death (IC50): DOX < micellar DOX), which was interpreted as delay in DOX release from the micelles.94
3.1.2. Mechanisms of cellular uptake
The mechanism by which NPs enter cells is as much important as the quantity of the internalized NPs because the subsequent intracellular events are dependent on the uptake pathway. Depending on their physicochemical properties, NPs can enter cells via various pathways.95–98 For example, particles with a size ranging from a fraction of a micron to ~10 μm depend on phagocytosis, performed by specialized phagocytic cells.96, 99 Smaller NPs may be taken up by macropinocytosis,96, 99, 100 clathrin- or caveolae-mediated endocytosis.95, 96, 101–103 Cells can internalize NPs up to 300 nm in diameter by macropinocytosis, where the cell membrane protrudes and fuses back with another part of the membrane to produce large vesicles around the NPs.5,9 Clathrin-mediated endocytosis occurs as clathrin proteins in the cell membrane polymerize and form a vesicle (~100 nm) around a NP, which is then transported to an early endosome.96 Some NPs may utilize a caveolar route,103 where the cell membrane is coated with caveolin along with cholesterol and lipids and forms a flask-shaped invagination called caveolae.96 In particular, clathrin- or caveolae-mediated endocytosis involves cellular receptors for specific ligands,100 such as folic acid,104 transferrin,105 or albumin,106 which facilitate endo- or transcytosis of these molecules. For this reason, NPs incorporating these ligands have been widely explored as a way of achieving cell-specific NP delivery.
For studying the NP uptake pathway, cells are treated with specific inhibitors of specific internalization pathways107–109 prior to incubation with fluorescently labeled NPs. Chlorpromazine is an inhibitor of clathrin-mediated endocytosis, and filipin and methyl-β-cylcodextrin (MbCD) are inhibitors of caveloae-mediated endocytosis.107 Macropinocytosis and phagocytosis can be inhibited by pre- treatment with amiloride (inhibitor of Na-K exchange) or cytochalasin D (F-actin-depolymerizing drug).107 Following the pre-treatment, cells are cultured with NPs and analyzed with flow cytometry or confocal microscopy combined with quantitative imaging analysis software to determine the sensitivity of the NP uptake to each inhibitor. For instance, the uptake pathways of mesoporous silica and carboxyl-terminated polystyrene NPs were compared by investigating their responses to pre-treatment of MbCD.88 MbCD has high affinity for cholesterol and forms inclusion complexes with cholesterol when added to cells at 5–10 mM.107 This way, MbCD removes cholesterol from the plasma membrane, interfering with cholesterol-dependent uptake pathways such as carveolae-mediated endocytosis.107 The two NPs showed opposite responses to the MbCD treatment. The uptake of mesoporous silica NPs was hindered by the MbCD treatment, whereas that of polystyrene NPs did not change, indicating that mesoporous silica NPs, but not polystyrene NPs, were taken up via caveolae-mediated endocytosis.88
The cellular entry of NPs incorporating specific ligands, the so-called targeted NPs, mirrors the interaction between the ligands and corresponding receptors.110 Moreover, NPs act as a scaffold on which multiple ligands are concentrated, thus enabling simultaneous interactions with multiple receptors on the cells (i.e., multivalent effect).110 As a result, the binding strength of ligand-modified NPs to cell receptors is often orders of magnitude higher than that of free ligands.111, 112 Several studies demonstrate that the NPs modified with receptor-specific ligands achieve a greater cytotoxicity than non-modified ones due to the enhancement of cellular binding and uptake. For example, Liu et al. produced folate-receptor targeted polymeric micelles, where folic acid was conjugated to the hydrophilic block.113 Cellular uptake of the targeted micelles by mouse breast cancer (4T1) and human epidermal carcinoma (KB) cells was significantly enhanced by the presence of folic acid on the micelles compared to the untargeted micelles.113 Consequently, the folate- targeted micelles carrying DOX were more cytotoxic than untargeted NPs due to folate-receptor mediated endocytosis.113 A similar result was reported with another folate-receptor targeted micelle based on a different polymer.114 In addition to naturally overexpressed targets, cells may be pre-treated to induce overexpression of specific receptors, such as p32 receptors that are upregulated by thermal treatment. Park et al. reported that magnetic nanoworms and DOX-loaded liposomes, decorated with a peptide ligand targeted to p32 receptors, showed increased binding and internalization by MDA-MB-435 human carcinoma cells, which were pre-heated with gold nanorods to induce expression of p32 receptors.115
Once NPs targeted to specific cellular receptors are developed, it is important to confirm whether the cellular uptake is indeed mediated by the intended receptor-ligand interactions. One common way is to compare cellular uptake of the targeted NPs in cells that express the specific receptors to different degrees. For example, Kim et al. produced PLGA NPs targeted to folate receptors and compared their uptake in KB cells (folate receptor overexpressing cell line) and A549 lung cancer cells (folate receptor-deficient cell line) to find that the NP uptake was much higher for KB cells than for A549 cells.116 Additionally, cells are pre-treated or co-incubated with free ligands in addition to the targeted NPs to investigate whether the NP uptake is competitively inhibited. If the NP uptake is receptor-mediated, cellular uptake and/or bioactivity of a drug delivered by the NPs is diminished by the presence of excess free ligands in a concentration-dependent manner.113, 117, 118
In addition to the presence of ligands, several other factors affect the endocytic pathway that NPs take to enter a cell.95, 119, 120 Particle size has a direct influence on NP uptake pathway. It is assumed that NPs carried via the receptor-mediated endocytic pathways have average hydrodynamic diameters close to the sizes of vesicles formed during clathrin- or caveolae-mediated endocytosis, which are 100 nm or 60 nm, respectively. 96, 101 Macropinocytosis has a greater flexibility in the upper limit of particle size.96 Particle size has an additional role in targeted NPs as a main determinant of ligand density on the NP surface.110, 112, 119 A small particle has a high surface curvature that limits relative orientation between ligands, leaving large background area without ligand coverage.112 Relatively large NPs can have a higher ligand density on the surface, but if the membrane cannot catch up with the high demand for receptors within the area of binding, NP uptake is also limited.112 Therefore, Jiang et al. concludes that 40 – 50 nm is an optimal size for receptor-mediated endocytosis.112 According to Gratton et al., the cellular uptake pathway is also influenced by the particle shape.93 They used the PRINT technique to produce particles with different aspect ratios and observed that the particles had different sensitivity to inhibitors of various endocytosis pathways.93 Another factor to influence endocytic pathway is the surface charge of NPs. Typically cationic NPs are internalized more readily than anionic ones, due to the ability to interact with negatively-charged cell membrane and clathrin-coated pits in the membrane.95, 96, 110, 119, 121
3.1.3. Intracellular trafficking
Once a NP is internalized by cells, its intracellular fate critically influences its therapeutic effect, especially when the drug target is localized in a particular organelle and/or the drug is unstable in a specific intracellular environment (e.g., acidic pH or lysozyme in late endo/lysosomes). To track the intracellular trafficking of the NPs, markers of intracellular organelles are co-localized with NPs and observed over a period of time. Alternatively, the organelles are located using fluorescently labeled antibodies after fixation and permeabilization of cells. One pitfall of the latter technique is potential artifacts resulting from the fixation/permeabilization process, such as protein extraction or relocalization.122, 123
In the study of mesoporous silica and polystyrene NPs discussed earlier, the NPs were incubated with cells which were pre-labeled with LysoTracker, a fluorescent probe that accumulates in acidic organelles.88 The mesoporous silica NPs and the LysoTracker signals colocalized in 5 min, indicating the residence of silica NPs in lysosomal vesicles. With time the fluorescence of the silica NPs and LysoTracker signals separated, which suggested the escape of NPs from the acidic vesicles.88 On the other hand, carboxyl-terminated polystyrene NPs did not show colocalization with LysoTracker signals at any time, indicating their residence in recycling vesicles.88 This result is consistent with the limited intracellular accumulation of the polystyrene NPs.88
3.1.4. Bioactivity of NPs
When a NP is developed for drug delivery, it is of utmost interest whether the potency and efficacy of a drug are changed and/or target specificity of the drug is enhanced due to the delivery system. When an anti-cancer drug is delivered via NPs, various methods measuring the metabolic activity or cell membrane integrity are used to estimate the viability of the treated cells. For example, colorimetric assays such as MTT, MTS, and XTT assays measure mitochondrial function of live cells, according to the ability to reduce these tetrazolium salts to intensely colored formazan dyes.124 Bioluminescence assays measure ATP produced by live cells using luciferase. Since luciferase metabolizes luciferin in an energy-dependent manner, the luciferase activity (luminescence intensity) is proportional to the amount of ATP, i.e., cell viability.124 Dye/stain exclusion assays utilize chemicals such as trypan blue, propidium iodide, and calcein-AM, which are selectively excluded from or trapped in live cells according to membrane integrity or esterase activity. Lactate dehydrogenase (LDH) assays also reflect the integrity of the cell membrane. LDH is a constitutive cytoplasmic enzyme, which is released when the cell membrane is compromised. Therefore, the LDH activity in cell medium indicates the proportion of non-viable cells.125
In the CMCS/MMT-covered MNPs introduced earlier, doxorubicin (DOX) was electrostatically complexed to the NP at pH 6, forming DOX/CMCS/MMT-MNPs.90 DOX release from the NP was faster at pH 5 versus pH 7.4 due to the protonation of CMCS at pH 5, where DOX was no longer retained via electrostatic interactions.90 The cytotoxic effect of the NPs was observed in MCF-7 cells via MTT assay in comparison with free DOX and the vehicle.90 Cytotoxicity in MCF-7 cells increased in the order of CMCS-MNPs (vehicle), CMCS/MMT-MNPs (vehicle), free DOX, and DOX/CMCS/MMT-MNPs.90 Interestingly, the toxicity of DOX/CMCS/MMT-MNPs in H9c2 cardiomyocytes was less than that of free DOX, due to the antioxidant effects of CMCS.90
Lei et al. studied the cytotoxicity and cellular uptake of DOX-loaded poly(lactic-co-glycolic acid) (PLGA) NPs in drug-sensitive and resistant cell lines: SKOV-3 ovarian carcinoma cells (drug resistant, p53 mutation, HER2+), MES-SA uterine sarcoma cells (drug sensitive), and its drug resistant variant MES-SA/Dx5 cells (P-glycoprotein (P-gp)-overexpressing).126 DOX-PLGA NPs (DNPs) and antibody-conjugated DOX-PLGA NPs (ADNPs) were comparable in particle size (163 nm and 213 nm) and drug loading (2.7% and 2.3%), except that ADNPs had 9.3 μg anti-HER2 antibody per mg NPs.126 Due to the HER2-mediated endocytosis, ADNPs were taken up better than DNPs by SKOV-3 cells. In contrast, no difference in cellular uptake was observed between the two NPs in MES-SA and MES-SA/Dx5 cells, which did not express HER-2. Notably, both NPs showed higher uptake than free DOX in MES-SA/Dx5 cells, indicating that the NPs were not subject to P-gp efflux. In this study, cytotoxicity of the NPs reflected the cellular uptake profile: ADNPs were more cytotoxic than DNPs or free DOX in SKOV-3 cells, although the difference did not reach statistical significance. Both ADNPs and DNPs showed higher toxicity than free DOX in MES-SA/Dx5 cells.126
On the other hand, there are examples where bioactivity of NPs does not necessarily match their cellular uptake.127 In our recent study, PLGA NPs conjugated to a cell-penetrating peptide, TAT, were used to increase intracellular delivery of paclitaxel (PTX) to multidrug resistant (MDR) cells. As expected, the PLGA-TAT NPs were more efficiently taken up by MDR cells than PLGA NPs, but they did not increase PTX delivery to the MDR cells (hence their killing). This discrepancy can be interpreted as indirect evidence of extracellular drug release from the NPs, which may not be observed in typical in vitro release kinetics studies using a buffered saline.127
3.2. Three-dimensional (3D) approaches
In in vitro 2D cell culture, target cells are directly and uniformly exposed to NPs for a desired period of time with no limitation in the higher end of a concentration range. However, this condition may not be an accurate reflection of in vivo events occurring at 3-dimensional (3D) masses such as solid tumors, where NPs face various access barriers to target cells.128 Moreover, due to the highly unnatural geometric and mechanical properties, there is a good possibility that the 2D-cultured cells have limited potential to represent the phenotype and genetic functions of living tissues, which can drastically affect their responses to chemical stimuli.129–131 The fact that in vivo efficacy often betray the drug screening results obtained in 2D cell culture is not irrelevant to the artificial nature of 2D culture models.132 Therefore, several efforts have been made to develop 3D cell models, which can better mimic cell-cell and cell-extracellular matrix (ECM) interactions seen in a living organism, as a test bed of NP systems. This section introduces various 3D models and examples in which 3D models were used for evaluation of the efficacy of a drug or drug-loaded NPs.
Commonly used models include (i) cells encapsulated in scaffolds, (ii) multicellular spheroids,133 (iii) a combination of spheroids and scaffolds,134 and (iv) multilayer cell models132 (Fig. 2). Other 3D models include excised tissues or tissue components135 and a microfluidic device based on polydimethylsiloxane template.136–138
Figure 2.

Commonly used 3D tumor models to determine NP efficacy.
In the cells-in scaffold model, which has been widely studied in the context of tissue engineering, cancer cells are grown in either synthetic or natural scaffolds such as hydrogels of ECM components,139–141 PEG hydrogels,142 peptide nanofiber scaffolds,143 multilayered paper scaffolds,144 or polymers.130 An advantage of this model is that cells are exposed to a microenvironment similar to their native ECM and reflect its influence on the cell growth. In one example, 3D tumor models were created by seeding MDA-MB-231 human breast cancer cells in collagen I hydrogels.140 The 3D tumor models expressed a phenotype reflecting in vivo tumor progression, such as hypoxia, necrosis, and angiogenic gene upregulation, in a manner dependent on the thickness of the collagen gels.140 In another study, C4-2B bone metastatic prostate cancer cells were cultured in in-situ crosslinkable hyaluronic acid (HA) hydrogels, where cancer cells grew forming clustered structures similar to real tumors.141 This model was used in testing the efficacy of anti-cancer drugs to show that cells in the HA gels were more sensitive to camptothecin than those in 2D culture.141 The increased drug sensitivity of cells in the HA hydrogel is attributed to the biological activities of HA on cancer cells, which may be reflective of the ECM-cell interactions in vivo.141 Conversely, epithelial ovarian cancer cells grown in PEG hydrogels showed a reduced sensitivity to PTX treatment than those in 2D culture.142 These results suggest that a scaffold is not simply a space-filler but plays an active role in expression of phenotypes relevant to drug sensitivity.
Mitra et al. developed a cells-in scaffold model of Y79 retinoblastoma for the study of NP efficacy. Here, large and porous PLGA microparticles (~150 μm) were produced as a scaffold, in which dispersed cells were seeded and allowed to grow.130 The porous microparticles were produced by the double emulsion method using sucrose as a porogen. Gelatin, polyvinyl alcohol, and chitosan were incorporated to promote cell attachment to the microparticle scaffold.130 Cells in the 3D model not only attached to the microparticle surface, but infiltrated the particles over time.130 Compared to the cells grown in 2D, those grown in the 3D model exhibited higher ECM production and altered gene regulation.130 When dosed with carboplatin-, etoposide-, or DOX-loaded NPs or their free drug counterparts, 4.5–21.8 fold higher IC50 values were observed in the 3D model as compared with 2D.130 These results indicate that 3D culture condition can greatly change the chemical and biological environment of the cells, thereby therapeutic outcomes of the tested drug.130
Multicellular spheroids refer to spherical aggregates of cancer cells that can reflect tight junctions between cells and ECM synthesis.145 A multicellular spheroid model was used in the evaluation of DOX-loaded micelles based on a poly(ethylene oxide)-poly[(R)-3-hydroxybutyrate]-poly-(ethylene oxide) (PEO-PHB-PEO/DOX) with respect to their ability to penetrate the spheroids.94 Here, the 37 nm micelles or free DOX were incubated with SiHa cell spheroids with a diameter of 400 μm.94 In 30 minutes, DOX-loaded micelles penetrated spheroid cores to a greater extent than free DOX, although this difference disappeared in 2 hours.94 This difference was explained by the ability of the PEGylated micelles to avoid non-specific binding to ECM and immediate cellular uptake. 94 In another example, Kim et al. used a multicellular cylindroid model to investigate how surface charges control the penetration and cellular uptake of gold NPs in tumor matrix.146 Gold NPs (6 nm) were modified with trimethyl ammonium- or carboxylate-terminated tetra(ethylene glycol) to produce cationic or anionic surfaces, respectively. The NPs were additionally conjugated with fluorescein and incubated with a cylindroid, and the fluorescence in the cylindroid was quantified according to time and radial position. The results showed that gold NPs with a cationic surface were readily consumed by actively proliferating cells at the periphery, whereas negative NPs penetrated into the apoptoic/necrotic interior of the cylindroid at a higher rate than cationic ones.146
Ho et al. used a spheroid/scaffold combination model of U251 human glioma cells to study the effect of the geometry of a cell model on DOX and irinotecan drug resistance.134 Spheroids were first formed by growing cells in a plate coated with poly(2-hydroxylethyl methacrylate), which prevented cell attachment to the well bottom. Subsequently, the spheroids were seeded into a porous PLGA scaffold coated with collagen.134 The spheroids maintained their structure for 2 days, allowing for a time window in which the drug effect could be tested. Drug resistance was highest for the spheroids seeded in the scaffold, followed by those seeded as dispersed cells in a comparable scaffold, with cells grown in 2D having the least drug resistance.134 Lactate production was highest in the spheroid-seeded scaffold model, while the 2D cell culture model produced the lowest lactate per cell.134 The authors attributed the increased drug resistance in the 3D model to the tendency to form hypoxic regions, supported by the high lactate production, rather than the limitation in drug transport.
In the multilayer cell models, cancer cells are grown on a permeable membrane support to reach 200 – 250 μm thick cell layers.145 Hosoya et al. created multilayer cell models simulating pancreatic cancer with fibrotic tissue to study intratumoral transport of different macromolecules.132 These models consisted of alternating layers of fibroblasts and fibronectin-gelatin films on transwell inserts.132 The thickness of the models with 5 layers of cultured cells was 30 – 50 μm. Transport of FITC-dextrans across the cell model was quantified by measuring the fluorescence of the medium below the transwell.132 As readily expected, the dextran transport decreased as the number of cell layers increased or the molecular size of dextran increased. Approximately 29% of the 250 kDa FITC-dextran conjugate (12 nm) permeated the K643f monolayer over 24 hours.132 During the same time, the 12 nm dextran had approximately 21% and 19% permeability through 2 and 5 layer models, respectively.132
4. In vivo studies
Once NPs demonstrate a proof of concept in vitro, their safety and therapeutic effectiveness are tested in animal models. The results of animal studies play a pivotal role in decision making toward clinical trials. An animal model that can reflect pathophysiology of a human disease is an invaluable tool for predicting therapeutic outcomes in human. This section discusses the currently available experimental animal models, their strengths and weakness, and emerging trends in the animal model development. Given that the majority of in vivo NP studies have been performed in the context of cancer therapy, the discussion focuses on animal models of tumor unless specified otherwise.
4.1. Evaluation of NPs in animal models of tumor: state of the art
Mouse models with allograft or human xenograft tumors are widely used in in vivo evaluation of NPs due to the relatively low cost and well-established protocols. In these models, cancer cells are inoculated or tumor tissues are implanted (typically subcutaneously) in immunodeficient mice (athymic nude or severe combined immunodeficient mice), allowed to grow to visible tumors (Fig. 3), and treated with experimental therapeutics to examine the pharmacokinetics, biodistribution, and the pharmacological effects.
Figure 3.
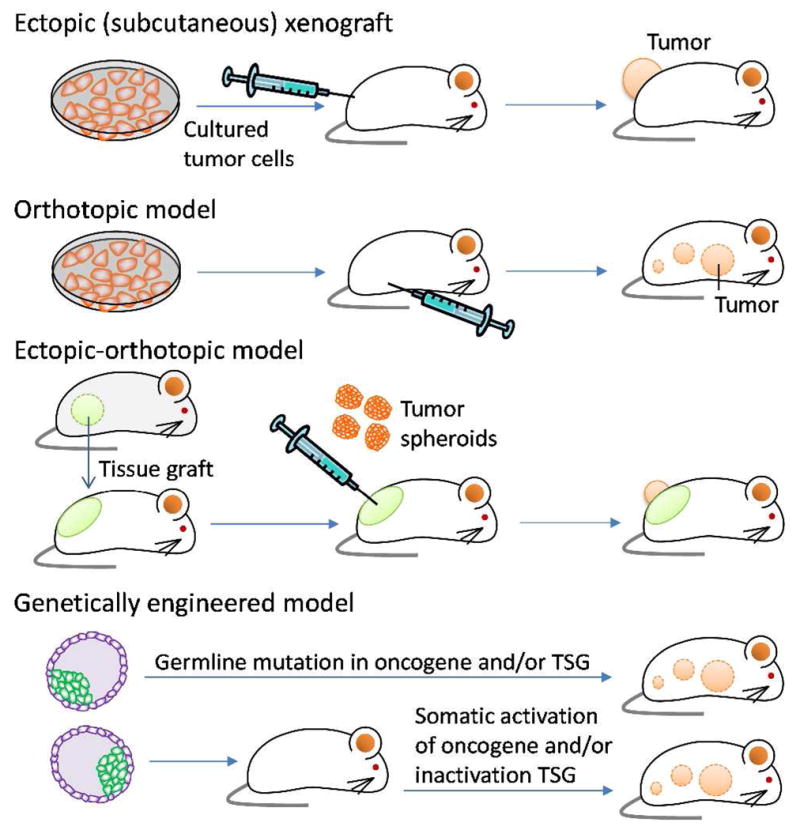
Animal models of tumor used in the evaluation of in vivo efficacy of NPs. TSG: Tumor suppressor genes.
For example, therapeutic efficacy of PEGylated liposomal DOX (PLD) was tested in a mouse model of cancer.147 C-26 mouse colon carcinoma cells were inoculated subcutaneously in the left flank of a BALB/c mouse, and the response to a treatment was monitored by measuring the size of tumors.147 Here, free DOX at a dose of 6 mg/kg only slightly delayed tumor growth compared with the saline control, whereas with PLD at doses at 6 or 9 mg/kg tumors regressed to non-measurable sizes.147 Consequently, all animals receiving PLD groups survived 120 days (duration of the experiment), whereas those receiving saline and free DOX groups survived a mean of 50 and 49 days, respectively.147 The therapeutic benefit of PLD is attributable to its high bioavailability and preferential accumulation in tumors.147 Similarly, Vaage et al. used human prostate carcinoma PC-3 implanted subcutaneously into mice and reported that the therapeutic efficacy of DOX was increased and its toxic side effects reduced when delivered as PEGylated liposomes.148 The superior efficacy of PLD over free DOX was further demonstrated in mouse models of murine mammary carcinomas,149 xenografted human ovarian,150 and pancreatic carcinomas.151
However, the performance of NPs in these animal models is not always predictive of clinical outcomes. The PLD that demonstrated 100% survival of tumor-bearing mice147 was at best equivalent to free DOX in clinical efficacy (progression-free survival (PFS) and overall survival) in a randomized Phase III trial with metastatic breast cancer patients.152 Here, women with metastatic breast cancer (n = 509) were randomly assigned to either PLD 50 mg/m2 (every 4 weeks) or DOX 60 mg/m2 (every 3 weeks). PLD and DOX were comparable with respect to PFS (6.9 versus 7.8 months) and overall survival (21 versus 22 months).152 In a Phase II study with metastatic breast cancer patients, the overall response rate of the PLD-treated group (45 to 60 mg/m2 every 3 to 4 weeks for a maximum of six cycles) was 31% (95% confidence interval, 20% to 43%),153 comparable to the response rates (25 to 40%) for free DOX at conventional doses (50 to 75 mg/m2 every 3 weeks) in advanced breast cancer patients with similar characteristics.154–157
The discrepancy between preclinical in vivo results and clinical outcomes is found in another example. PK1, a covalent conjugate of DOX and N-(2-hydroxypropyl) methacrylamide (HPMA) copolymer via biodegradable (Gly-Phe-Leu-Gly) oligopeptide, was evaluated in various animal models.158, 159 The models were created by intraperitoneal (i.p.) injection of L1210 leukemia cells, subcutaneous (s.c.) injections of B16F10 melanoma cells, Walker sarcoma cells, P388 leukemia cells, and M5076 cells, or subcutaneous implantation of LS174T human colon xenograft.158 When administered i.p. to mice bearing L1210 ascitic tumor, PK1 showed relatively good antitumor activity as compared to free DOX.158 The highest T/C, a ratio of median survival of the test group (T) to that of untreated control (C), seen in the PK1-treated group was >762%, as opposed to 214%, that of free DOX-treated group.158 In the case of solid tumor models (B16F10, Walker, P388, M5076; and LS174T xenograft), i.p. administration of PK1 resulted in an increase in survival rate as compared to free DOX. In particular, P388 and Walker sarcoma showed remarkable regression after the treatment.158 On the other hand, the phase II studies of PK1 in patients with non-small cell lung (NSCLC, n=29), colorectal (n=16) and breast (n=17) cancer showed less exciting outcomes.160 Of 26 evaluable patients with NSCLC, 3 chemotherapy-naïve patients had partial responses, and none of the 16 evaluable patients with colorectal cancer showed responses.160 Of 14 evaluable patients with breast cancer, only 3 anthracycline-naïve patients had partial responses.160
Another example is a macromolecular conjugate of PTX and poly(L-glutamic acid) (PTX poliglumex). PTX poliglumex demonstrated a prolonged circulation half-life and greater tumor uptake as compared to Taxol (PTX solubilized with Cremophor EL) in a mouse model.161 Consequently, PTX poliglumex exhibited significant tumor growth delay after a single intravenous (i.v.) injection at 80 mg/kg (as PTX equivalent) compared with Taxol at the same dose in mice bearing syngeneic ovarian OCA-1 carcinoma.161 A similar antitumor effect was shown in a rat model with 13762F rat mammary adenocarcinoma.161 Clinical outcomes in Phase II trials were modest. In women with recurrent epithelial ovarian, primary peritoneal, or fallopian tube carcinoma, the response rate and median time to disease progression of PTX poliglumex (175 mg/m2, every 21 days) were 10% and 2.1 months, respectively,162 and the median PFS was 2.8 months.163 Even considering variability due to prior treatment history, these responses were not favorable as compared with those of the standard regimen based on PTX (135 mg/m2) and platinum (75 mg/m2) based chemotherapy,164 which showed >70% of response rate and 18 months of median PFS.165 The lack of advantages over existing regimens, combined with unexplained toxicity, led the developer to officially withdraw the application for a marketing authorization of PTX poliglumex in 2009.166
4.2. Limitations of current tumor models in predicting clinical efficacy
In explaining the gap between the results of rodent models and clinical outcomes, several limitations of current animal models may be considered. First, the frequently used s.c. tumor implants do not represent the primary human cancers (e.g., lung, colon, breast) nor the preferred sites of metastasis (e.g., liver for colon cancer metastasis).167 Instead, allograft or xenograft tumors are artificially implanted s.c. (mostly for the sake of convenience), where the tumors grow in an environment different from the primary organs, with much reduced potential for metastasis.168, 169 Second, immortalized cancer cell lines used in many models as the source of xenografts have been maintained over many passages in culture and may have lost architectural and cellular properties unique to the original tumors.170, 171 Even though grafted tumors can represent important attributes of the original tumors, it is uncertain whether it captures the genetic and epigenetic variability of tumors in its entirety.170 Third, when human xenografts are inoculated in mouse models, the tumors build stroma and vasculature out of murine sources.168, 170, 171 The potential impact of this artificial arrangement on the architecture of stroma, cell-stroma interactions, and tumor propagation is barely considered in the establishment of models and interpretation of preclinical studies. Fourth, due to the foreign origin of tumors, it is inevitable to use mice with compromised immune systems, such as athymic or severe combined immunedeficient (SCID) mice.172 Consequently, potential immune responses to NPs, which directly influence their bioavailability,61, 173–175 are not properly evaluated in these models. Fifth, the size and growth rate of tumors in mice are not comparable to those of human patients. While human tumors typically develop over a number of years, tumors in murine models are designed to grow in days or weeks for high throughput evaluation.170 In addition, typical s.c. tumors can be as large as ~1 cm3 for a 25 g mouse (4% of the body weight). Human patients with tumors that can be visibly identified would be candidates for surgical debulking rather than chemotherapy. One of the likely reasons to favor rapidly growing tumor models in the evaluation of NPs is the positive correlation between tumor growth rate and the EPR effect,176 the main driving force for tumor-selective NP accumulation.177, 178 Nonetheless, one should be aware that the clinical significance of vascular permeability effect in drug delivery is much debated 60, 179 and little is known about the effectiveness of the EPR effect in metastatic or microscopic residual tumors, where targeted chemotherapy is most desired.
4.3. Alternative animal models of tumor
To make a reliable and clinically relevant evaluation tool, an animal model of tumor must fulfill several requirements. It should faithfully recapitulate the pathophysiology of human cancer, reproduce the problems associated with a specific type and location of primary and metastatic cancer, and allow for evaluation of biological events associated with tumor progression.180 In addition, the model should be reproducible and affordable and provide a quantitative endpoint of therapeutic responses.180 It may not be possible to develop a single model that meets all the requirements and works for all, but several efforts are currently made to develop models that better addresses each requirement. Based on an understanding of these models, one may choose an experimental model that is most appropriate for the specific questions asked in each study.
4.3.1. Orthotopic tumor models
In an orthotopic model, a tumor allo- or xenograft is grown in proximity to the tissues or organs that the tumor cells were derived from (Fig. 3).170 The orthotropic model is advantageous over ectopic s.c. models in that it provides a host environment closer to a normal milieu of the tumor, where the cells can grow in the same manner as in human cancer. The histological, biochemical, and immunological properties of primary tumors determine their metastatic potential.180 In many cases, orthotopically implanted tumor cells have a greater potential for metastasis compared to the same cells implanted s.c.181; therefore, when the desired effect of a new drug product is against metastasis, it is desirable to use an orthotopic model. The microenvironment also influences responses of tumors to a therapeutic agent.181–183 Fidler et al. reported that a s.c. human colon cancer xenograft was relatively non-invasive and sensitive to DOX, whereas the same tumor implanted in the cecal wall was less responsive.182 The difference in therapeutic responses was attributed to differential expression of Pgp in the tissues.182 In another example, human small-cell lung cancer (SCLC) cells were grown orthotopically (in the lung) or ectotopically (s.c.) in SCID mice and administered with cisplatin and mitomycin C (MMC). The two models displayed opposite response profiles: while an orthotopic SCLC model was responsive to cisplatin but not to MMC, similar to clinical situation, s.c. model showed sensitivity to MMC but not to cisplatin.183 According to this model, an orthotopic model better reflects the clinical effects of drugs on human SCLC than the tumors growing s.c.183
On the other hand, one challenge of an orthotopic model is that tumor burden is not readily detectable as in s.c. models.167, 181 Except for breast tumor models, which develop superficial tumors, most orthotopic tumors are located in internal organs such as prostate, kidney, brain, lungs, liver and are not conducive to caliper measurement.181 One way to monitor therapeutic responses is to assess tumor burden terminally after serial sacrifice of animals. In this case, group size needs to be determined considering potential “non-takers” (animals that have not developed tumors at the time of treatment).181 The main disadvantage of this approach is that it is labor-intensive and necessitates a large number of animals. Alternatively, non-invasive imaging techniques may be used together with cancer cells producing fluorescent or luminescent signals.181, 184 Genes encoding fluorescent proteins and/or luciferase are introduced to human or murine cell lines in vitro to stably express the proteins in living animals.185–188 Optical imaging tools, such as fluorescence or bioluminescence, are used to monitor the growth of orthotopic tumors and metastasis in host organs externally in real time.188–191 The two techniques are often used in combination: fluorescence imaging for high throughput in vitro tests or superficial tumor imaging and bioluminescence imaging for detection of relatively deep tissues.188 Orthotopic models of pancreatic cancer192 and bladder cancer193 expressing luciferase have been used for evaluating therapeutic efficacy of the targeted gold NPs and hyperbranched polyglycerol NPs, respectively.
4.3.2. Ectopic-orthotopic tumor models
The ectopic-orthotopic model is a hybrid of s.c. and orthotopic model.194–196 In this system, an exogenous tissue sample is first implanted ectopically (in the skin), and a tumor sample is then implanted within the tissue graft (Fig. 3).195, 196 For example, mammary fat pad from a lactating female mouse, prostate tissue from a male mouse, lung, or liver are prepared as minced tissue fragments and implanted in the skin of a host animal. Tumor tissues grown as spheroids are then placed upon the engrafted tissue stroma, which provides orthotopic environment essential for tumor-mesenchymal interactions.195 To visualize the extent of vascularization and tumor progression, the tissues may be grown in a window chamber implanted into a dorsal skinfold in the host animal.
The presence of orthotopic tissue environment is shown to play a critical role in the growth and vascularization of tumors. For example, Transgenic Adenocarcinoma Mouse Prostate-C2 prostate tumors were poorly angiogenic and showed no significant growth in the absence of prostate tissue, whereas tumors grown with prostate stroma were highly angiogenic and proliferative.196 On the other hand, tumor spheroids implanted on the orthotopic tissue stroma showed less vascular permeability than those directly implanted on the skinfold.195 Consequently, a single i.v. administration of DOX was much less effective on the ectotopic-orthotopic tumors than the s.c. tumors, consistent with clinical outcomes.195 This result suggests that many pre-clinical results obtained in the subcutaneous animal models may have been exaggerated due to the pervasive vascular leakiness less natural to human tumors.195
4.3.3. Humanized mice
When narrowly defined, the term humanized mice refers to animal models in which human immune cells or hematopoietic stem cells are adoptively transferred to mice so that human immune systems are established in the mice at least partly.197–199 Zhou et al. used a BALB/c-Rag2−/−γc−/− humanized mouse (RAG-hu) model in the evaluation of cationic PAMAM dendrimers carrying a small interfering RNA (siRNA) for the therapy of HIV-1 infection.200 The RAG-hu model was prepared by injecting human fetal liver-derived CD34+ hematopoietic progenitor cells into the liver of a neonatal mouse, pre-conditioned by irradiation. When the animals no longer produce antibodies to a human antigen, they were infected with HIV-1 and then treated with dendrimer-siRNA NPs. Upon systemic application, the NPs decreased viral loads in animals by several orders of magnitude and protected CD4+ T-cells from virus-induced depletion.200 In the context of cancer research, the humanized mice are used in studying human immune responses to tumors and their roles in tumor progression and metastasis.172 Although the human histocompatibility alleles that can be expressed in a mouse are currently limited,199 humanized mice is a useful tool for evaluating drugs that provide protection against cancer by controlling the immune system.
4.3.4. Genetically engineered mouse models
In genetically engineered mouse (GEM) models, tumor formation is driven by genetic manipulation of animals. GEMs are created by activating clinically relevant oncogenes or inactivating tumor suppressor genes (TSG) via germline or somatic mutations, which predispose animals to certain types of tumors (Fig. 3).172, 201–203 The main advantage of GEM models is that they reflect genetic changes responsible for specific tumors and syngeneic tumor-host interactions181; therefore, they are very useful for studying the roles of oncogenes of interest and interactions between tumor cells and microenvironment.172 GEMs have not been used as widely as other models in routine evaluation of NPs due to the high cost, time, and intellectual property issues. The challenges in tumor monitoring discussed in the orthotopic models also apply to the GEM models.204
Recently, Sengupta et al. used a GEM with somatic PTEN and K-Ras mutations (K-rasLSL/+/Ptenfl/fl), which predispose the animals to ovarian cancer,205 to demonstrate the antitumor efficacy of cholesterol-tethered platinum II-based supramolecular NPs.206 Ovarian tumors were induced by intrabursal injection of adenovirus carrying Cre recombinase (Adeno-Cre) and luciferase.206 Tumor growth in animals receiving treatments was quantified by monitoring bioluminescence resulting from tumor luciferase expression.206 In another example, Dibirdik et al. studied the anti-cancer activity of a PEGylated liposomal NP carrying a multifunctional tyrosine kinase inhibitor in a MMTV/Neu transgenic mouse model of metastatic ErbB2/HER-2+ chemotherapy-resistant breast cancer.207 In MMTV/Neu transgenic mice, the wild-type neu gene is overexpressed in the mammary gland under the control of the MMTV long terminal repeat,208 which induces progressive and metastatic breast cancer.209 The PEGylated liposomal formulation of the multifunctional tyrosine kinase inhibitor was more effective than standard chemotherapy against the chemotherapy-resistant breast cancer in the MMTV/Neu transgenic mice.207
There is also an increasing appreciation of GEM as a valuable model for identifying biomarkers related to human diseases and developing therapeutics targeted to the biomarkers. For example, Kelly et al. used pancreatic ductal adenocarcinoma (PDAC) cell lines isolated from GEM to screen peptides specifically binding to cell surface antigens on the cells.210 A magnetofluorescent NP was modified with the identified peptide and used as an imaging agent to locate incipient PDAC in GEM.210
5. Future Perspectives
The field of nanomedicine has grown enormously in the past few decades. Nanoparticulate drug carriers are now created in various forms based on organic and inorganic material platforms with an unprecedented control over the size, shape, surface properties, drug loading and release. On the other hand, their clinical translation is relatively slow, with only a handful of commercial products from the early time, such as liposomes or micelles. One of the main reasons is that the knowledge obtained from in vitro and preclinical studies has little value in predicting clinical outcomes of new NP products. It may not be an exaggeration to say that it is not the talent to create NPs but the technology to evaluate them that currently limits further advancement of nanomedicine. For example, new NPs are routinely characterized with respect to surface charge and ligand density, which are then correlated with their behaviors in cell models. On the other hand, in blood or other physiological fluids, NPs are easily covered with protein corona, which ultimately dictates in vivo fates and therapeutic outcomes of the NPs.110,119 In recognition of the disparity between in vitro properties and in vivo outcomes, many groups now migrate to research models that involve early in vivo proof of concept studies. However, the majority of investigators in academia may not be able to afford this approach, nor is it necessarily acceptable in an ethical perspective. Moreover, clinical predictive values of some animal models are recently revisited, with respect to their relevance to human diseases and the ability to recapitulate disease progression. Therefore, it is important for the investigators to initiate an open discussion of the limitations and challenges of current methodologies and explore a new avenue of nanomedicine characterization, which can predict the clinical outcomes in the early stage of product development with a greater reliability. These methods may include new cell models, labeling and detection methods, analytical technologies, mathematical modeling, and animal models that portray critical attributes of human diseases. The need for new NP evaluation method is another reason to pay attention to recent advances in microfluidic technologies, which have emerged as a promising tool to create in vitro microenvironments that mimic in vivo conditions.211
Acknowledgments
This work was supported by NSF DMR-1056997, NIH R21 CA135130, and a grant from the Lilly Endowment, Inc. to College of Pharmacy. This study was also partly supported by the NIH/NCRR-Indiana Clinical and Translational Sciences Institute Pre-doctoral Fellowship (TL1 RR025759, PI: A. Shekhar) to KCL and the Egyptian Government Ministry of Higher Education Missions Sector to SAA
References
- 1.Dynamic Light Scattering. An introduction in 30 minutes. Malvern Instruments; [Accessed on 11/1/12]. http://www.malvern.com/malvern/kbase.nsf/allbyno/KB000734?opendocument. [Google Scholar]
- 2.Dynamic Light Scattering. Common Terms Defined (MRK 1764-01) [Accessed on 11/1/12];Malvern Instruments. http://www.malvern.com/common/downloads/campaign/MRK1764-01.pdf.
- 3.Lu XY, Wu DC, Li ZJ, Chen GQ. Polymer nanoparticles. Prog Mol Biol Transl Sci. 2011;104:299–323. doi: 10.1016/B978-0-12-416020-0.00007-3. [DOI] [PubMed] [Google Scholar]
- 4.Hoo CM, Starostin N, West P, Mecartney ML. A comparison of atomic force microscopy (AFM) and dynamic light scattering (DLS) methods to characterize nanoparticle size distributions. J Nanopart Res. 2008;10:89–96. [Google Scholar]
- 5.Mahl D, Diendorf J, Meyer-Zaika W, Epple M. Possibilities and limitations of different analytical methods for the size determination of a bimodal dispersion of metallic nanoparticles. Colloid Surface A. 2011;377:386–392. [Google Scholar]
- 6.Boyd RD, Pichaimuthu SK, Cuenat A. New approach to inter-technique comparisons for nanoparticle size measurements; using atomic force microscopy, nanoparticle tracking analysis and dynamic light scattering. Colloid Surface A. 2011;387:35–42. [Google Scholar]
- 7.Saveyn H, De Baets B, Thas O, Hole P, Smith J, Van der Meeren P. Accurate particle size distribution determination by nanoparticle tracking analysis based on 2-D Brownian dynamics simulation. J Colloid Interf Sci. 2010;352:593–600. doi: 10.1016/j.jcis.2010.09.006. [DOI] [PubMed] [Google Scholar]
- 8.Finder C, Wohlgemuth M, Mayer C. Analysis of particle size distribution by particle tracking. Part Part Syst Char. 2004;21:372–378. [Google Scholar]
- 9. [Accessed on 10/30/12];Disc Centrifuge. http://www.cpsinstruments.eu/centrifuge.html.
- 10.Murdock RC, Braydich-Stolle L, Schrand AM, Schlager JJ, Hussain SM. Characterization of nanomaterial dispersion in solution prior to in vitro exposure using dynamic light scattering technique. Toxicol Sci. 2008;101:239–253. doi: 10.1093/toxsci/kfm240. [DOI] [PubMed] [Google Scholar]
- 11.Jiang J, Oberdörster G, Biswas P. Characterization of size, surface charge, and agglomeration state of nanoparticle dispersions for toxicological studies. J Nanopart Res. 2009;11:77–89. [Google Scholar]
- 12.Vasylkiv O, Sakka Y. Synthesis and colloidal processing of zirconia nanopowder. J Am Ceram Soc. 2001;84:2489–2494. [Google Scholar]
- 13.Bowen P. Particle size distribution measurement from millimeters to nanometers and from rods to platelets. J Disper Sci Technol. 2002;23:631–662. [Google Scholar]
- 14.Brant J, Lecoanet H, Wiesner MR. Aggregation and deposition characteristics of fullerene nanoparticles in aqueous systems. J Nanopart Res. 2005;7:545–553. [Google Scholar]
- 15.Widegren J, Bergström L. Electrostatic stabilization of ultrafine titania in ethanol. J Am Ceram Soc. 2002;85:523–528. [Google Scholar]
- 16.Kirby BJ, Hasselbrink EF., Jr Zeta potential of microfluidic substrates: 1. Theory, experimental techniques and effects on separations. Electrophoresis. 2004;25:187–202. doi: 10.1002/elps.200305754. [DOI] [PubMed] [Google Scholar]
- 17.Kirby BJ, Hasselbrink EF., Jr Zeta potential of microfluidic substrates: 2. Data for polymers. Electrophoresis. 2004;25:203–13. doi: 10.1002/elps.200305755. [DOI] [PubMed] [Google Scholar]
- 18.Hoffman AS. The origins and evolution of “controlled” drug delivery systems. J Control Release. 2008;132:153–63. doi: 10.1016/j.jconrel.2008.08.012. [DOI] [PubMed] [Google Scholar]
- 19.Anhalt K, Geissler S, Harms M, Weigandt M, Fricker G. Development of a new method to assess nanocrystal dissolution based on light scattering. Pharm Res. 2012;29:2887–901. doi: 10.1007/s11095-012-0795-4. [DOI] [PubMed] [Google Scholar]
- 20.Accardo A, Tesauro D, Roscigno P, Gianolio E, Paduano L, D’Errico G, Pedone C, Morelli G. Physicochemical properties of mixed micellar aggregates containing CCK peptides and Gd complexes designed as tumor specific contrast agents in MRI. J Am Chem Soc. 2004;126:3097–3107. doi: 10.1021/ja039195b. [DOI] [PubMed] [Google Scholar]
- 21.Cheng C, Wei H, Zhang XZ, Cheng SX, Zhuo RX. Thermo-triggered and biotinylated biotin-P(NIPAAm-co-HMAAm)-b-PMMA micelles for controlled drug release. J Biomed Mater Res A. 2009;88A:814–822. doi: 10.1002/jbm.a.31770. [DOI] [PubMed] [Google Scholar]
- 22.Toncheva V, Schacht E, Ng SY, Barr J, Heller J. Use of block copolymers of poly(ortho esters) and poly(ethylene glycol) micellar carriers as potential tumour targeting systems. J Drug Target. 2003;11(6):345–353. doi: 10.1080/10611860310001633839. [DOI] [PubMed] [Google Scholar]
- 23.Yang X, Li L, Wang Y, Tan Y. Preparation, pharmacokinetics and tissue distribution of micelles made of reverse thermo-responsive polymers. Int J Pharm. 2009;370:210–215. doi: 10.1016/j.ijpharm.2008.11.028. [DOI] [PubMed] [Google Scholar]
- 24.Kalyanasundaram K, Thomas JK. Environmental effects on vibronic band intensities in pyrene monomer fluorescence and their application in studies of micellar systems. J Am Chem Soc. 1977;99:2039–2044. [Google Scholar]
- 25.Chen W, Cheng Y, Wang B. Dual-responsive boronate crosslinked micelles for targeted drug delivery. Angew Chem Int Ed Engl. 2012;51:5293–5295. doi: 10.1002/anie.201201179. [DOI] [PubMed] [Google Scholar]
- 26.Lin WJ, Juang LW, Lin CC. Stability and release performance of a series of pegylated copolymeric micelles. Pharm Res. 2003;20:668–673. doi: 10.1023/a:1023215320026. [DOI] [PubMed] [Google Scholar]
- 27.Xu P, Tang H, Li S, Ren J, Van Kirk E, Murdoch WJ, Radosz M, Shen Y. Enhanced stability of core-surface cross-linked micelles fabricated from amphiphilic brush copolymers. Biomacromolecules. 2004;5:1736–1744. doi: 10.1021/bm049874u. [DOI] [PubMed] [Google Scholar]
- 28.Yang C, Ebrahim Attia AB, Tan JPK, Ke X, Gao S, Hedrick JL, Yang YY. The role of non-covalent interactions in anticancer drug loading and kinetic stability of polymeric micelles. Biomaterials. 2012;33:2971–2979. doi: 10.1016/j.biomaterials.2011.11.035. [DOI] [PubMed] [Google Scholar]
- 29.Zhao X, Poon Z, Engler AC, Bonner DK, Hammond PT. Enhanced stability of polymeric micelles based on postfunctionalized poly(ethylene glycol)-b-poly(γ-propargyl L-glutamate): the substituent effect. Biomacromolecules. 2012;13:1315–1322. doi: 10.1021/bm201873u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Surfactant micelle characterization using dynamic light scattering (MRK809–01) Malvern Instruments; [Accessed on 11/15/12]. http://www.malvern.com/malvern/kbase.nsf/allbyno/KB001097/$file/MRK809-01.pdf. [Google Scholar]
- 31.Zhang XZ, Wu DQ, Chu CC. Synthesis, characterization and controlled drug release of thermosensitive IPN–PNIPAAm hydrogels. Biomaterials. 2004;25:3793–3805. doi: 10.1016/j.biomaterials.2003.10.065. [DOI] [PubMed] [Google Scholar]
- 32.Needham D, Anyarambhatla G, Kong G, Dewhirst MW. A new temperature-sensitive liposome for use with mild hyperthermia: characterization and testing in a human tumor xenograft model. Cancer Res. 2000;60:1197–1201. [PubMed] [Google Scholar]
- 33.Nakayama M, Okano T, Miyazaki T, Kohori F, Sakai K, Yokoyama M. Molecular design of biodegradable polymeric micelles for temperature-responsive drug release. J Control Release. 2006;115:46–56. doi: 10.1016/j.jconrel.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 34.Pişkin E. Molecularly designed water soluble, intelligent, nanosize polymeric carriers. Int J Pharm. 2004;277:105–118. doi: 10.1016/j.ijpharm.2003.06.003. [DOI] [PubMed] [Google Scholar]
- 35.Kim YH, Kwon IC, Bae YH, Kim SW. Saccharide effect on the lower critical solution temperature of thermosensitive polymers. Macromolecules. 1995;28:939–944. [Google Scholar]
- 36.Zhang L, Chan JM, Gu FX, Rhee JW, Wang AZ, Radovic-Moreno AF, Alexis F, Langer R, Farokhzad OC. Self-assembled lipid--polymer hybrid nanoparticles: a robust drug delivery platform. ACS Nano. 2008;2:1696–1702. doi: 10.1021/nn800275r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sun TM, Du JZ, Yan LF, Mao HQ, Wang J. Self-assembled biodegradable micellar nanoparticles of amphiphilic and cationic block copolymer for siRNA delivery. Biomaterials. 2008;29:4348–4355. doi: 10.1016/j.biomaterials.2008.07.036. [DOI] [PubMed] [Google Scholar]
- 38.Isaacs SR, Choo H, Ko WB, Shon YS. Chemical, thermal, and ultrasonic stability of hybrid nanoparticles and nanoparticle multilayer films. Chem Mater. 2005;18:107–114. [Google Scholar]
- 39.Na K, Bae YH. Self-assembled hydrogel nanoparticles responsive to tumor extracellular pH from pullulan derivative/sulfonamide conjugate: characterization, aggregation, and adriamycin release in vitro. Pharm Res. 2002;19:681–688. doi: 10.1023/a:1015370532543. [DOI] [PubMed] [Google Scholar]
- 40.Yokoyama M, Sugiyama T, Okano T, Sakurai Y, Naito M, Kataoka K. Analysis of micelle formation of an adriamycin-conjugated poly(ethylene glycol)-poly(aspartic acid) block copolymer by gel permeation chromatography. Pharm Res. 1993;10:895–899. doi: 10.1023/a:1018921513605. [DOI] [PubMed] [Google Scholar]
- 41.Masayuki Y, Kwon GS, Teruo O, Yasuhisa S, Mayumi N, Kazunori K. Influencing factors on in vitro micelle stability of adriamycin-block copolymer conjugates. J Control Release. 1994;28:59–65. [Google Scholar]
- 42.Opanasopit P, Yokoyama M, Watanabe M, Kawano K, Maitani Y, Okano T. Influence of serum and albumins from different species on stability of camptothecin-loaded micelles. J Control Release. 2005;104:313–321. doi: 10.1016/j.jconrel.2005.02.014. [DOI] [PubMed] [Google Scholar]
- 43.Akiyoshi K, Kobayashi S, Shichibe S, Mix D, Baudys M, Wan Kim S, Sunamoto J. Self-assembled hydrogel nanoparticle of cholesterol-bearing pullulan as a carrier of protein drugs: Complexation and stabilization of insulin. J Control Release. 1998;54:313–320. doi: 10.1016/s0168-3659(98)00017-0. [DOI] [PubMed] [Google Scholar]
- 44.Iijima M, Nagasaki Y, Okada T, Kato M, Kataoka K. Core-Polymerized Reactive micelles from heterotelechelic amphiphilic block copolymers. Macromolecules. 1999;32:1140–1146. [Google Scholar]
- 45.Jones MC, Leroux JC. Polymeric micelles – a new generation of colloidal drug carriers. Eur J Pharm Biopharm. 1999;48:101–111. doi: 10.1016/s0939-6411(99)00039-9. [DOI] [PubMed] [Google Scholar]
- 46.Opanasopit P, Yokoyama M, Watanabe M, Kawano K, Maitani Y, Okano T. Block copolymer design for camptothecin incorporation into polymeric micelles for passive tumor targeting. Pharm Res. 2004;21:2001–2008. doi: 10.1023/b:pham.0000048190.53439.eb. [DOI] [PubMed] [Google Scholar]
- 47.Watanabe M, Kawano K, Yokoyama M, Opanasopit P, Okano T, Maitani Y. Preparation of camptothecin-loaded polymeric micelles and evaluation of their incorporation and circulation stability. Int J Pharm. 2006;308:183–189. doi: 10.1016/j.ijpharm.2005.10.030. [DOI] [PubMed] [Google Scholar]
- 48.Lu J, Owen SC, Shoichet MS. Stability of self-assembled polymeric micelles in serum. Macromolecules. 2011;44:6002–6008. doi: 10.1021/ma200675w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen H, Kim S, Li L, Wang S, Park K, Cheng JX. Release of hydrophobic molecules from polymer micelles into cell membranes revealed by Förster resonance energy transfer imaging. Proc Natl Acad Sci U S A. 2008;105:6596–6601. doi: 10.1073/pnas.0707046105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen H, Kim S, He W, Wang H, Low PS, Park K, Cheng JX. Fast release of lipophilic agents from circulating PEG-PDLLA micelles revealed by in vivo forster resonance energy transfer imaging. Langmuir. 2008;24:5213–5217. doi: 10.1021/la703570m. [DOI] [PubMed] [Google Scholar]
- 51.Miller T, Rachel R, Besheer A, Uezguen S, Weigandt M, Goepferich A. Comparative investigations on in vitro serum stability of polymeric micelle formulations. Pharm Res. 2012;29:448–459. doi: 10.1007/s11095-011-0555-x. [DOI] [PubMed] [Google Scholar]
- 52.Piston DW, Kremers GJ. Fluorescent protein FRET: the good, the bad and the ugly. Trends Biochem Sci. 2007;32:407–414. doi: 10.1016/j.tibs.2007.08.003. [DOI] [PubMed] [Google Scholar]
- 53.Walkey CD, Chan WC. Understanding and controlling the interaction of nanomaterials with proteins in a physiological environment. Chem Soc Rev. 2012;41:2780–2799. doi: 10.1039/c1cs15233e. [DOI] [PubMed] [Google Scholar]
- 54.Dobrovolskaia M, Aggarwal P, Hall J, McNeil S. Preclinical studies to understand nanoparticle interaction with the immune system and its potential effects on nanoparticle biodistribution. Mol Pharmaceut. 2008;5:487–495. doi: 10.1021/mp800032f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tan J, Butterfield D, Voycheck C, Caldwell K, Li J. Surface modification of nanoparticles by PEO/PPO block copolymers to minimize interactions with blood components and prolong blood circulation in rats. Biomaterials. 1993;14:823–833. doi: 10.1016/0142-9612(93)90004-l. [DOI] [PubMed] [Google Scholar]
- 56.Vonarbourg A, Passirani C, Saulnier P, Simard P, Leroux J, Benoit J. Evaluation of pegylated lipid nanocapsules versus complement system activation and macrophage uptake. J Biomed Mater Res A. 2006;78:620–628. doi: 10.1002/jbm.a.30711. [DOI] [PubMed] [Google Scholar]
- 57.Senior J, Delgado C, Fisher D, Tilcock C, Gregoriadis G. Influence of surface hydrophilicity of liposomes on their interaction with plasma protein and clearance from the circulation: studies with poly(ethylene glycol)-coated vesicles. Biochim Biophys Acta. 1991;1062:77–82. doi: 10.1016/0005-2736(91)90337-8. [DOI] [PubMed] [Google Scholar]
- 58.Torchilin V, Omelyanenko V, Papisov M, Bogdanov A, Trubetskoy V, Herron J, Gentry C. Poly(ethylene glycol) on the liposome surface: on the mechanism of polymer-coated liposome longevity. Biochim Biophys Acta. 1994;1195:11–20. doi: 10.1016/0005-2736(94)90003-5. [DOI] [PubMed] [Google Scholar]
- 59.Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986;46:6387–6392. [PubMed] [Google Scholar]
- 60.Bae YH, Park K. Targeted drug delivery to tumors: myths, reality and possibility. J Control Release. 2011;153:198–205. doi: 10.1016/j.jconrel.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dams E, Laverman P, Oyen W, Storm G, Scherphof G, van Der Meer J, Corstens F, Boerman O. Accelerated blood clearance and altered biodistribution of repeated injections of sterically stabilized liposomes. J Pharmacol Exp Ther. 2000;292:1071–1079. [PubMed] [Google Scholar]
- 62.Wang X, Ishida T, Kiwada H. Anti-PEG IgM elicited by injection of liposomes is involved in the enhanced blood clearance of a subsequent dose of PEGylated liposomes. J Control Release. 2007;119:236–244. doi: 10.1016/j.jconrel.2007.02.010. [DOI] [PubMed] [Google Scholar]
- 63.Garay R, El-Gewely R, Armstrong J, Garratty G, Richette P. Antibodies against polyethylene glycol in healthy subjects and in patients treated with PEG-conjugated agents. Expert Opin Drug Del. 2012;9:1319–1323. doi: 10.1517/17425247.2012.720969. [DOI] [PubMed] [Google Scholar]
- 64.Amoozgar Z, Park J, Lin Q, Yeo Y. Low molecular-weight chitosan as a pH-sensitive stealth coating for tumor-specific drug delivery. Mol Pharmaceut. 2012;9:1262–1270. doi: 10.1021/mp2005615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Passirani C, Barratt G, Devissaguet J, Labarre D. Long-circulating nanoparticles bearing heparin or dextran covalently bound to poly(methyl methacrylate) Pharm Res. 1998;15:1046–1050. doi: 10.1023/a:1011930127562. [DOI] [PubMed] [Google Scholar]
- 66.Doh KO, Yeo Y. Application of polysaccharides for surface modification of nanomedicines. Therapeutic Delivery. 2012;3:1447–1456. doi: 10.4155/tde.12.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fang C, Shi B, Pei YY, Hong MH, Wu J, Chen HZ. In vivo tumor targeting of tumor necrosis factor-alpha-loaded stealth nanoparticles: effect of MePEG molecular weight and particle size. Eur J Pharm Sci. 2006;27:27–36. doi: 10.1016/j.ejps.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 68.Goppert TM, Muller RH. Protein adsorption patterns on poloxamer- and poloxamine-stabilized solid lipid nanoparticles (SLN) Eur J Pharm Biopharm. 2005;60:361–372. doi: 10.1016/j.ejpb.2005.02.006. [DOI] [PubMed] [Google Scholar]
- 69.Gessner A, Waicz R, Lieske A, Paulke B, Mader K, Muller RH. Nanoparticles with decreasing surface hydrophobicities: influence on plasma protein adsorption. Int J Pharm. 2000;196:245–249. doi: 10.1016/s0378-5173(99)00432-9. [DOI] [PubMed] [Google Scholar]
- 70.Zhang W, Liu J, Li S, Chen M, Liu H. Preparation and evaluation of stealth Tashinone IIA-loaded solid lipid nanoparticles: Influence of Poloxamer 188 coating on phagocytic uptake. J Microencapsul. 2008;25:203–209. doi: 10.1080/02652040701852181. [DOI] [PubMed] [Google Scholar]
- 71.Bocca C, Caputo O, Cavalli RB, Gabriel L, Miglietta A, Gasco MR. Phagocytic uptake of fluorescent stealth and non-stealth solid lipid nanoparticles. Int J Pharm. 1998;175:185–193. [Google Scholar]
- 72.Rolland A, Merdrignac G, Gouranton J, Bourel D, Le Verge R, Genetet B. Flow cytometric quantitative evaluation of phagocytosis by human mononuclear and polymorphonuclear cells using fluorescent nanoparticles. J Immunol Methods. 1987;96:185–193. doi: 10.1016/0022-1759(87)90313-9. [DOI] [PubMed] [Google Scholar]
- 73.Vonarbourg A, Passirani C, Saulnier P, Benoit JP. Parameters influencing the stealthiness of colloidal drug delivery systems. Biomaterials. 2006;27:4356–4373. doi: 10.1016/j.biomaterials.2006.03.039. [DOI] [PubMed] [Google Scholar]
- 74.Champion J, Mitragotri S. Role of target geometry in phagocytosis. Proc Natl Acad Sci U S A. 2006;103:4930–4934. doi: 10.1073/pnas.0600997103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Arnida, Janat-Amsbury MM, Ray A, Peterson CM, Ghandehari H. Geometry and surface characteristics of gold nanoparticles influence their biodistribution uptake by macrophages. Eur J Pharm Biopharm. 2011;77:417–423. doi: 10.1016/j.ejpb.2010.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Longmire MR, Ogawa M, Choyke PL, Kobayashi H. Biologically optimized nanosized molecules and particles: more than just size. Bioconjug Chem. 2011;22:993–1000. doi: 10.1021/bc200111p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Geng Y, Dalhaimer P, Cai S, Tsai R, Tewari M, Minko T, Discher DE. Shape effects of filaments versus spherical particles in flow and drug delivery. Nat Nano. 2007;2:249–255. doi: 10.1038/nnano.2007.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bertholon I, Vauthier C, Labarre D. Complement activation by core-shell poly(isobutylcyanoacrylate)-polysaccharide nanoparticles: influences of surface morphology, length, and type of polysaccharide. Pharm Res. 2006;23:1313–1323. doi: 10.1007/s11095-006-0069-0. [DOI] [PubMed] [Google Scholar]
- 79.Sahu A, Lambris J. Structure and biology of complement protein C3, a connecting link between innate and acquired immunity. Immunol Rev. 2001;180:35–48. doi: 10.1034/j.1600-065x.2001.1800103.x. [DOI] [PubMed] [Google Scholar]
- 80.Laurell C. Quantitative estimation of proteins by electrophoresis in agarose gel containing antibodies. Anal Biochem. 1966;15:45–52. doi: 10.1016/0003-2697(66)90246-6. [DOI] [PubMed] [Google Scholar]
- 81.Boackle R, Caughman G, Vesely J, Medgyesi G, Fudenberg H. Potentiation of factor H by heparin: a rate-limiting mechanism for inhibition of the alternative complement pathway. Mol Immunol. 1983;20:1157–1164. doi: 10.1016/0161-5890(83)90139-6. [DOI] [PubMed] [Google Scholar]
- 82.Alhareth K, Vauthier C, Bourasset F, Gueutin C, Ponchel G, Moussa F. Conformation of surface-decorating dextran chains affects the pharmacokinetics and biodistribution of doxorubicin-loaded nanoparticles. Eur J Pharm Biopharm. 2012;81:453–457. doi: 10.1016/j.ejpb.2012.03.009. [DOI] [PubMed] [Google Scholar]
- 83.Vauthier C, Persson B, Lindner P, Cabane B. Protein adsorption and complement activation for di-block copolymer nanoparticles. Biomaterials. 2011;32:1646–1656. doi: 10.1016/j.biomaterials.2010.10.026. [DOI] [PubMed] [Google Scholar]
- 84.Labarre D, Vauthier C, Chauvierre C, Petri B, Muller R, Chehimi MM. Interactions of blood proteins with poly(isobutylcyanoacrylate) nanoparticles decorated with a polysaccharidic brush. Biomaterials. 2005;26:5075–5084. doi: 10.1016/j.biomaterials.2005.01.019. [DOI] [PubMed] [Google Scholar]
- 85.Suzuki Y, Miyatake K, Okamoto Y, Muraki E, Minami S. Influence of the chain length of chitosan on complement activation. Carbohyd Polym. 2003;54:465–469. [Google Scholar]
- 86. [Accessed on 10/5/12]; http://ncl.cancer.gov/assay_cascade.asp.
- 87.Xu P, Gullotti E, Tong L, Highley CB, Errabelli DR, Hasan T, Cheng JX, Kohane DS, Yeo Y. Intracellular drug delivery by poly(lactic-co-glycolic acid) nanoparticles, revisited. Mol Pharmaceut. 2009;6:190–201. doi: 10.1021/mp800137z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ekkapongpisit M, Giovia A, Follo C, Caputo G, Isidoro C. Biocompatibility, endocytosis, and intracellular trafficking of mesoporous silica and polystyrene nanoparticles in ovarian cancer cells: effects of size and surface charge groups. Int J Nanomed. 7:4147–4158. doi: 10.2147/IJN.S33803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Brown M, Wittwer C. Flow cytometry: Principles and clinical applications in hematology. Clin Chem. 2000;46:1221–1229. [PubMed] [Google Scholar]
- 90.Anirudhan TS, Sandeep S. Synthesis, characterization, cellular uptake and cytotoxicity of a multifunctional magnetic nanocomposite for the targeted delivery and controlled release of doxorubicin to cancer cells. J Mater Chem. 22:12888–12899. [Google Scholar]
- 91.Anirudhan TS, Sandeep S. Synthesis and characterization of a novel pH-controllable composite hydrogel for anticancer drug delivery. New J Chem. 35:2869–2876. [Google Scholar]
- 92.Fujii K, Kuroda T, Sakoda K, Iyi N. Fluorescence resonance energy transfer and arrangements of fluorophores in integrated coumarin/cyanine systems within solid-state two-dimensional nanospace. J Photoch Photobio A. 225:125–134. [Google Scholar]
- 93.Gratton SEA, Ropp PA, Pohlhaus PD, Luft JC, Madden VJ, Napier ME, DeSimone JM. The effect of particle design on cellular internalization pathways. Proc Natl Acad Sci U S A. 2008;105:11613–11618. doi: 10.1073/pnas.0801763105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kim TH, Mount CW, Gombotz WR, Pun SH. The delivery of doxorubicin to 3-D multicellular spheroids and tumors in a murine xenograft model using tumor-penetrating triblock polymeric micelles. Biomaterials. 31:7386–7397. doi: 10.1016/j.biomaterials.2010.06.004. [DOI] [PubMed] [Google Scholar]
- 95.Huang RB, Mocherla S, Heslinga MJ, Charoenphol P, Eniola-Adefeso O. Dynamic and cellular interactions of nanoparticles in vascular-targeted drug delivery. Mol Membr Biol. 2010;27:312–327. doi: 10.3109/09687688.2010.522117. [DOI] [PubMed] [Google Scholar]
- 96.Harush-Frenkel O, Altschuler Y, Benita S. Nanoparticle-cell interactions: drug delivery implications. Crit Rev Ther Drug Carrier Syst. 2008;25:485–544. doi: 10.1615/critrevtherdrugcarriersyst.v25.i6.10. [DOI] [PubMed] [Google Scholar]
- 97.Soldati T, Schliwa M. Powering membrane traffic in endocytosis and recycling. Nat Rev Mol Cell Biol. 2006;7:897–908. doi: 10.1038/nrm2060. [DOI] [PubMed] [Google Scholar]
- 98.Vandeurs B, Petersen OW, Olsnes S, Sandvig K. The ways of endocytosis. Int Rev Cytol. 1989;117:131–177. doi: 10.1016/s0074-7696(08)61336-4. [DOI] [PubMed] [Google Scholar]
- 99.Garnett MC, Kallinteri P. Nanomedicines and nanotoxicology: some physiological principles. Occup Med (Lond) 2006;56:307–311. doi: 10.1093/occmed/kql052. [DOI] [PubMed] [Google Scholar]
- 100.Polo S, Di Fiore PP. Endocytosis conducts the cell signaling orchestra. Cell. 2006;124:897–900. doi: 10.1016/j.cell.2006.02.025. [DOI] [PubMed] [Google Scholar]
- 101.Conner SD, Schmid SL. Regulated portals of entry into the cell. Nature. 2003;422:37–44. doi: 10.1038/nature01451. [DOI] [PubMed] [Google Scholar]
- 102.Roth MG. Clathrin-mediated endocytosis before fluorescent proteins. Nat Rev Mol Cell Biol. 2006;7:63–68. doi: 10.1038/nrm1783. [DOI] [PubMed] [Google Scholar]
- 103.Rejman J, Oberle V, Zuhorn IS, Hoekstra D. Size-dependent internalization of particles via the pathways of clathrin-and caveolae-mediated endocytosis. Biochem J. 2004;377:159–169. doi: 10.1042/BJ20031253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Sabharanjak S, Mayor S. Folate receptor endocytosis and trafficking. Adv Drug Deliv Rev. 2004;56:1099–1109. doi: 10.1016/j.addr.2004.01.010. [DOI] [PubMed] [Google Scholar]
- 105.Grant BD, Donaldson JG. Pathways and mechanisms of endocytic recycling. Nat Rev Mol Cell Biol. 2009;10:597–608. doi: 10.1038/nrm2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Carver LA, Schnitzer JE. Caveolae: Mining little caves for new cancer targets. Nat Rev Cancer. 2003;3:571–581. doi: 10.1038/nrc1146. [DOI] [PubMed] [Google Scholar]
- 107.Ivanov AI. Pharmacological inhibition of endocytic pathways: is it specific enough to be useful? Methods Mol Biol. 2008;440:15–33. doi: 10.1007/978-1-59745-178-9_2. [DOI] [PubMed] [Google Scholar]
- 108.Perumal OP, Inapagolla R, Kannan S, Kannan RM. The effect of surface functionality on cellular trafficking of dendrimers. Biomaterials. 2008;29:3469–3476. doi: 10.1016/j.biomaterials.2008.04.038. [DOI] [PubMed] [Google Scholar]
- 109.Ranjan A, Pothayee N, Seleem MN, Sriranganathan N, Kasimanickam R, Makris M, Riffle JS. In vitro trafficking and efficacy of core-shell nanostructures for treating intracellular Salmonella infections. Antimicrob Agents Chemother. 2009;53:3985–3988. doi: 10.1128/AAC.00009-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Albanese A, Tang PS, Chan WCW. The effect of nanoparticle size, shape, and surface chemistry on biological systems. Annu Rev Biomed Eng. 2012;14:1–16. doi: 10.1146/annurev-bioeng-071811-150124. [DOI] [PubMed] [Google Scholar]
- 111.Myung JH, Gajjar KA, Saric J, Eddington DT, Hong S. Dendrimer-mediated multivalent binding for the enhanced capture of tumor cells. Angew Chem Int Ed Engl. 2011;50:11769–11772. doi: 10.1002/anie.201105508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Jiang W, KimBetty YS, Rutka JT, ChanWarren CW. Nanoparticle-mediated cellular response is size-dependent. Nat Nano. 2008;3:145–150. doi: 10.1038/nnano.2008.30. [DOI] [PubMed] [Google Scholar]
- 113.Liu SQ, Wiradharma N, Gao SJ, Tong YW, Yang YY. Bio-functional micelles self-assembled from a folate-conjugated block copolymer for targeted intracellular delivery of anticancer drugs. Biomaterials. 2007;28:1423–1433. doi: 10.1016/j.biomaterials.2006.11.013. [DOI] [PubMed] [Google Scholar]
- 114.Bae Y, Nishiyama N, Kataoka K. In vivo antitumor activity of the folate-conjugated pH-Sensitive polymeric micelle selectively releasing adriamycin in the intracellular acidic compartments. Bioconjug Chem. 2007;18:1131–1139. doi: 10.1021/bc060401p. [DOI] [PubMed] [Google Scholar]
- 115.Park JH, von Maltzahn G, Xu MJ, Fogal V, Kotamraju VR, Ruoslahti E, Bhatia SN, Sailor MJ. Cooperative nanomaterial system to sensitize, target, and treat tumors. Proc Natl Acad Sci U S A. 2010;107:981–986. doi: 10.1073/pnas.0909565107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kim SH, Jeong JH, Chun KW, Park TG. Target-specific cellular uptake of PLGA nanoparticles coated with poly(L-lysine)-poly(ethylene glycol)-folate conjugate. Langmuir. 2005;21:8852–8857. doi: 10.1021/la0502084. [DOI] [PubMed] [Google Scholar]
- 117.Jiang G, Park K, Kim J, Kim KS, Hahn SK. Target specific intracellular delivery of siRNA/PEI-HA complex by receptor mediated endocytosis. Mol Pharmaceut. 2009;6:727–737. doi: 10.1021/mp800176t. [DOI] [PubMed] [Google Scholar]
- 118.Han SE, Kang H, Shim GY, Kim SJ, Choi HG, Kim J, Hahn SK, Oh YK. Cationic derivatives of biocompatible hyaluronic acids for delivery of siRNA and antisense oligonucleotides. J Drug Target. 2009;17:123–132. doi: 10.1080/10611860802472461. [DOI] [PubMed] [Google Scholar]
- 119.Zhao F, Zhao Y, Liu Y, Chang XL, Chen CY, Zhao YL. Cellular uptake, intracellular trafficking, and cytotoxicity of nanomaterials. Small. 2011;7:1322–1337. doi: 10.1002/smll.201100001. [DOI] [PubMed] [Google Scholar]
- 120.Huang JG, Leshuk T, Gu FX. Emerging nanomaterials for targeting subcellular organelles. Nano Today. 2011;6:478–492. [Google Scholar]
- 121.Harush-Frenkel O, Debotton N, Benita S, Altschuler Y. Targeting of nanoparticles to the clathrin-mediated endocytic pathway. Biochem Biophys Res Commun. 2007;353:26–32. doi: 10.1016/j.bbrc.2006.11.135. [DOI] [PubMed] [Google Scholar]
- 122.Schnell U, Dijk F, Sjollema KA, Giepmans BNG. Immunolabeling artifacts and the need for live-cell imaging. Nat Methods. 2012;9:152–158. doi: 10.1038/nmeth.1855. [DOI] [PubMed] [Google Scholar]
- 123.Melan MA, Sluder G. Redistribution and differential extraction of soluble proteins in permeabilized cultured cells. Implications for immunofluorescence microscopy. J Cell Sci. 1992;101:731–743. doi: 10.1242/jcs.101.4.731. [DOI] [PubMed] [Google Scholar]
- 124.Slater K. Cytotoxicity tests for high-throughput drug discovery. Curr Opin Biotech. 2001;12:70–74. doi: 10.1016/s0958-1669(00)00177-4. [DOI] [PubMed] [Google Scholar]
- 125.Decker T, Lohmann-Matthes ML. A quick and simple method for the quantitation of lactate dehydrogenase release in measurements of cellular cytotoxicity and tumor necrosis factor (TNF) activity. J Immunol Methods. 1988;115:61–69. doi: 10.1016/0022-1759(88)90310-9. [DOI] [PubMed] [Google Scholar]
- 126.Lei TJ, Srinivasan S, Tang Y, Manchanda R, Nagesetti A, Fernandez-Fernandez A, McGoron AJ. Comparing cellular uptake and cytotoxicity of targeted drug carriers in cancer cell lines with different drug resistance mechanisms. Nanomed-Nanotechnol. 7:324–332. doi: 10.1016/j.nano.2010.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Gullotti E, Yeo Y. Beyond the imaging: Limitations of cellular uptake study in the evaluation of nanoparticles. J Control Release. 2012;164:170–176. doi: 10.1016/j.jconrel.2012.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Holback H, Yeo Y. Intratumoral drug delivery with nanoparticulate carriers. Pharm Res. 2011;28:1819–1830. doi: 10.1007/s11095-010-0360-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Lee GY, Kenny PA, Lee EH, Bissell MJ. Three-dimensional culture models of normal and malignant breast epithelial cells. Nat Methods. 2007;4:359–365. doi: 10.1038/nmeth1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Mitra M, Mohanty C, Harilal A, Maheswari UK, Sahoo SK, Krishnakumar S. A novel in vitro three-dimensional retinoblastoma model for evaluating chemotherapeutic drugs. Mol Vis. 18:1361–1378. [PMC free article] [PubMed] [Google Scholar]
- 131.Sun T, Jackson S, Haycock JW, MacNeil S. Culture of skin cells in 3D rather than 2D improves their ability to survive exposure to cytotoxic agents. J Biotechnol. 2006;122:372–381. doi: 10.1016/j.jbiotec.2005.12.021. [DOI] [PubMed] [Google Scholar]
- 132.Hosoya H, Kadowaki K, Matsusaki M, Cabral H, Nishihara H, Ijichi H, Koike K, Kataoka K, Miyazono K, Akashi M, Kano MR. Engineering fibrotic tissue in pancreatic cancer: A novel three-dimensional model to investigate nanoparticle delivery. Biochem Bioph Res Co. 419:32–37. doi: 10.1016/j.bbrc.2012.01.117. [DOI] [PubMed] [Google Scholar]
- 133.Khademhosseini A, Eng G, Yeh J, Fukuda J, Blumling J, III, Langer R, Burdick JA. Micromolding of photocrosslinkable hyaluronic acid for cell encapsulation and entrapment. J Biomed Mater Res A. 2006;79:522–532. doi: 10.1002/jbm.a.30821. [DOI] [PubMed] [Google Scholar]
- 134.Ho WJ, Pham EA, Kim JW, Ng CW, Kim JH, Kamei DT, Wu BM. Incorporation of multicellular spheroids into 3-D polymeric scaffolds provides an improved tumor model for screening anticancer drugs. Cancer Sci. 101:2637–2643. doi: 10.1111/j.1349-7006.2010.01723.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Astashkina AI, Mann BK, Prestwich GD, Grainger DW. A 3-D organoid kidney culture model engineered for high-throughput nephrotoxicity assays. Biomaterials. 33:4700–4711. doi: 10.1016/j.biomaterials.2012.02.063. [DOI] [PubMed] [Google Scholar]
- 136.Ng CP, Pun SH. A perfusable 3D cell-matrix tissue culture chamber for in situ evaluation of nanoparticle vehicle penetration and transport. Biotechnol Bioeng. 2008;99:1490–1501. doi: 10.1002/bit.21698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Blake AJ, Pearce TM, Rao NS, Johnson SM, Williams JC. Multilayer PDMS microfluidic chamber for controlling brain slice microenvironment. Lab Chip. 2007;7:842–849. doi: 10.1039/b704754a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Chang R, Emami K, Wu H, Sun W. Biofabrication of a three-dimensional liver micro-organ as an in vitro drug metabolism model. Biofabrication. 2010;2:045004. doi: 10.1088/1758-5082/2/4/045004. [DOI] [PubMed] [Google Scholar]
- 139.Zhang H, Lee MY, Hogg MG, Dordick JS, Sharfstein ST. Gene delivery in three-dimensional cell cultures by superparamagnetic nanoparticles. ACS Nano. 2010;4:4733–4743. doi: 10.1021/nn9018812. [DOI] [PubMed] [Google Scholar]
- 140.Szot CS, Buchanan CF, Freeman JW, Rylander MN. 3D in vitro bioengineered tumors based on collagen I hydrogels. Biomaterials. 32:7905–7912. doi: 10.1016/j.biomaterials.2011.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Gurski LA, Jha AK, Zhang C, Jia XQ, Farach-Carson MC. Hyaluronic acid-based hydrogels as 3D matrices for in vitro evaluation of chemotherapeutic drugs using poorly adherent prostate cancer cells. Biomaterials. 2009;30:6076–6085. doi: 10.1016/j.biomaterials.2009.07.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Loessner D, Stok KS, Lutolf MP, Hutmacher DW, Clements JA, Rizzi SC. Bioengineered 3D platform to explore cell-ECM interactions and drug resistance of epithelial ovarian cancer cells. Biomaterials. 31:8494–8506. doi: 10.1016/j.biomaterials.2010.07.064. [DOI] [PubMed] [Google Scholar]
- 143.Zhang SG, Gelain F, Zhao XJ. Designer self-assembling peptide nanofiber scaffolds for 3D tissue cell cultures. Semin Cancer Biol. 2005;15:413–420. doi: 10.1016/j.semcancer.2005.05.007. [DOI] [PubMed] [Google Scholar]
- 144.Derda R, Laromaine A, Mammoto A, Tang SKY, Mammoto T, Ingber DE, Whitesides GM. Paper-supported 3D cell culture for tissue-based bioassays. Proc Natl Acad Sci U S A. 2009;106:18457–18462. doi: 10.1073/pnas.0910666106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Minchinton AI, Tannock IF. Drug penetration in solid tumours. Nat Rev Cancer. 2006;6:583–592. doi: 10.1038/nrc1893. [DOI] [PubMed] [Google Scholar]
- 146.Kim B, Han G, Toley BJ, Kim C-k, Rotello VM, Forbes NS. Tuning payload delivery in tumour cylindroids using gold nanoparticles. Nat Nano. 2010;5:465–472. doi: 10.1038/nnano.2010.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Huang SK, Mayhew E, Gilani S, Lasic DD, Martin FJ, Papahadjopoulos D. Pharmacokinetics and therapeutics of sterically stabilized liposomes in mice bearing C-26 colon carcinoma. Cancer Res. 1992;52:6774–81. [PubMed] [Google Scholar]
- 148.Vaage J, Barberaguillem E, Abra R, Huang A, Working P. Tissue distribution and therapeutic effect of intravenous free or encapsulated liposomal doxorubicin on human prostate carcinoma xenografts. Cancer. 1994;73:1478–1484. doi: 10.1002/1097-0142(19940301)73:5<1478::aid-cncr2820730526>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 149.Vaage J, Mayhew E, Lasic D, Martin F. Therapy of primary and metastatic mouse mammary carcinomas with doxorubicin encapsulated in long circulating liposomes. Int J Cancer. 1992;51:942–948. doi: 10.1002/ijc.2910510618. [DOI] [PubMed] [Google Scholar]
- 150.Vaage J, Donovan D, Mayhew E, Abra R, Huang A. Therapy of human ovarian carcinoma xenografts using doxorubicin encapsulated in sterically stabilized liposomes. Cancer. 1993;72:3671–3675. doi: 10.1002/1097-0142(19931215)72:12<3671::aid-cncr2820721219>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 151.Vaage J, Donovan D, Uster P, Working P. Tumour uptake of doxorubicin in polyethylene glycol-coated liposomes and therapeutic effect against a xenografted human pancreatic carcinoma. Brit J Cancer. 1997;75:482–486. doi: 10.1038/bjc.1997.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.O’Brien ME, Wigler N, Inbar M, Rosso R, Grischke E, Santoro A, Catane R, Kieback DG, Tomczak P, Ackland SP, Orlandi F, Mellars L, Alland L, Tendler C. Reduced cardiotoxicity and comparable efficacy in a phase III trial of pegylated liposomal doxorubicin HCl (CAELYX/Doxil) versus conventional doxorubicin for first-line treatment of metastatic breast cancer. Ann Oncol. 2004;15:440–449. doi: 10.1093/annonc/mdh097. [DOI] [PubMed] [Google Scholar]
- 153.Ranson MR, Carmichael J, Obyrne K, Stewart S, Smith D, Howell A. Treatment of advanced breast cancer with sterically stabilized liposomal doxorubicin: Results of a multicenter phase II trial. J Clin Oncol. 1997;15:3185–3191. doi: 10.1200/JCO.1997.15.10.3185. [DOI] [PubMed] [Google Scholar]
- 154.Van Oosterom AT, Mouridsen HT, Wildiers J, Paridaens R, Cocconi G, Rotmensz N, Sylvester R. Carminomycin versus doxorubicin in advanced breast cancer, a randomized phase II study of the E.O.R.T.C. Breast Cancer Cooperative Group. Eur J Cancer Clin Oncol. 1986;22:601–605. doi: 10.1016/0277-5379(86)90050-7. [DOI] [PubMed] [Google Scholar]
- 155.Lawton PA, Spittle MF, Ostrowski MJ, Young T, Madden F, Folkes A, Hill BT, MacRae K. A comparison of doxorubicin, epirubicin and mitozantrone as single agents in advanced breast carcinoma. Clin Oncol (R Coll Radiol) 1993;5:80–84. doi: 10.1016/s0936-6555(05)80851-9. [DOI] [PubMed] [Google Scholar]
- 156.Cowan JD, Osborne CK, Neidhart JA, Von Hoff DD, Constanzi JJ, Vaughn CB. A randomized trial of doxorubicin, mitoxantrone and bisantrene in advanced breast cancer (a South West Oncology Group Study) Invest New Drugs. 1985;3:149–152. doi: 10.1007/BF00174162. [DOI] [PubMed] [Google Scholar]
- 157.Pfeiffer P, Cold S, Rose C. Cytotoxic treatment of metastatic breast cancer. Which drugs and drug combinations to use? Acta Oncol. 1992;31:219–224. doi: 10.3109/02841869209088906. [DOI] [PubMed] [Google Scholar]
- 158.Duncan R, Seymour LW, O’Hare KB, Flanagan PA, Wedge S, Hume IC, Ulbrich K, Strohalm J, Subr V, Spreafico F, Grandi M, Ripamonti M, Farao M, Suarato A. Preclinical evaluation of polymer-bound doxorubicin. J Control Release. 1992;19:331–346. [Google Scholar]
- 159.Duncan R. Drug-polymer conjugates: potential for improved chemotherapy. Anticancer Drugs. 1992;3:175–210. doi: 10.1097/00001813-199206000-00001. [DOI] [PubMed] [Google Scholar]
- 160.Seymour LW, Ferry DR, Kerr DJ, Rea D, Whitlock M, Poyner R, Boivin C, Hesslewood S, Twelves C, Blackie R, Schatzlein A, Jodrell D, Bissett D, Calvert H, Lind M, Robbins A, Burtles S, Duncan R, Cassidy J. Phase II studies of polymer-doxorubicin (PK1, FCE28068) in the treatment of breast, lung and colorectal cancer. Int J Oncol. 2009;34:1629–1636. doi: 10.3892/ijo_00000293. [DOI] [PubMed] [Google Scholar]
- 161.Li C, Yu DF, Newman RA, Cabral F, Stephens LC, Hunter N, Milas L, Wallace S. Complete regression of well-established tumors using a novel water-soluble poly(L-glutamic acid)-paclitaxel conjugate. Cancer Res. 1998;58:2404–2409. [PubMed] [Google Scholar]
- 162.Sabbatini P, Aghajanian C, Dizon D, Anderson S, Dupont J, Brown JV, Peters WA, Jacobs A, Mehdi A, Rivkin S, Eisenfeld AJ, Spriggs D. Phase II study of CT-2103 in patients with recurrent epithelial ovarian, fallopian tube, or primary peritoneal carcinoma. J Clin Oncol. 2004;22:4523–4531. doi: 10.1200/JCO.2004.12.043. [DOI] [PubMed] [Google Scholar]
- 163.Sabbatini P, Sill MW, O’Malley D, Adler L, Secord AA. A phase II trial of paclitaxel poliglumex in recurrent or persistent ovarian or primary peritoneal cancer (EOC): A gynecologic oncology group study. Gynecol Oncol. 2008;111:455–460. doi: 10.1016/j.ygyno.2008.07.049. [DOI] [PubMed] [Google Scholar]
- 164.Yang D, Yu L, Van S. Clinically relevant anticancer polymer paclitaxel therapeutics. Cancers. 2010;3:17–42. doi: 10.3390/cancers3010017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.McGuire WP, Hoskins WJ, Brady MF, Kucera PR, Partridge EE, Look KY, ClarkePearson DL, Davidson M. Cyclophosphamide and cisplatin compared with paclitaxel and cisplatin in patients with stage III and stage IV ovarian cancer. New Engl J Med. 1996;334:1–6. doi: 10.1056/NEJM199601043340101. [DOI] [PubMed] [Google Scholar]
- 166. [Accessed on 10/15/12];European Medicines Agency Questions and answers on the withdrawal of the marketing authorisation application for Opaxio (Paclitaxel poliglumex) http://www.ema.europa.eu/docs/en_GB/document_library/Medicine_QA/2010/01/WC500060347.pdf.
- 167.Bibby MC. Orthotopic models of cancer for preclinical drug evaluation: advantages and disadvantages. Eur J Cancer. 2004;40:852–857. doi: 10.1016/j.ejca.2003.11.021. [DOI] [PubMed] [Google Scholar]
- 168.Kelland LR. “Of mice and men”: values and liabilities of the athymic nude mouse model in anticancer drug development. Eur J Cancer. 2004;40:827–836. doi: 10.1016/j.ejca.2003.11.028. [DOI] [PubMed] [Google Scholar]
- 169.Damia G, D’Incalci M. Contemporary pre-clinical development of anticancer agents - What are the optimal preclinical models? Eur J Cancer. 2009;45:2768–2781. doi: 10.1016/j.ejca.2009.08.008. [DOI] [PubMed] [Google Scholar]
- 170.Kamb A. What’s wrong with our cancer models? Nat Rev Drug Discov. 2005;4:161–165. doi: 10.1038/nrd1635. [DOI] [PubMed] [Google Scholar]
- 171.Sausville EA, Burger AM. Contributions of human tumor xenografts to anticancer drug development. Cancer Res. 2006;66:3351–3354. doi: 10.1158/0008-5472.CAN-05-3627. [DOI] [PubMed] [Google Scholar]
- 172.Talmadge JE, Singh RK, Fidler IJ, Raz A. Murine models to evaluate novel and conventional therapeutic strategies for cancer. Am J Pathol. 2007;170:793–804. doi: 10.2353/ajpath.2007.060929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Ishida T, Masuda K, Ichikawa T, Ichihara M, Irimura K, Kiwada H. Accelerated clearance of a second injection of PEGylated liposomes in mice. Int J Pharm. 2003;255:167–174. doi: 10.1016/s0378-5173(03)00085-1. [DOI] [PubMed] [Google Scholar]
- 174.Ishida T, Maeda R, Ichihara M, Irimura K, Kiwada H. Accelerated clearance of PEGylated liposomes in rats after repeated injections. J Control Release. 2003;88:35–42. doi: 10.1016/s0168-3659(02)00462-5. [DOI] [PubMed] [Google Scholar]
- 175.Ishida T, Ichihara M, Wang X, Yamamoto K, Kimura J, Majima E, Kiwada H. Injection of PEGylated liposomes in rats elicits PEG-specific IgM, which is responsible for rapid elimination of a second dose of PEGylated liposomes. J Control Release. 2006;112:15–25. doi: 10.1016/j.jconrel.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 176.Karathanasis E, Chan L, Karumbaiah L, McNeeley K, D’Orsi CJ, Annapragada AV, Sechopoulos I, Bellamkonda RV. Tumor vascular permeability to a nanoprobe correlates to tumor-specific expression levels of angiogenic markers. PLoS One. 2009;4:e5843. doi: 10.1371/journal.pone.0005843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Greish K. Enhanced permeability and retention of macromolecular drugs in solid tumors: A royal gate for targeted anticancer nanomedicines. J Drug Target. 2007;15:457–464. doi: 10.1080/10611860701539584. [DOI] [PubMed] [Google Scholar]
- 178.Fang J, Nakamura H, Maeda H. The EPR effect: Unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv Drug Deliv Rev. 2011;63:136–151. doi: 10.1016/j.addr.2010.04.009. [DOI] [PubMed] [Google Scholar]
- 179.McDonald DM, Baluk P. Significance of blood vessel leakiness in cancer. Cancer Res. 2002;62:5381–5385. [PubMed] [Google Scholar]
- 180.Killion JJ, Radinsky R, Fidler IJ. Orthotopic models are necessary to predict therapy of transplantable tumors in mice. Cancer Metastasis Rev. 1998;17:279–284. doi: 10.1023/a:1006140513233. [DOI] [PubMed] [Google Scholar]
- 181.Kung AL. Practices and pitfalls of mouse cancer models in drug discovery. Adv Cancer Res. 2007;96:191–212. doi: 10.1016/S0065-230X(06)96007-2. [DOI] [PubMed] [Google Scholar]
- 182.Fidler IJ, Wilmanns C, Staroselsky A, Radinsky R, Dong Z, Fan D. Modulation of tumor cell response to chemotherapy by the organ environment. Cancer Metastasis Rev. 1994;13:209–222. doi: 10.1007/BF00689637. [DOI] [PubMed] [Google Scholar]
- 183.Kuo TH, Kubota T, Watanabe M, Furukawa T, Kase S, Tanino H, Saikawa Y, Ishibiki K, Kitajima M, Hoffman RM. Site-specific chemosensitivity of human small-cell lung carcinoma growing orthotopically compared to subcutaneously in SCID mice: the importance of orthotopic models to obtain relevant drug evaluation data. Anticancer Res. 1993;13:627–630. [PubMed] [Google Scholar]
- 184.Hoffman RM. Orthotopic metastatic (MetaMouse) models for discovery and development of novel chemotherapy. Methods Mol Med. 2005;111:297–322. doi: 10.1385/1-59259-889-7:297. [DOI] [PubMed] [Google Scholar]
- 185.Chishima T, Miyagi Y, Wang XE, Yamaoka H, Shimada H, Moossa AR, Hoffman RM. Cancer invasion and micrometastasis visualized in live tissue by green fluorescent protein expression. Cancer Res. 1997;57:2042–2047. [PubMed] [Google Scholar]
- 186.Kobayashi M, Murakami T, Uchibori R, Chun NA, Kobayashi E, Morita T, Ozawa K. Establishment and characterization of transplantable, luminescence labeled rat renal cell carcinoma cell lines. J Urol. 2010;183:2029–2035. doi: 10.1016/j.juro.2009.12.090. [DOI] [PubMed] [Google Scholar]
- 187.Yamamoto N, Jiang P, Yang M, Xu M, Yamauchi K, Tsuchiya H, Tomita K, Wahl GM, Moossa AR, Hoffman RM. Cellular dynamics visualized in live cells in vitro and in vivo by differential dual-color nuclear-cytoplasmic fluorescent-protein expression. Cancer Res. 2004;64:4251–4256. doi: 10.1158/0008-5472.CAN-04-0643. [DOI] [PubMed] [Google Scholar]
- 188.Yan W, Xiao D, Yao K. Combined bioluminescence and fluorescence imaging visualizing orthotopic lung adenocarcinoma xenograft in vivo. Acta Biochim Biophys Sin. 2011;43:595–600. doi: 10.1093/abbs/gmr054. [DOI] [PubMed] [Google Scholar]
- 189.Liu J, Wang Y, Qu X, Li X, Ma X, Han R, Hu Z, Chen X, Sun D, Zhang R, Chen D, Chen D, Chen X, Liang J, Cao F, Tian J. In vivo quantitative bioluminescence tomography using heterogeneous and homogeneous mouse models. Opt Express. 2010;18:13102–13113. doi: 10.1364/OE.18.013102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.El Hilali N, Rubio N, Martinez-Villacampa M, Blanco J. Combined noninvasive imaging and luminometric quantification of luciferase-labeled human prostate tumors and metastases. Lab Invest. 2002;82:1563–1571. doi: 10.1097/01.lab.0000036877.36379.1f. [DOI] [PubMed] [Google Scholar]
- 191.Rosol TJ, Tannehill-Gregg SH, LeRoy BE, Mandl S, Contag CH. Animal models of bone metastasis. Cancer. 2003;97:748–757. doi: 10.1002/cncr.11150. [DOI] [PubMed] [Google Scholar]
- 192.Patra CR, Bhattacharya R, Wang E, Katarya A, Lau JS, Dutta S, Muders M, Wang S, Buhrow SA, Safgren SL, Yaszemski MJ, Reid JM, Ames MM, Mukherjee P, Mukhopadhyay D. Targeted delivery of gemcitabine to pancreatic adenocarcinoma using cetuximab as a targeting agent. Cancer Res. 2008;68:1970–1978. doi: 10.1158/0008-5472.CAN-07-6102. [DOI] [PubMed] [Google Scholar]
- 193.Mugabe C, Matsui Y, So AI, Gleave ME, Baker JH, Minchinton AI, Manisali I, Liggins R, Brooks DE, Burt HM. In vivo evaluation of mucoadhesive nanoparticulate docetaxel for intravesical treatment of non-muscle-invasive bladder cancer. Clin Cancer Res. 2011;17:2788–2798. doi: 10.1158/1078-0432.CCR-10-2981. [DOI] [PubMed] [Google Scholar]
- 194.Phil Oh PB, Czarny Malgorzata, Schnitzer Jan. Host microenvironment dictates tumor growth, angiogenesis and vascular permeability. FASEB J. 2009;23:438.11. Meeting Abstract Supplement. [Google Scholar]
- 195.Schnitzer J, Philip EOH. Borgstrom, Per. Ectopic, orthotopic model for resvascularization and tumor assessment. WO2008108993. International Patent Publication. 2008
- 196.Frost GI, Lustgarten J, Dudouet B, Nyberg L, Hartley-Asp B, Borgstrom P. Novel syngeneic pseudo-orthotopic prostate cancer model: vascular, mitotic and apoptotic responses to castration. Microvasc Res. 2005;69:1–9. doi: 10.1016/j.mvr.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 197.Thomsen M, Yacoub-Youssef H, Marcheix B. Reconstitution of a human immune system in immunodeficient mice: models of human alloreaction in vivo. Tissue Antigens. 2005;66:73–82. doi: 10.1111/j.1399-0039.2005.00409.x. [DOI] [PubMed] [Google Scholar]
- 198.Legrand N, Weijer K, Spits H. Experimental models to study development and function of the human immune system in vivo. J Immunol. 2006;176:2053–8. doi: 10.4049/jimmunol.176.4.2053. [DOI] [PubMed] [Google Scholar]
- 199.Macchiarini F, Manz MG, Palucka AK, Shultz LD. Humanized mice: are we there yet? J Exp Med. 2005;202:1307–11. doi: 10.1084/jem.20051547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Zhou JH, Neff CP, Liu XX, Zhang J, Li HT, Smith DD, Swiderski P, Aboellail T, Huang YY, Du Q, Liang ZC, Peng L, Akkina R, Rossi JJ. Systemic administration of combinatorial dsiRNAs via nanoparticles efficiently suppresses HIV-1 infection in humanized mice. Mol Ther. 2011;19:2228–2238. doi: 10.1038/mt.2011.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Sharpless NE, DePinho RA. Model organisms - The mighty mouse: genetically engineered mouse models in cancer drug development. Nat Rev Drug Discov. 2006;5:741–754. doi: 10.1038/nrd2110. [DOI] [PubMed] [Google Scholar]
- 202.Adams JM, Cory S. Transgenic models of tumor development. Science. 1991;254:1161–1167. doi: 10.1126/science.1957168. [DOI] [PubMed] [Google Scholar]
- 203.Heyer J, Kwong LN, Lowe SW, Chin L. Non-germline genetically engineered mouse models for translational cancer research. Nat Rev Cancer. 2010;10:470–480. doi: 10.1038/nrc2877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Mohammed A, Janakiram NB, Lightfoot S, Gali H, Vibhudutta A, Rao CV. Early detection and prevention of pancreatic cancer: use of genetically engineered mouse models and advanced imaging technologies. Curr Med Chem. 2012;19:3701–13. doi: 10.2174/092986712801661095. [DOI] [PubMed] [Google Scholar]
- 205.Dinulescu DM, Ince TA, Quade BJ, Shafer SA, Crowley D, Jacks T. Role of K-ras and Pten in the development of mouse models of endometriosis and endometrioid ovarian cancer. Nat Med. 2005;11:63–70. doi: 10.1038/nm1173. [DOI] [PubMed] [Google Scholar]
- 206.Sengupta P, Basu S, Soni S, Pandey A, Roy B, Oh MS, Chin KT, Paraskar AS, Sarangi S, Connor Y, Sabbisetti VS, Kopparam J, Kulkarni A, Muto K, Amarasiriwardena C, Jayawardene I, Lupoli N, Dinulescu DM, Bonventre JV, Mashelkar RA, Sengupta S. Cholesterol-tethered platinum II-based supramolecular nanoparticle increases antitumor efficacy and reduces nephrotoxicity. Proc Natl Acad Sci US A. 2012;109:11294–11299. doi: 10.1073/pnas.1203129109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Dibirdik I, Yiv S, Qazi S, Uckun FM. In vivo anti-cancer activity of a liposomal nanoparticle construct of multifunctional tyrosine kinase inhibitor 4-(4′-hydroxyphenyl)-amino-6, 7-dimethoxyquinazoline. J Nanomedic Nanotechnol. 2010;1:001–004. [Google Scholar]
- 208.Uckun FM, Dibirdik I, Qazi S, Vassilev A, Ma H, Mao C, Benyumov A, Emami KH. Anti-breast cancer activity of LFM-A13, a potent inhibitor of Polo-like kinase (PLK) Bioorgan Med Chem. 2007;15:800–814. doi: 10.1016/j.bmc.2006.10.050. [DOI] [PubMed] [Google Scholar]
- 209.Uckun FM, Vassilev AO, Dibirdik I, Liu XP, Erbeck D, Tibbles HE, Qazi S, Venkatachalam TK. Anti-cancer activity profile of 3′-azidothymidine 5′-[p-methoxyphenyl methoxyalaninyl phosphate] (Compound 003), a novel nucleoside analog. Arzneimittelforschung. 2004;54:715–31. doi: 10.1055/s-0031-1297029. [DOI] [PubMed] [Google Scholar]
- 210.Kelly KA, Bardeesy N, Anbazhagan R, Gurumurthy S, Berger J, Alencar H, Depinho RA, Mahmood U, Weissleder R. Targeted nanoparticles for imaging incipient pancreatic ductal adenocarcinoma. PLoS Med. 2008;5:e85. doi: 10.1371/journal.pmed.0050085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Valencia PM, Farokhzad OC, Karnik R, Langer R. Microfluidic technologies for accelerating the clinical translation of nanoparticles. Nat Nanotechnol. 2012;7:623–629. doi: 10.1038/nnano.2012.168. [DOI] [PMC free article] [PubMed] [Google Scholar]