

Abstract
Traumatic injury ranks as the number one cause of death for the under 44 year old age group and 5th leading cause of death overall (www.nationaltraumainstitute.org/home/trauma_statistics.html). Although improved resuscitation of trauma patients has dramatically reduced immediate mortality from hemorrhagic shock, long-term morbidity and mortality continue to be unacceptably high during the post-resuscitation period, particularly as a result of impaired host immune responses to subsequent challenges such as surgery or infection. Acute alcohol intoxication (AAI) is a significant risk factor for traumatic injury; with intoxicating blood alcohol levels present in more than 40% of injured patients (1–5). Severity of trauma, hemorrhagic shock and injury is higher in intoxicated individuals than that of sober victims, resulting in higher mortality rates in this patient population. Necessary invasive procedures (surgery, anesthesia) and subsequent challenges (infection) that intoxicated trauma victims are frequently subjected to are additional stresses to an already compromised inflammatory and neuroendocrine milieu and further contribute to their morbidity and mortality. Thus, dissecting the dynamic imbalance produced by AAI during trauma is of critical relevance for a significant proportion of injured victims. This review outlines how AAI at the time of hemorrhagic shock not only prevents adequate responses to fluid resuscitation but also impairs the ability of the host to overcome a secondary infection. Moreover, it discusses the neuroendocrine mechanisms underlying alcohol-induced hemodynamic dysregulation and its relevance to host defense restoration of homeostasis following injury.
Introduction
The incidence of traumatic injury in alcohol-intoxicated individuals has continued to escalate during recent years. Center for Disease Control-derived data indicates that 15% of Americans that consume alcohol (51% of the population) are "binge drinkers" who have consumed five or more drinks on the same occasion at least once in the past month. This scenario is frequently conducive to traumatic injury; which according to the National Center for Health Statistics is responsible for the greatest number of years of potential life lost before age 65; higher than that attributed to cancer, heart disease, and HIV. Data from the National Center for Injury Prevention and Control as well as that derived from prospective and retrospective studies indicate a 40% incidence of positive blood alcohol concentrations in trauma victims, with 35% presenting with blood alcohol levels above 100 mg/dl and a significant number (75%) of these individuals presenting evidence of chronic alcohol abuse (6, 7). The likelihood of alcohol-related emergency department visits to be injury-related is estimated to be 1.6 times greater than that of the non-intoxicated population. In addition to increasing the incidence of traumatic injury, alcohol intoxication also increases the severity of the injury reflected in greater injury severity scores, as well as an increased need for intensive care, blood transfusions and surgery (8). This contributes to worsened outcomes in the intoxicated trauma patient including increased mortality and acute medical complications such as respiratory failure (9).
Alcohol intoxication aggravates traumatic injury-related hemodynamic instability (10). Alcohol-intoxicated trauma patients are more hypotensive at the time of arrival to the emergency department compared to non-intoxicated individuals (8, 10). Furthermore, intoxicated patients require greater 24 hr intravenous crystalloid fluid resuscitation volumes and significantly more blood products than non-intoxicated patients (11). Low mean arterial blood pressure (MABP) at the time of arrival into the emergency room has been reported to be a predictor of poor patient outcome from traumatic injury and blood loss (12). Thus, alcohol-mediated dysregulation of hemodynamic counteregulatory responses following trauma-hemorrhage is a potentially significant mechanism contributing to the increased morbidity and mortality seen in the alcohol-intoxicated trauma patient.
This review describes a body of work examining the impact of AAI on the neuroendocrine and inflammatory counteregulatory responses to hemorrhagic shock. Collectively, the results from pre-clinical experiments provide substantial evidence for a role of impaired central modulation of the neuroendocrine system in response to hemorrhage as an important mechanism underlying the worsened outcome from injury in the AAI host. Moreover, we discuss how those observations can lead to development of resuscitation strategies tailored to improving clinical outcomes in the AAI hemorrhaged host.
Hemodynamic counteregulatory response to injury involves neuroendocrine and autonomic activation
Understanding of the mechanisms affected by alcohol intoxication that can interfere with the adequate recovery from traumatic injury and hemorrhagic shock requires identification of the principal responses that re-establish homeostasis, their mediators, and how those are triggered by events associated with injury and shock. Immediate alterations in blood volume, hypoxia, pain, and tissue injury trigger the firing of both sensory and motor afferent fibers (Figure 1). These afferent signals have their first relay in the nucleus of the tractus solitarius (NTS) and are predominantly integrated at the level of the periventricular nucleus (PVN) of the hypothalamus. The activation of the descending efferent signals, results in neurotransmitter and hormonal release at the target effector organs where they elicit specific responses. Though several parallel pathways are activated by hypotension and tissue hypoperfusion; the efferent pathways involved in cardiovascular regulation and pressor responses to traumatic injury have been predominantly identified to be of sympatho-motor nature (13, 14). Activation of the sympathetic nervous system (SNS) in response to blood loss is centrally mediated through the balance of inhibitory and excitatory pathways integrated at the hypothalamic PVN (14). SNS activation results in increased adrenal-derived catecholamine release and increased norepinephrine turnover in peripheral organs. In addition, blood loss stimulates effector neurons in the hypothalamus, increasing secretion of corticotropin-releasing hormone (CRH) (15, 16), oxytocin (17) and arginine vasopressin (AVP) (18–20). In concert, these neurotransmitters and neuropeptides, particularly AVP (21), exert dynamic counterregulation aimed at restoring tissue perfusion and MABP. Disruption of hormone activation, release, or target organ responsiveness to their action results in impaired homeostatic mechanisms and delayed restoration of MABP and thus, perfusion pressure leading to tissue hypoperfusion during AAI.
Figure 1. Counteregulatory response to hemorrhagic shock in AAI.
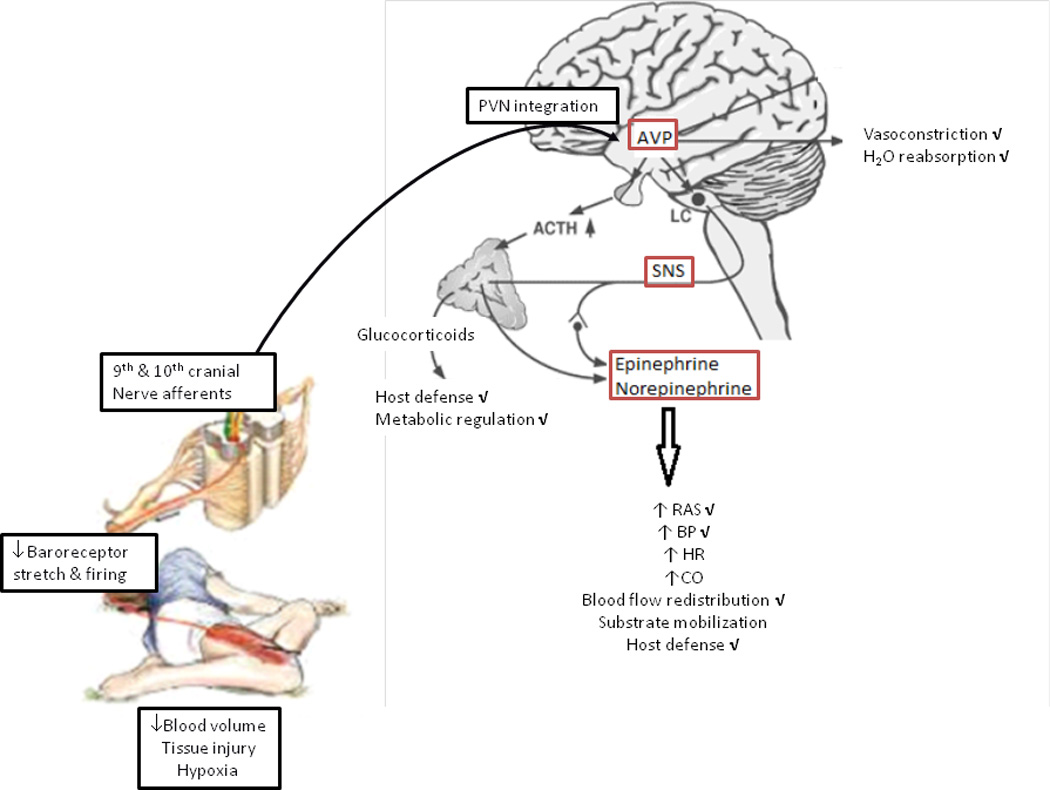
Alterations in blood volume and tissue injury trigger afferent signals that are predominantly integrated at the level of the PVN of the hypothalamus. The activation of the descending efferent signals including the SNS and the HPA results in neurotransmitter and hormone release (i.e., AVP, angiotensin, epinephrine, norepinephrine, and glucocorticoids) that orchestrate a counteregulatory response characterized by increased blood pressure, heart rate, cardiac output, blood flow redistribution, substrate mobilization, modulation of host defense, and substrate mobilization. AAI alters the counteregulatory response at multiple levels shown by the check mark next to each of the specific components. The underlying mechanisms affected by AAI (indicated by the boxes) are primarily due to disruption of the central mechanisms involved in triggering AVP release and SNS activation. ACTH, adrenocorticotropin hormone; AVP, arginine vasopressin; SNS, sympathetic nervous system; RAS, renin angiotensin system; BP, blood pressure; HR, heart rate; CO, cardiac output; HPA, hypothalamo-pituitary axis; PVN, periventricular nucleus.
Impact of acute alcohol intoxication on hemodynamic counteregulation to shock
Studies from our laboratory utilizing a conscious, chronically catheterized rodent model of binge-like alcohol consumption preceding hemorrhagic shock have shown that AAI decreases basal MABP, exacerbates hypotension during hemorrhage, and attenuates blood pressure recovery during fluid resuscitation (Figure 2) (22, 23). In response to a fixed-volume hemorrhage, AAI animals are significantly more hypotensive throughout the hemorrhage and resuscitation period. Hemodynamic deficits are observed in response to fixed-pressure (40 mmHg) hemorrhage when examining the total volume of blood removed from each group to achieve the target pressure. A significantly lesser amount of blood is removed from the AAI animals to achieve a fixed-pressure of 40 mmHg. Furthermore, as was seen with fixed-volume hemorrhage, AAI animals demonstrate an impaired response to fluid resuscitation following fixed-pressure hemorrhage despite having lower amounts of blood removed.
Figure 2. Alcohol intoxication impairs hemodynamic counteregulatory response to hemorrhagic shock and disrupts the integrity of host defense mechanisms in the trauma/hemorrhage host.
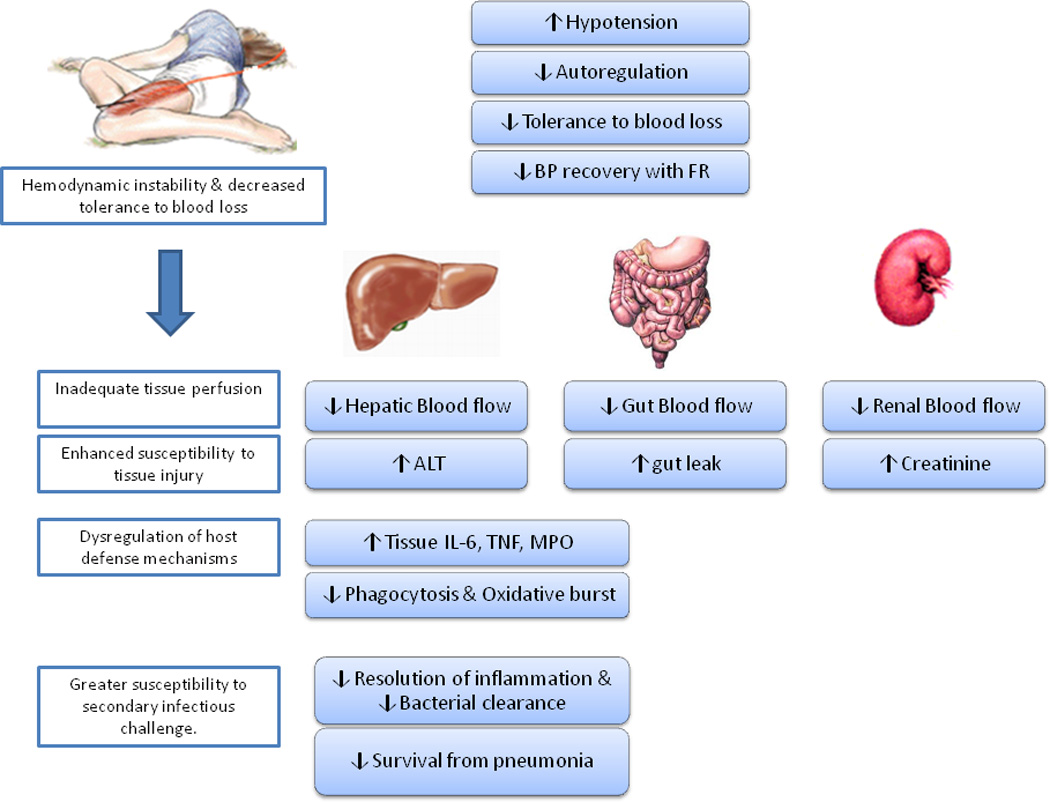
Alcohol intoxicated rodents present with lower blood pressure at time of injury, have decreased tolerance to blood loss, and have impaired blood pressure recovery during fluid resuscitation. The accentuated hypotension leads to tissue hypoperfusion, which enhances susceptibility to tissue injury reflected in greater elevation in circulating alanine ALT and creatinine levels as well as increased intestinal permeability. Moreover, it is associated with increased tissue inflammation and neutrophil recruitment and activation, suppressed phagocytic and oxidative burst activity in circulating mononuclear cells, and early suppression followed by late (5 days post injury) exacerbation of inflammatory response. These alterations in host defense mechanisms are directly related to the inability of the host to effectively resolve a bacterial infection during the post-injury period. ALT, alanine aminotransferase.
This disruption of the hemodynamic response and recovery was predicted to also lead to alterations in the hemorrhage-induced redistribution of blood flow. Hemorrhagic shock results in a selective redistribution of blood flow to vital organs such as the heart and brain and away from others, particularly the splanchnic circulation (24). The impact of AAI on end-organ blood flow has been studied using fluorescent microspheres to determine tissue perfusion 2 hours post-resuscitation (Figure 2). We demonstrated that hemorrhagic shock in AAI animals results in greater reduction of blood flow to the liver, kidney, small and large intestines than that seen in non-intoxicated animals (25). These studies confirmed marked decreases in end-organ blood flow despite fluid resuscitation.
Macro- and microcirculatory changes during trauma and hemorrhage have been implicated in the subsequent development of sepsis and multiple organ failure (26). Thus, the greater hypotension in the AAI animals and the associated tissue hypoperfusion would be expected to contribute to an increased host susceptibility to infection and tissue injury during recovery. Support for this prediction comes from the observation that circulating levels of alanine aminotransferase (ALT) and creatinine, are significantly increased in AAI-hemorrhaged animals (27).
The intoxicated trauma patient may be particularly vulnerable to organ injury following traumatic injury and resulting hemorrhage and resuscitation (25). A large proportion of acutely intoxicated trauma victims are also chronic alcohol abusers (26). Therefore, preexisting and ongoing injury resulting chronic alcohol abuse may compound with the effects of hypotension predisposing them to an even worsened outcome (25, 27). These clinical findings have been confirmed in animal models which have begun to investigate the complex mechanisms of liver injury following hypotension during alcohol intoxication (28).
Additionally, ischemia and injury following hemorrhagic shock leads to a loss of intestinal barrier function (28). Increased intestinal permeability and the resulting enhanced translocation of commensal and pathologic bacteria, bacterial products, and pro-inflammatory mediators has been strongly associated with worse outcomes following traumatic injury and blood loss (29, 30). Both clinical and animals studies have provided evidence for a link between increased intestinal permeability and risk of complications during recovery from trauma and hemorrhage (31–33). The incidence of commonly associated complications including multiple organ dysfunction syndrome and acute lung injury are reported to be increased in intoxicated trauma victims (34). Chronic alcohol is well known to disrupt intestinal barrier function and induce gut leak (35). A detrimental impact of AAI on injury-induced gut leak during recovery has also been demonstrated following burn injury (36). Potential mechanisms for the impact of alcohol on this process following injury and hemorrhage have been delineated by other labs utilizing a variety of models. Common mechanism identified with changes seen during AAI include increased NO and subsequent free radical production (37) and disruption of the protective effects of prostaglandin E2 (PGE2) (38).
At the level of the intestinal epithelial cell, injury and increased inflammatory products lead to production and activation of NOS and subsequent NO production (33, 35). NO has been demonstrated to directly disrupt barrier function in intestinal epithelial cell monolayers in vitro (28). NO can also combine with other free radicals from neutrophils and other inflammatory cells leading to modification, namely carbonylation and thiolation, of structural and junctional proteins and loss of barrier function (35). Alcohol intoxication increases NO content in plasma and tissues (39–41). Thus, one can identify a role for the AAI-induced increase in NO that may exacerbate the effects of hemorrhage and provide a potential mechanism by which AAI can worsen gut leak following injury and hemorrhage.
An additional mechanism of increased intestinal injury during AAI could be a decrease in gut mucosal protective factors, namely PGE2. PGE2 is continuously produced by cyclooxygenase, which is constituently expressed in the lamina propria of the small intestine (42, 43). Under normal conditions, PGE2 serves to regulate motility, secretion, and growth and repair in the gut (44). PGE2 also plays a critical role in the response to and repair from injury and has been shown to be important in the response to infection, colitis, and following radiation injury (45). Furthermore, cyclooxygenase -2 is rapidly induced following shock states (37). Pharmacologic inhibition of cyclooxygenase during HS, and thus production of PGE2leads to worsened gut injury as well as enhanced hepatic injury (46). Using an Ussing chamber to examine barrier integrity ex vivo following ischemia/reperfusion in swine, PGE2 application was demonstrated to recover of barrier function lost following cyclooxygenase inhibition (47, 48). These studies suggest a significant protective role for PGE2 in the maintenance and recovery of intestinal barrier function following injury including shock or ischemia. In vitro studies on the effects of alcohol on intestinal epithelial cell function and morphology have also demonstrated a significant protective role for PGE2 in the development of AAI-induced barrier dysfunction (49). Pretreatment of intestinal epithelial cells with PGE2 prevents the change in cellular morphology and loss of barrier function observed following alcohol exposure (50, 51).
Disruption of intestinal barrier function was found in our model of hemorrhage as reflected by increased FITC-labeled dextran translocation during the early recovery period (Figure 2). This finding was associated with increased intestinal NO content and a decrease in PGE2 content. Thus, alcohol- induced hemodynamic dysregulation during trauma-hemorrhage results in inadequate tissue perfusion and during the resuscitation period leading to enhanced susceptibility to tissue injury.
Impact of acute alcohol intoxication on host defense responses during traumatic injury
While the vital function of autonomic and neuroendocrine pathways activated by traumatic injury is to restore cardiovascular stability following blood loss and during fluid resuscitation, these pathways also exert immunomodulatory effects on host response during recovery (52). The post-injury period has been shown to be characterized by a significant biphasic modulation of host defense mechanisms resulting in an early pro-inflammatory response often followed by a protracted immunosuppressive period. A dysregulated pattern of post-injury pro- and anti-inflammatory responses has been identified to be a main contributor to infection, sepsis, multiple organ failure and death resulting from traumatic injury (52). Several lines of evidence indicate that the mechanisms underlying hemodynamic counteregulation to trauma-hemorrhage are closely intertwined with those involved in modulating host defense mechanisms. Alcohol intoxication has been shown to impact multiple aspects of host defense (53) and to produce marked dysregulation of host defense mechanisms during the post-injury period (Figure 2). Several studies have shown that acute alcohol exposure results in suppressed pro-inflammatory cytokine release in response to an inflammatory challenge (54–58); decreased neutrophil recruitment and phagocytic function (56, 59); impaired chemotaxis (60); and oxidative burst capacity (61). Moreover, we demonstrated that even 24 hours post-hemorrhagic shock alcohol-intoxicated animals had a marked suppression in neutrophil responsiveness to an inflammatory challenge (62). The lack of change in the magnitude of IL-10 production suggests a preferential suppression of inflammatory cytokine release that may contribute to enhanced susceptibility to infection during the recovery period.
Contrasting findings were observed at the tissue level, where alcohol intoxication enhanced the pro-inflammatory milieu at completion of hemorrhage and fluid resuscitation (22). The relevance of the combined tissue inflammation and immune cell hyporesponsiveness was demonstrated by worsened outcomes following infection with Klebsiella pneumonia during the post-injury recovery period (61). AAI animals demonstrated an overall impairment in pulmonary bacterial clearance and resolution of tissue inflammation following infection contributing to a greater mortality. Thus, alcohol intoxication markedly impairs hemodynamic recovery following traumatic injury, decreases tissue perfusion, enhances tissue injury, and in addition dysregulates host defense mechanisms leading to increased susceptibility to a second-hit infectious challenge. Hence, dissecting the mechanisms responsible for impaired restoration of hemodynamic homeostasis in the alcohol-intoxicated host is of critical relevance.
Mechanisms of acute alcohol intoxication mediated hemodynamic instability
Several possibilities could account for the greater hypotension and impaired hemodynamic stability observed in the alcohol-intoxicated hemorrhaged host including and alcohol-mediated decrease in blood volume, an impaired responsiveness to vasopressors released in response to blood loss, or a disrupted neuroendocrine response to blood loss and hypotension.
Several investigators suggest a significant suppression of AVP release by AAI alone that could have contributed to a decreased blood volume during the period of alcohol infusion (63, 64). The potential contribution of an alcohol-mediated decrease in circulating blood volume at the time of hemorrhage was examined using an indicator dilution technique. Results from those studies demonstrated that alcohol-induced basal hypotension and impaired hemodynamic counteregulation were not associated with a decrease in circulating blood volume prior to hemorrhage. Moreover, the impact of alcohol intoxication on vascular responsiveness was examined utilizing both in vitro and in vivo techniques. Studies using isolated vessels (aortic and mesenteric arterioles) demonstrated that neither alcohol, hemorrhage alone, or their combination impair vascular reactivity to pressors (65). Results from those studies showed that second messenger signaling in response to changes in membrane potential is intact, as reflected by preserved KCl-mediated contraction in vessels isolated from both control and alcohol-intoxicated animals. Moreover, receptor-mediated contraction, tested with the alpha-adrenergic agonist phenylephrine also was shown to be unaffected by alcohol. These findings suggested that impaired vascular responsiveness to pressor agents such as adrenergic agonists was not the underlying mechanism for greater hypotension or blunted recovery of blood pressure in AAI animals. This conclusion is further supported by similar findings from in vivo studies showing that AAI did not impair the vascular responsiveness to intravenous administration of AVP during fluid resuscitation. Taken together, these studies suggest that circulating blood volume and vascular responsiveness to counter-regulatory hormones are not significant contributors to the AAI-induced impairment in hemodynamic stability.
In contrast, blunted neuroendocrine activation was identified as an important mechanism for disregulation of hemodynamic counteregulation. Our studies demonstrated that alcohol intoxication at the time of injury results in significantly attenuated release of the counter-regulatory hormones and potent vasoconstrictors AVP, epinephrine and norepinephrine in response to fixed-pressure hemorrhage (Figure 3) (22). Additionally, when examining the neuroendocrine response to a fixed-volume hemorrhage, AAI was shown to result in an inappropriate neuroendocrine response for the exacerbated hypotension produced by a given blood loss. These findings provided evidence for the role of disruption of the counter-regulatory neuroendocrine response to blood loss in the intoxicated host. We predicted that these effects were the result of impairment in central neuroendocrine activation. Further investigations focused on potential mechanisms of this disruption and strategies to overcome the detrimental impact of AAI.
Figure 3. Alcohol intoxication disrupts neuroendocrine compensatory mechanisms in response to blood loss.
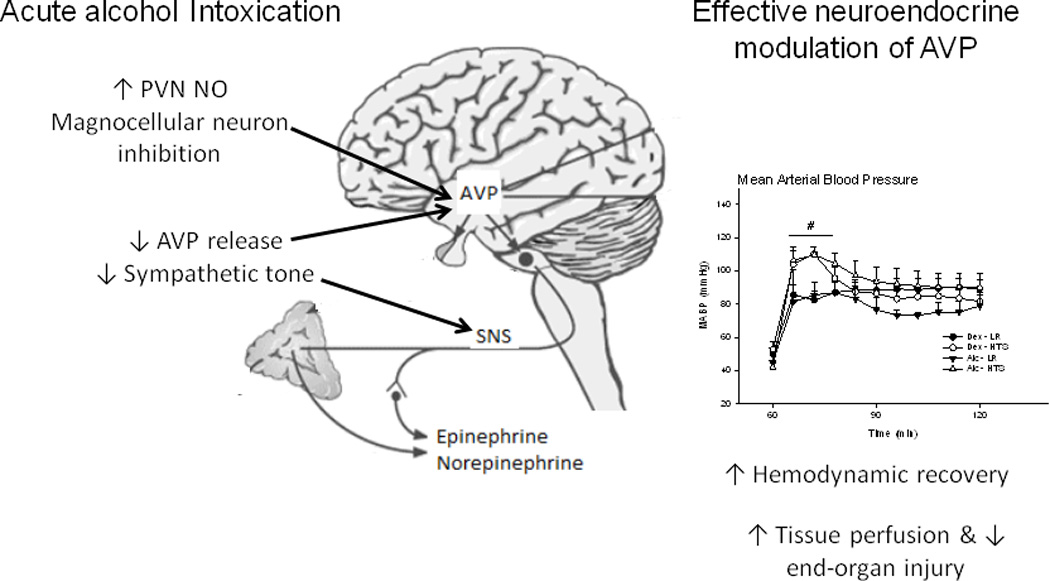
AAI upregulates PVN NO expression exerting an inhibitory tone on AVP release and attenuation of epinephrine, norepinephrine, and angiotensin II release in response to blood loss. Increased resuscitation fluid osmolarity sensitizes the AVP response to blood loss, improves hemodynamic recovery in the AAI host, This modulation of neuroendocrine response was associated with marked improvement in restoration of blood pressure, improved tissue perfusion, and attenuated end organ injury reflected in decreased gut leak. AAI, acute alcohol intoxication; AVP, arginine vasopressin; NO, nitric oxide, PVN, periventricular nucleus.
Acute alcohol intoxication disrupts central AVP controlling mechanisms
Pharmacologic central cholinergic stimulation had previously been demonstrated to lead to activation of descending sympathetic pathways in non-intoxicated animals (66). This activation was evidenced by increases in circulating levels of counter-regulatory hormones such as AVP and epinephrine as well as enhanced blood pressure following a range of cardiovascular challenges (67, 68). As had been reported in other models, intracerebroventricular (ICV) administration of choline, a pre-cursor of acetylcholine, produced an immediate activation of the SNS as evidenced by an increase in MABP and a rise in plasma epinephrine, norepinephrine, and AVP within five minutes of injection in dextrose control animals (69). While we confirmed that SNS activation could be achieved with this approach, ICV choline did not reverse the hemorrhage-induced hypotension or produce a sustained increase in vasoactive hormone release throughout the duration of hemorrhage in AAI animals. We speculated that this was due to the short duration of action of choline. Subsequent studies utilized central administration of the acetylcholinesterase inhibitor neostigmine to produce a sustained increase in acetylcholine and subsequent central sympathetic stimulation. Our results showed marked improvement in hemodynamic compensation and outcome from hemorrhagic shock in neostigmine-treated AAI rodents (70).
Further support for the ability to restore the neuroendocrine response was provided by studies utilizing the peripherally administered acetylcholinesterase inhibitor physostigmine at the time of resuscitation. Beneficial effects of intravenous administration of physostigmine in the non-intoxicated host following hemorrhage have been reported by others (71). Whether phsyostigmine could restore neuroendocrine activation in the intoxicated host was not known. As was observed with central cholinergic activation, physostigmine improved hemodynamic recovery and this was associated with attenuation of the rise in the makers of liver and renal damage (alanine aminotransferase and blood urea nitrogen) in AAI animals (27).
The relevance of the neuroendocrine system was confirmed by studies demonstrating that the pressor response produced by ICV neostigmine is primarily mediated through enhanced AVP release. Moreover, the pressor response appears to be triggered through Central Nervous System (CNS) nicotinic receptors. Combined, these studies provided strong evidence that alcohol exerts its detrimental effects on hemodynamic counterregulation to severe blood loss through central cholinergic-mediated sympathetic nervous system activation. In addition, they identified a defective triggering signal for AVP release in the alcohol-intoxicated host. The observation that enhancing central nervous system cholinergic activity restores the neuroendocrine response to hemorrhagic shock in AAI suggested that alcohol interferes with the central signaling mechanisms regulating AVP release during hemorrhagic shock (69).
AVP: A Critical Counter-regulatory Hormone during Shock
AVP is a peptide hormone secreted from the posterior pituitary that plays a central role in water homeostasis and blood pressure regulation by exerting a pressor and anti-diuretic effect (72). AVP exerts its effects by binding to two distinct primary G protein-coupled receptors, the V1 receptor expressed in the vasculature and the V2 receptor expressed in the basolateral membrane of the principal cells in the collecting duct of the kidney resulting in vasoconstriction and increased water reabsorption, respectively (72). Both the increase in vascular resistance and water reabsorption ultimately contribute to restoration of MABP, an essential response in maintaining tissue perfusion to vital organs following hemorrhagic shock.
Two lines of evidence have provided strong support for the important role of AVP in restoration of MABP following hemorrhagic shock. Administration of an AVP V1 receptor antagonist during hemorrhage significantly attenuates the restoration of blood pressure during the resuscitation period (73). Studies from our laboratory have demonstrated the importance of AVP in blood pressure recovery during fluid resuscitation with hypertonic saline (25). The immediate pressor effect of hypertonic saline resuscitation was demonstrated to be mediated by an increase in circulating concentration of AVP. Peripheral antagonism of the V1a receptor, the receptor responsible for the pressor effects of AVP, prevented the early blood pressure recovery to hemorrhage achieved with hypertonic saline resuscitation without affecting the blood pressure response to hemorrhage achieved with traditional lactated Ringer’s resuscitation providing additionally evidence supporting the importance of AVP to blood pressure recovery during hemorrhagic shock.
In addition, clinical case reports and animal studies have demonstrated that exogenous AVP administration plays an important role in blood pressure recovery following hemorrhagic shock (74, 75). Exogenous AVP administration to hypotensive patients not only restores vascular tone, but also significantly improves MABP in subjects unresponsive to fluid resuscitation or other vasopressors following severe blood loss (75). Additional benefits of AVP include increased vascular reactivity, decreased acidosis and decreased resuscitation volume necessary to maintain a target MABP (76–78). Yang et al. demonstrated that AVP administration following hemorrhagic shock increases vascular reactivity and increases the pressor effect and contractile responses of superior mesenteric arteries to other vasopressors such as norepinephrine (78). Furthermore, following prolonged hemorrhagic shock, resuscitation with AVP has been shown to improve MABP and significantly reduce the volume of resuscitation fluid necessary to maintain MABP, suggesting that AVP improves cardiovascular function and the delayed progression to cardiovascular collapse (77). SNS activation and subsequent release of epinephrine and norepinephrine are important in the restoration of MABP following hemorrhagic shock; however, after prolonged hypotension, patients may become unresponsive to fluid replacement and catecholamines such as epinephrine (75). During the late phases of hemorrhagic shock, compensatory mechanisms and aggressive fluid resuscitation strategies fail, making additional strategies to restore MABP and tissue perfusion critical. Moreover, catecholamine administration has been shown to have untoward effects such as cardiac arrhythmias and metabolic acidosis (79). When compared to epinephrine as a vasopressor following hemorrhage, AVP produces a more sustained increase in MABP and improved survival compared to untreated animals (76). Taken together, the results from these studies indicate that AVP is a critical counter-regulatory hormone in response to blood loss. Therefore, subsequent focused on determining the central signaling mechanisms involved in the impaired release of AVP in response to hemorrhage during AAI.
Mechanisms of the impaired AVP release during hemorrhage in AAI
AVP release is regulated by a number of different mediators including angiotensin (ANG) II and nitric oxide (NO) (80–86). ANG II acts both peripherally and centrally to restore blood pressure during hypovolemia. Peripherally, ANG II binds to the primary angiotensin (AT1) receptor, resulting in constriction of the vasculature (85). Centrally produced ANG II stimulates AVP release by binding to AT1 receptors in the PVN and SON (82, 83). This is particularly important during hemorrhage as demonstrated by a greater decrease in MABP and markedly attenuated AVP concentrations during hemorrhage following central administration of Losartan, an AT1 receptor antagonist (81).
NO is a gas produced from the conversion of L-arginine to L-citrulline by the enzyme nitric oxide synthase (NOS) (87, 88). NO is produced in the CNS where it functions as an important signaling molecule regulating neuroendocrine function in the hypothalamus (86, 89). All three isoforms of NOS are present in the magnocellular neurons in the PVN and SON of the hypothalamus, the primary sites of AVP production (86). AVP is produced in both magnocellular and parvocellular neurons; however, NO exerts inhibitory effects on the magnocellular neurons, but stimulates parvocellular neuronal activity in hypothalamic slice preparations (89–91). Similarly, patch clamp recordings from magnocellular and parvocellular neurons show that administration of the NO donor, N-acetyl-S-nitroso-D-penicillamine (SNAP), inhibits the activity of magnocellular neurons while it produces reversible membrane depolarization in parvocellular neurons (89–91). The contribution of NO to regulation of AVP release has been confirmed in in vivo studies by Kadekaro and others, which show that central administration of nitro-L-arginine-methylester (L-NAME), a NOS inhibitor, produces a significant increase in plasma AVP as well as a significant pressor effect under baseline conditions in normovolumic rats (92). However, during hemorrhage L-NAME does not further increase AVP release suggesting that NO inhibits the release of AVP under normovolumic and iso-osmotic conditions and this inhibitory tone is removed during hemorrhage (92). Additionally, using whole-cell patch clamp recordings, Ludwig and others first demonstrated that the NO-mediated inhibition of AVP neuronal activity is the result of increased GABA-ergic synaptic activity (89). Alcohol intoxication has been reported to result in increased NO in plasma, anterior pituitary, and PVN of the hypothalamus as well as increased nitric oxide synthase activity in the anterior pituitary and PVN identifying a potential mechanism of the blunted AVP response to hemorrhage during AAI (74). Thus, we predicted that increased NO production in the PVN and SON during AAI and hemorrhage may attenuate AVP release directly through inhibition of AVP-producing magnocellular neuronal activity or indirectly via NO-mediated inhibition of noradrenergic activation or stimulation of GABA-ergic activation regulating the PVN.
Our studies demonstrated that the blunted AVP release following hemorrhagic shock in AAI animals is associated with a significant increase in PVN NO content; suggesting excess NO tone as a potential mechanism for suppressed AVP release (94). The inhibitory role of PVN NO in the alcohol-induced inhibition of AVP release during hemorrhage was confirmed by the results of studies that used central administration of the NOS inhibitor L-NAME. The significant reduction in PVN NOS activity and NO content increased the blood volume removed necessary to achieve the target hypotension in AAI hemorrhaged animals (94). Thus, NOS inhibition enhanced the hemodynamic compensatory response in AAI animals, reflected as well by restoration of circulating AVP and ANG II levels following hemorrhage. These findings confirmed that AAI-enhanced central nitric oxide production inhibits AVP release during hemorrhage.
Strategies to sensitize control mechanisms of AVP release following shock in AAI
Release of AVP is triggered not only by decreased blood volume, but more importantly by hyperosmolality. In contrast to the attenuated release of AVP in response to blood loss, our studies show that alcohol intoxication does not impair AVP release in response to an osmotic challenge elicited by intravenous hypertonic (7%) saline (HTS) administration (94). Furthermore, HTS administration was associated with decreased PVN NO content in intoxicated animals, suggesting the possibility that fluid resuscitation with HTS could reduce PVN NO content and restore the AVP response to blood loss in AAI rodents (Figure 3).
Indeed, subsequent studies examining resuscitation with HTS demonstrated enhanced MABP recovery and elevated circulating levels of AVP in AAI animals (25). The pressor response to HTS was found to be the result of circulating AVP, and specifically through AVP’s actions at the V1 receptor. Treatment with an AVP V1 antagonist abolished the pressor response to HTS without impairing blood pressure recovery in animals resuscitated with lactate Ringer’s. As predicted, the enhanced levels of circulating AVP following resuscitation with HTS are associated with decreased PVN NO content. Thus, increased resuscitation fluid osmolarity sensitizes the AVP response to blood loss improving outcome from hemorrhagic shock in the alcohol-intoxicated host.
Targeted resuscitation strategies for hemorrhagic shock in the acute alcohol intoxicated host
Enhanced MABP recovery does not always directly improve tissue perfusion. Indeed, in a previous study we demonstrated that despite improved MABP during fluid resuscitation, delayed peripheral neostigmine administration markers of organ injury in the intoxicated host (95). Additional studies have indicated selective redistribution of blood flow following pressor use during shock states that may exacerbate organ injury (96, 97). This was of importance as the pressor effects of HTS in our model are mainly due to increased circulating levels of AVP. While AVP has been consistently reported to increase blood pressure, its effects on end-organ and tissue perfusion have been more varied. For example, AVP decreases brain and renal perfusion following cardiogenic shock (96). The increase in mean arterial pressures, therefore, may be at the expense of tissue perfusion. HTS resuscitation, however, has been associated with more rapid return of tissue perfusion and oxygenation in non-intoxicated animals. If these benefits would be prevented by AAI was unknown. Our studies demonstrated that HTS resuscitation improves blood flow to certain vital organs following hemorrhagic shock in AAI rodents (Figure 3). The persistence of this effect 2 hours after the administration of a bolus of HTS suggests that while the elevation in MABP compared to Lactated Ringers (LR) was transient, the microvascular benefit extends well into the recovery period.
It is also possible that HTS contributes to attenuation of organ injury independent of effects on tissue blood flow and/or redistribution. HTS has been reported to decrease inflammation and neutrophil influx following hemorrhagic shock in the non-intoxicated host resulting in lessened organ damage and dysfunction when compared to LR resuscitation. Enhanced neutrophil influx and sequestration have been strongly implicated in the increased incidence and severity of hepatic, pulmonary, and gut injury following injury or hemorrhage during AAI (36, 98). Neutrophil influx and the resulting tissue injury following injury and HS are tissue and time-dependent (99). Increased intestinal neutrophil influx has been observed following burn injury during acute alcohol intoxication (36). Furthermore, in addition to the increased chemotaxis, AAI suppresses neutrophil apoptosis potentially leading to increased time for neutrophil-induced tissue damage (100). Neutrophil depletion by anti-rat neutrophil antiserum ameliorates the injury and resulting gut leak induced by AAI and burn injury (36). In addition to intestinal injury, infiltrating neutrophils have been implicated in the production of lung and hepatic injury (30, 98). In a study of hemorrhagic shock following chronic alcohol administration, increased chemokine production, adhesion molecule expression, and the resulting neutrophil recruitment and sequestration were strongly implicated in the worsened hepatic injury in alcohol-treated animals (98, 101). Thus, the attenuated neutrophil influx observed following HTS resuscitation may serve as a new target for resuscitation strategies aimed at limiting adverse secondary outcomes such as infection and organ injury.
Furthermore, improvement in organ function following HTS resuscitation was demonstrated by prevention of the rise in markers of hepatic injury including alanine aminotransferase in AAI animals. HTS also led to decreased oxidative stress measured by protein carbonylation in the gut, attenuated depletion of gut prostaglandin E2and decreased NO content in AAI animals. These changes were associated with functional improvements as reflected by attenuation in gut leak at 24 hours post-hemorrhagic shock. The observed negative correlation between PGE2 levels and gut leak suggests that even a small increase in PGE2 could have led to significant protective effects.
These findings provide additional support to the idea that improved splanchnic tissue perfusion is central to protection from tissue injury following hemorrhagic shock. In particular, our findings show marked ability for hypertonic saline to improve splanchnic circulation. Studies from Hauser’s labortory have also shown that resuscitation with hypertonic saline ameliorates hemorrhage-induced gut and lung injury; further supporting the correlation between gut and lung injury (102). The significant impact of improved splanchnic circulation and tissue perfusion to overall outcome from hemorrhagic shock is also supported by studies from Chaudry’s laboratory showing estradiol-mediated improvement of intestinal blood flow (103). Moreover, similar findings have been reported by Deitch’s group, where castrated and flutamide-treated male rats were significantly protected against trauma hemorrhagic shock-induced gut injury and in turn, mesenteric lymph of these animals had a decreased capacity of producing distant organ injury (104).
Clinical trials with HTS have failed to demonstrate benefit over traditional crystalloid resuscitation when using 28-day mortality as an outcome (105). Although no benefit has been found on 28-day mortality, the primary outcome measure for these trials, HTS does limit the incidence of negative secondary outcomes such as need for dialysis and renal failure. Additionally, smaller clinical studies demonstrate that the immnomodulatory effects of HTS observed in animals studies can be replicated in humans (106). Random and blinded assignment to different resuscitation protocols in these studies was necessary to investigate the global impact of fluid choice. However, this approach does not address the proposed strategy of applying HTS selectively to the intoxicated host or other subpopulations. In the non-intoxicated trauma victim, HTS may not provide additional benefit as counterregulatory responses to injury and blood loss are already at maximal or optimal levels. In contrast, the impaired hemodynamic and neuroendocrine responses to HS during AAI may be corrected or enhanced by HTS resuscitation and lead to improved outcomes. Furthermore, the non-hemodynamic effects of HTS may be as important if not more important than enhanced hemodynamic stability in the intoxicated host. Thus, taken together our findings and reports in the literature suggest that the alcohol-intoxicated trauma-hemorrhage victim may be particularly benefited by HTS.
Summary and conclusion
AAI is a significant risk factor for trauma and subsequent blood loss. In addition to the increased incidence of injury, alcohol leads to a dysregulation of the hemodynamic, neuroendocrine, inflammatory, and immune responses to hemorrhage. This disruption of the normal neuroendocrine counteregulatory response impairs hemodynamic stability and recovery contributing to compromised tissue perfusion and increased end-organ injury. We have demonstrated that this impaired counteregulatory response results from the blunted central neuroendocrine and autonomic activation. Furthermore, we identified an important role for elevated levels of NO in the PVN. Strategies to decrease PVN NO content utilizing both pharmacologic inhibition as well as selected fluid resuscitation with HTS confirmed both the significant role of PVN NO content in the impaired responses and demonstrated potential therapeutic avenues for further exploration. Importantly, resuscitation with HTS not only improved the hemodynamic recovery, but improved tissue perfusion leading to decreased organ damage. The impact of HTS on other outcome measures including response to infection following hemorrhage during intoxication certainly warrant future studies and may hold great promise for targeted strategies to limit negative secondary outcomes in this vulnerable population.
ACKNOWLEDGEMENTS
The authors would like to thank Drs. Keisa Mathis, Patrick Greffeinstein, Herb Phelan, and Kris Norenberg for contributions to this body of work. This research was supported by DOD-PR-054196, LA BoR-110350057A, NIAAA-F30AA019587-01A1, AMA Foundation, NIAAA-AA7577, and the American Physiological Society Porter Physiology Development Award.
Abbreviations used throughout the manuscript
- AAI
Acute Alcohol Intoxication
- MABP
Mean Arterial Blood Pressure
- NTS
Nucleus of the Tractus Solitarius
- PVN
Paraventricular nucleus
- CRH
Corticotropin-Releasing Hormone
- AVP
Arginine Vasopressin
- ALT
Alanine Aminotransferase
- ICV
Intracerebroventricular
- SNS
Sympathetic Nervous System
- CNS
Central Nervous System
- ANG II
Angiotensin II
- NO
Nitric Oxide
- NOS
Nitric Oxide Synthase
- HTS
Hypertonic Saline
- LR
Lactated Ringers
- PGE2
Prostaglandin E2
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Cherpitel CJ. Alcohol and casualties: a comparison of emergency room and coroner data. Alcohol Alcohol. 1994;29:211–218. [PubMed] [Google Scholar]
- 2.Cherpitel CJ, Ye Y, Bond J, et al. The causal attribution of injury to alcohol consumption: a cross-national meta-analysis from the emergency room collaborative alcohol analysis project. Alcohol Clin Exp Res. 2003;27:1805–1812. doi: 10.1097/01.ALC.0000095863.78842.F0. [DOI] [PubMed] [Google Scholar]
- 3.Cherpitel CJ, Bond J, Ye Y, et al. Alcohol-related injury in the ER: a cross-national meta-analysis from the Emergency Room Collaborative Alcohol Analysis Project (ERCAAP) J Stud Alcohol. 2003;64:641–649. doi: 10.15288/jsa.2003.64.641. [DOI] [PubMed] [Google Scholar]
- 4.Madan AK, Yu K, Beech DJ. Alcohol and drug use in victims of life-threatening trauma. J Trauma. 1999;47:568–571. doi: 10.1097/00005373-199909000-00026. [DOI] [PubMed] [Google Scholar]
- 5.Pories SE, Gamelli RL, Vacek P, et al. Intoxication and injury. J Trauma. 1992;32:60–64. doi: 10.1097/00005373-199201000-00013. [DOI] [PubMed] [Google Scholar]
- 6.Rivara FP, Jurkovich GJ, Gurney JG, et al. The magnitude of acute and chronic alcohol abuse in trauma patients. Arch Surg. 1993;128:907–912. doi: 10.1001/archsurg.1993.01420200081015. discussion 912–903. [DOI] [PubMed] [Google Scholar]
- 7.Beech DJ, Mercadel R. Correlation of alcohol intoxication with life-threatening assaults. J Natl Med Assoc. 1998;90:761–764. [PMC free article] [PubMed] [Google Scholar]
- 8.Fabbri 2001 [Google Scholar]
- 9.Tien 2006 [Google Scholar]
- 10.Shih HC, Hu SC, Yang CC, et al. Alcohol intoxication increases morbidity in drivers involved in motor vehicle accidents. Am J Emerg Med. 2003;21:91–94. doi: 10.1053/ajem.2003.50025. [DOI] [PubMed] [Google Scholar]
- 11.Bilello J, McCray V, Davis J, et al. Acute ethanol intoxication and the trauma patient: hemodynamic pitfalls. World J Surg. 2011;35:2149–2153. doi: 10.1007/s00268-011-1191-7. [DOI] [PubMed] [Google Scholar]
- 12.Heckbert SR, Vedder NB, Hoffman W, et al. Outcome after hemorrhagic shock in trauma patients. J Trauma. 1998;45:545–549. doi: 10.1097/00005373-199809000-00022. [DOI] [PubMed] [Google Scholar]
- 13.Schmid PG, Sharabi FM, Guo GB, et al. Vasopressin and oxytocin in the neural control of the circulation. Fed Proc. 1984;43:97–102. [PubMed] [Google Scholar]
- 14.Kannan H, Hayashida Y, Yamashita H. Increase in sympathetic outflow by paraventricular nucleus stimulation in awake rats. Am J Physiol. 1989;256:R1325–R1330. doi: 10.1152/ajpregu.1989.256.6.R1325. [DOI] [PubMed] [Google Scholar]
- 15.Darlington DN, Barraclough CA, Gann DS. Hypotensive hemorrhage elevates corticotropin-releasing hormone messenger ribonucleic acid (mRNA) but not vasopressin mRNA in the rat hypothalamus. Endocrinology. 1992;130:1281–1288. doi: 10.1210/endo.130.3.1311234. [DOI] [PubMed] [Google Scholar]
- 16.Brown MR, Gray TS, Fisher LA. Corticotropin-releasing factor receptor antagonist: effects on the autonomic nervous system and cardiovascular function. Regul Pept. 1986;16:321–329. doi: 10.1016/0167-0115(86)90032-7. [DOI] [PubMed] [Google Scholar]
- 17.Schiltz JC, Hoffman GE, Stricker EM, et al. Decreases in arterial pressure activate oxytocin neurons in conscious rats. Am J Physiol. 1997;273:R1474–R1483. doi: 10.1152/ajpregu.1997.273.4.R1474. [DOI] [PubMed] [Google Scholar]
- 18.Benetos A, Gavras H, Stewart JM, et al. Vasodepressor role of endogenous bradykinin assessed by a bradykinin antagonist. Hypertension. 1986;8:971–974. doi: 10.1161/01.hyp.8.11.971. [DOI] [PubMed] [Google Scholar]
- 19.Share L. Role of vasopressin in cardiovascular regulation. Physiol Rev. 1988;68:1248–1284. doi: 10.1152/physrev.1988.68.4.1248. [DOI] [PubMed] [Google Scholar]
- 20.Khanna S, Sibbald JR, Smith DW, et al. Initiation of rat vasopressin cell responses to simulated hypotensive hemorrhage. Am J Physiol. 1994;267:R1142. doi: 10.1152/ajpregu.1994.267.4.R1142. [DOI] [PubMed] [Google Scholar]
- 21.Zerbe RL, Bayorh MA, Feuerstein G. Vasopressin: an essential pressor factor for blood pressure recovery following hemorrhage. Peptides. 1982;3:509–514. doi: 10.1016/0196-9781(82)90117-6. [DOI] [PubMed] [Google Scholar]
- 22.Phelan H, Stahls P, Hunt J, et al. Impact of alcohol intoxication on hemodynamic, metabolic, and cytokine responses to hemorrhagic shock. J Trauma. 2002;52:675–682. doi: 10.1097/00005373-200204000-00010. [DOI] [PubMed] [Google Scholar]
- 23.Mathis KW, Zambell K, Olubadewo JO, et al. Altered hemodynamic counter-regulation to hemorrhage by acute moderate alcohol intoxication. Shock. 2006;26:55–61. doi: 10.1097/01.shk.0000215320.06866.30. [DOI] [PubMed] [Google Scholar]
- 24.Wang P, Ba ZF, Burkhardt J, et al. Trauma-hemorrhage and resuscitation in the mouse: effects on cardiac output and organ blood flow. Am J Physiol. 1993;264:H1166–H1173. doi: 10.1152/ajpheart.1993.264.4.H1166. [DOI] [PubMed] [Google Scholar]
- 25.Sulzer JK, Whitaker AM, Molina PE. Hypertonic saline resuscitation enhances blood pressure recovery and decreases organ injury following hemorrhage in acute alcohol intoxicated rodents. J Trauma Acute Care Surg. 2012 doi: 10.1097/TA.0b013e31826fc747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Peitzman AB, Billiar TR, Harbrecht BG, et al. Hemorrhagic shock. Curr Probl Surg. 1995;32:925–1002. doi: 10.1016/s0011-3840(05)80008-5. [DOI] [PubMed] [Google Scholar]
- 27.Mathis KW, Sulzer J, Molina PE. Systemic administration of a centrally acting acetylcholinesterase inhibitor improves outcome from hemorrhagic shock during acute alcohol intoxication. Shock. 2010;34:162–168. doi: 10.1097/SHK.0b013e3181cff958. [DOI] [PubMed] [Google Scholar]
- 28.Xu DZ, Lu Q, Deitch EA. Nitric oxide directly impairs intestinal barrier function. Shock. 2002;17:139–145. doi: 10.1097/00024382-200202000-00010. [DOI] [PubMed] [Google Scholar]
- 29.Badami CD, Senthil M, Caputo FJ, et al. Mesenteric lymph duct ligation improves survival in a lethal shock model. Shock. 2008;30:680–685. doi: 10.1097/SHK.0b013e318173edd1. [DOI] [PubMed] [Google Scholar]
- 30.Lee MA, Yatani A, Sambol JT, et al. Role of gut-lymph factors in the induction of burn-induced and trauma-shock-induced acute heart failure. Int J Clin Exp Med. 2008;1:171–180. [PMC free article] [PubMed] [Google Scholar]
- 31.Spindler-Vesel A, Wraber B, Vovk I, et al. Intestinal permeability and cytokine inflammatory response in multiply injured patients. J Interferon Cytokine Res. 2006;26:771–776. doi: 10.1089/jir.2006.26.771. [DOI] [PubMed] [Google Scholar]
- 32.Zallen G, Moore EE, Johnson JL, et al. Posthemorrhagic shock mesenteric lymph primes circulating neutrophils and provokes lung injury. J Surg Res. 1999;83:83–88. doi: 10.1006/jsre.1999.5569. [DOI] [PubMed] [Google Scholar]
- 33.Han DW. Intestinal endotoxemia as a pathogenetic mechanism in liver failure. World J Gastroenterol. 2002;8:961–965. doi: 10.3748/wjg.v8.i6.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fleming M, Bhamb B, Schurr M, et al. Alcohol biomarkers in patients admitted for trauma. Alcohol Clin Exp Res. 2009;33:1777–1781. doi: 10.1111/j.1530-0277.2009.01016.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tang Y, Forsyth CB, Farhadi A, et al. Nitric oxide-mediated intestinal injury is required for alcohol-induced gut leakiness and liver damage. Alcohol Clin Exp Res. 2009;33:1220–1230. doi: 10.1111/j.1530-0277.2009.00946.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li X, Schwacha MG, Chaudry IH, et al. Acute alcohol intoxication potentiates neutrophil-mediated intestinal tissue damage after burn injury. Shock. 2008;29:377–383. doi: 10.1097/shk.0b013e31815abe80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang R, Gallo DJ, Baust JJ, et al. Effect of hemorrhagic shock on gut barrier function and expression of stress-related genes in normal and gnotobiotic mice. Am J Physiol Regul Integr Comp Physiol. 2002;283:R1263–R1274. doi: 10.1152/ajpregu.00278.2002. [DOI] [PubMed] [Google Scholar]
- 38.Gerstle JT, Seaton J, Kauffman GL, et al. The association between PGE2 activity and mucosal permeability in proximal small bowel. J Surg Res. 1994;57:579–583. doi: 10.1006/jsre.1994.1186. [DOI] [PubMed] [Google Scholar]
- 39.Wang JF, Greenberg SS, Spitzer JJ. Chronic alcohol administration stimulates nitric oxide formation in the rat liver with or without pretreatment by lipopolysaccharide. Alcohol Clin Exp Res. 1995;19:387–393. doi: 10.1111/j.1530-0277.1995.tb01520.x. [DOI] [PubMed] [Google Scholar]
- 40.Baraona E, Shoichet L, Navder K, et al. Mediation by nitric oxide of the stimulatory effects of ethanol on blood flow. Life Sci. 2002;70:2987–2995. doi: 10.1016/s0024-3205(02)01572-2. [DOI] [PubMed] [Google Scholar]
- 41.Baraona E, Zeballos GA, Shoichet L, et al. Ethanol consumption increases nitric oxide production in rats, and its peroxynitrite-mediated toxicity is attenuated by polyenylphosphatidylcholine. Alcohol Clin Exp Res. 2002;26:883–889. [PubMed] [Google Scholar]
- 42.Newberry RD, Stenson WF, Lorenz RG. Cyclooxygenase-2-dependent arachidonic acid metabolites are essential modulators of the intestinal immune response to dietary antigen. Nat Med. 1999;5:900–906. doi: 10.1038/11341. [DOI] [PubMed] [Google Scholar]
- 43.Newberry RD, McDonough JS, Stenson WF, et al. Spontaneous and continuous cyclooxygenase-2-dependent prostaglandin E2 production by stromal cells in the murine small intestine lamina propria: directing the tone of the intestinal immune response. J Immunol. 2001;166:4465–4472. doi: 10.4049/jimmunol.166.7.4465. [DOI] [PubMed] [Google Scholar]
- 44.Hawkey CJ, Rampton DS. Prostaglandins and the gastrointestinal mucosa: are they important in its function, disease, or treatment? Gastroenterology. 1985;89:1162–1188. doi: 10.1016/0016-5085(85)90225-2. [DOI] [PubMed] [Google Scholar]
- 45.Stenson WF. Prostaglandins and epithelial response to injury. Curr Opin Gastroenterol. 2007;23:107–110. doi: 10.1097/MOG.0b013e3280143cb6. [DOI] [PubMed] [Google Scholar]
- 46.Tsukada K, Hasegawa T, Tsutsumi S, et al. Roles of cyclooxygenase-2 in tissue injury during hemorrhagic shock. Shock. 2000;13:392–396. doi: 10.1097/00024382-200005000-00008. [DOI] [PubMed] [Google Scholar]
- 47.Blikslager AT, Zimmel DN, Young KM, et al. Recovery of ischaemic injured porcine ileum: evidence for a contributory role of COX-1 and COX-2. Gut. 2002;50:615–623. doi: 10.1136/gut.50.5.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gookin JL, Galanko JA, Blikslager AT, et al. PG-mediated closure of paracellular pathway and not restitution is the primary determinant of barrier recovery in acutely injured porcine ileum. Am J Physiol Gastrointest Liver Physiol. 2003;285:G967–G979. doi: 10.1152/ajpgi.00532.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Banan A, Smith GS, Rieckenberg CL, et al. Protection against ethanol injury by prostaglandin in a human intestinal cell line: role of microtubules. Am J Physiol. 1998;274:G111–G121. doi: 10.1152/ajpgi.1998.274.1.G111. [DOI] [PubMed] [Google Scholar]
- 50.Banan A, Smith GS, Deshpande Y, et al. Prostaglandins protect human intestinal cells against ethanol injury by stabilizing microtubules: role of protein kinase C and enhanced calcium efflux. Dig Dis Sci. 1999;44:697–707. doi: 10.1023/a:1026649422607. [DOI] [PubMed] [Google Scholar]
- 51.Banan A, Smith GS, Kokoska ER, et al. Role of actin cytoskeleton in prostaglandin-induced protection against ethanol in an intestinal epithelial cell line. J Surg Res. 2000;88:104–113. doi: 10.1006/jsre.1999.5786. [DOI] [PubMed] [Google Scholar]
- 52.Angele MK, Faist E. Clinical review: immunodepression in the surgical patient and increased susceptibility to infection. Crit Care. 2002;6:298–305. doi: 10.1186/cc1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Crews FT, Bechara R, Brown LA, et al. Cytokines and alcohol. Alcohol Clin Exp Res. 2006;30:720–730. doi: 10.1111/j.1530-0277.2006.00084.x. [DOI] [PubMed] [Google Scholar]
- 54.Gonzalez-Quintela A, Dominguez-Santalla MJ, Perez LF, et al. Influence of acute alcohol intake and alcohol withdrawal on circulating levels of IL-6, IL-8, IL-10 and IL-12. Cytokine. 2000;12:1437–1440. doi: 10.1006/cyto.2000.0715. [DOI] [PubMed] [Google Scholar]
- 55.Arbabi S, Garcia I, Bauer GJ, et al. Alcohol (ethanol) inhibits IL-8 and TNF: role of the p38 pathway. J Immunol. 1999;162:7441–7445. [PubMed] [Google Scholar]
- 56.Nelson S, Bagby G, Summer WR. Alcohol suppresses lipopolysaccharide-induced tumor necrosis factor activity in serum and lung. Life Sci. 1989;44:673–676. doi: 10.1016/0024-3205(89)90472-4. [DOI] [PubMed] [Google Scholar]
- 57.Szabo G, Mandrekar P, Girouard L, et al. Regulation of human monocyte functions by acute ethanol treatment: decreased tumor necrosis factor-alpha, interleukin-1 beta and elevated interleukin-10, and transforming growth factor-beta production. Alcohol Clin Exp Res. 1996;20:900–907. doi: 10.1111/j.1530-0277.1996.tb05269.x. [DOI] [PubMed] [Google Scholar]
- 58.Mandrekar P, Catalano D, White B, et al. Moderate alcohol intake in humans attenuates monocyte inflammatory responses: inhibition of nuclear regulatory factor kappa B and induction of interleukin 10. Alcohol Clin Exp Res. 2006;30:135–139. doi: 10.1111/j.1530-0277.2006.00012.x. [DOI] [PubMed] [Google Scholar]
- 59.Zhang P, Bagby GJ, Stoltz DA, et al. Granulocyte colony-stimulating factor modulates the pulmonary host response to endotoxin in the absence and presence of acute ethanol intoxication. J Infect Dis. 1999;179:1441–1448. doi: 10.1086/314763. [DOI] [PubMed] [Google Scholar]
- 60.Gluckman SJ, Dvorak VC, MacGregor RR. Host defenses during prolonged alcohol consumption in a controlled environment. Arch Intern Med. 1977;137:1539–1543. [PubMed] [Google Scholar]
- 61.Zambell KL, Phelan H, Vande Stouwe C, et al. Acute alcohol intoxication during hemorrhagic shock: impact on host defense from infection. Alcohol Clin Exp Res. 2004;28:635–642. doi: 10.1097/01.alc.0000122104.85971.55. [DOI] [PubMed] [Google Scholar]
- 62.Greiffenstein P, Mathis KW, Stouwe CV, et al. Alcohol binge before trauma/hemorrhage impairs integrity of host defense mechanisms during recovery. Alcohol Clin Exp Res. 2007;31:704–715. doi: 10.1111/j.1530-0277.2007.00355.x. [DOI] [PubMed] [Google Scholar]
- 63.Eisenhofer G, Johnson RH. Effect of ethanol ingestion on plasma vasopressin and water balance in humans. Am J Physiol. 1982;242:R522–R527. doi: 10.1152/ajpregu.1982.242.5.R522. [DOI] [PubMed] [Google Scholar]
- 64.Taivainen H, Laitinen K, Tähtelä R, et al. Role of plasma vasopressin in changes of water balance accompanying acute alcohol intoxication. Alcohol Clin Exp Res. 1995;19:759–762. doi: 10.1111/j.1530-0277.1995.tb01579.x. [DOI] [PubMed] [Google Scholar]
- 65.Molina MF, Whitaker A, Molina PE, et al. Alcohol does not modulate the augmented acetylcholine-induced vasodilatory response in hemorrhaged rodents. Shock. 2009;32:601–607. doi: 10.1097/SHK.0b013e31819e2b9a. [DOI] [PubMed] [Google Scholar]
- 66.Ulus IH, Arslan BY, Savci V, et al. Restoration of blood pressure by choline treatment in rats made hypotensive by haemorrhage. Br J Pharmacol. 1995;116:1911–1917. doi: 10.1111/j.1476-5381.1995.tb16682.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Savci V, Ulus IH. Central choline reverses hypotension caused by alpha-adrenoceptor or ganglion blockade in rats: the role of vasopressin. Eur J Pharmacol. 1996;311:153–161. doi: 10.1016/0014-2999(96)00424-4. [DOI] [PubMed] [Google Scholar]
- 68.Arslan BY, Ulus IH, Savci V, et al. Effects of intracerebroventricular injected choline on cardiovascular functions and sympathoadrenal activity. J Cardiovasc Pharmacol. 1991;17:814–821. doi: 10.1097/00005344-199105000-00018. [DOI] [PubMed] [Google Scholar]
- 69.Mathis KW, Molina PE. Transient central cholinergic activation enhances sympathetic nervous system activity but does not improve hemorrhage-induced hypotension in alcohol-intoxicated rodents. Shock. 2009;32:410–415. doi: 10.1097/SHK.0b013e31819e2d13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mathis KW, Molina PE. Central acetylcholinesterase inhibition improves hemodynamic counterregulation to severe blood loss in alcohol-intoxicated rats. Am J Physiol Regul Integr Comp Physiol. 2009;297:R437–R445. doi: 10.1152/ajpregu.00170.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Savic J, Varagic VM, Prokic D, et al. The life-saving effect of physostigmine in haemorrhagic shock. Resuscitation. 1991;21:57–60. doi: 10.1016/0300-9572(91)90078-d. [DOI] [PubMed] [Google Scholar]
- 72.Schrier RW, Berl T, Anderson RJ. Osmotic and nonosmotic control of vasopressin release. Am J Physiol. 1979;236:F321–F332. doi: 10.1152/ajprenal.1979.236.4.F321. [DOI] [PubMed] [Google Scholar]
- 73.Fujisawa Y, Miyatake A, Hayashida Y, et al. Role of vasopressin on cardiovascular changes during hemorrhage in conscious rats. Am J Physiol. 1994;267:H1713–H1718. doi: 10.1152/ajpheart.1994.267.5.H1713. [DOI] [PubMed] [Google Scholar]
- 74.Cohn SM. Potential benefit of vasopressin in resuscitation of hemorrhagic shock. J Trauma. 2007;62:S56–S57. doi: 10.1097/TA.0b013e318065ab06. [DOI] [PubMed] [Google Scholar]
- 75.Morales D, Madigan J, Cullinane S, et al. Reversal by vasopressin of intractable hypotension in the late phase of hemorrhagic shock. Circulation. 1999;100:226–229. doi: 10.1161/01.cir.100.3.226. [DOI] [PubMed] [Google Scholar]
- 76.Voelckel WG, Lurie KG, Lindner KH, et al. Vasopressin improves survival after cardiac arrest in hypovolemic shock. Anesth Analg. 2000;91:627–634. doi: 10.1097/00000539-200009000-00024. [DOI] [PubMed] [Google Scholar]
- 77.Johnson KB, Pearce FJ, Jeffreys N, et al. Impact of vasopressin on hemodynamic and metabolic function in the decompensatory phase of hemorrhagic shock. J Cardiothorac Vasc Anesth. 2006;20:167–172. doi: 10.1053/j.jvca.2005.11.015. [DOI] [PubMed] [Google Scholar]
- 78.Yang G, Liu L, Xu J, et al. Effect of arginine vasopressin on vascular reactivity and calcium sensitivity after hemorrhagic shock in rats and its relationship to Rho-kinase. J Trauma. 2006;61:1336–1342. doi: 10.1097/01.ta.0000197928.99745.22. [DOI] [PubMed] [Google Scholar]
- 79.Rudloff E, Kirby R. Fluid resuscitation and the trauma patient. Vet Clin North Am Small Anim Pract. 2008;38:645–652. xiii. doi: 10.1016/j.cvsm.2008.01.018. [DOI] [PubMed] [Google Scholar]
- 80.Ferguson AV, Washburn DL, Latchford KJ. Hormonal and neurotransmitter roles for angiotensin in the regulation of central autonomic function. Exp Biol Med (Maywood) 2001;226:85–96. doi: 10.1177/153537020122600205. [DOI] [PubMed] [Google Scholar]
- 81.Lee WJ, Yang EK, Ahn DK, et al. Central ANG II-receptor antagonists impair cardiovascular and vasopressin response to hemorrhage in rats. Am J Physiol. 1995;268:R1500–R1506. doi: 10.1152/ajpregu.1995.268.6.R1500. [DOI] [PubMed] [Google Scholar]
- 82.Mouw D, Bonjour JP, Malvin RL, et al. Central action of angiotensin in stimulating ADH release. Am J Physiol. 1971;220:239–242. doi: 10.1152/ajplegacy.1971.220.1.239. [DOI] [PubMed] [Google Scholar]
- 83.Qadri F, Culman J, Veltmar A, et al. Angiotensin II-induced vasopressin release is mediated through alpha-1 adrenoceptors and angiotensin II AT1 receptors in the supraoptic nucleus. J Pharmacol Exp Ther. 1993;267:567–574. [PubMed] [Google Scholar]
- 84.Ramsay DJ, Keil LC, Sharpe MC, et al. Angiotensin II infusion increases vasopressin, ACTH, and 11-hydroxycorticosteroid secretion. Am J Physiol. 1978;234:R66–R71. doi: 10.1152/ajpregu.1978.234.1.R66. [DOI] [PubMed] [Google Scholar]
- 85.Uhlich E, Weber P, Eigler J, et al. Angiotensin stimulated AVP-release in humans. Klin Wochenschr. 1975;53:177–180. doi: 10.1007/BF01466762. [DOI] [PubMed] [Google Scholar]
- 86.Kadekaro M. Nitric oxide modulation of the hypothalamo-neurohypophyseal system. Braz J Med Biol Res. 2004;37:441–450. doi: 10.1590/s0100-879x2004000400001. [DOI] [PubMed] [Google Scholar]
- 87.Alderton WK, Cooper CE, Knowles RG. Nitric oxide synthases: structure, function and inhibition. Biochem J. 2001;357:593–615. doi: 10.1042/0264-6021:3570593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Stuehr DJ. Enzymes of the L-arginine to nitric oxide pathway. J Nutr. 2004;134:2748S–2751S. doi: 10.1093/jn/134.10.2748S. discussion 2765S–2767S. [DOI] [PubMed] [Google Scholar]
- 89.Stern JE, Li Y, Zhang W. Nitric oxide: a local signalling molecule controlling the activity of pre-autonomic neurones in the paraventricular nucleus of the hypothalamus. Acta Physiol Scand. 2003;177:37–42. doi: 10.1046/j.1365-201X.2003.01045.x. [DOI] [PubMed] [Google Scholar]
- 90.Bains JS, Ferguson AV. Nitric oxide depolarizes type II paraventricular nucleus neurons in vitro. Neuroscience. 1997;79:149–159. doi: 10.1016/s0306-4522(96)00670-7. [DOI] [PubMed] [Google Scholar]
- 91.Liu QS, Jia YS, Ju G. Nitric oxide inhibits neuronal activity in the supraoptic nucleus of the rat hypothalamic slices. Brain Res Bull. 1997;43:121–125. doi: 10.1016/s0361-9230(96)00209-2. [DOI] [PubMed] [Google Scholar]
- 92.Kadekaro M, Terrell ML, Liu H, et al. Effects of L-NAME on cerebral metabolic, vasopressin, oxytocin, and blood pressure responses in hemorrhaged rats. Am J Physiol. 1998;274:R1070–R1077. doi: 10.1152/ajpregu.1998.274.4.R1070. [DOI] [PubMed] [Google Scholar]
- 93.Seo DO, Rivier C. Interaction between alcohol and nitric oxide on ACTH release in the rat. Alcohol Clin Exp Res. 2003;27:989–996. doi: 10.1097/01.ALC.0000071737.84882.C4. [DOI] [PubMed] [Google Scholar]
- 94.Whitaker AM, Sulzer JK, Molina PE. Augmented central nitric oxide production inhibits vasopressin release during hemorrhage in acute alcohol-intoxicated rodents. Am J Physiol Regul Integr Comp Physiol. 2011;301:R1529–R1539. doi: 10.1152/ajpregu.00035.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sulzer JK, Molina PE. Delayed resuscitation with physostigmine increases end organ damage in alcohol intoxicated rats. Shock. 2011;35:74–79. doi: 10.1097/SHK.0b013e3181e9aaaf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Muller S, How OJ, Hermansen SE, et al. Vasopressin impairs brain, heart and kidney perfusion: an experimental study in pigs after transient myocardial ischemia. Crit Care. 2008;12:R20. doi: 10.1186/cc6794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Westphal M, Ertmer C, Van Aken H, et al. Terlipressin in patients with septic shock: friend or foe? Intensive Care Med. 2004;30:992. doi: 10.1007/s00134-004-2243-3. author reply 993. [DOI] [PubMed] [Google Scholar]
- 98.Ono M, Yu B, Hardison EG, et al. Increased susceptibility to liver injury after hemorrhagic shock in rats chronically fed ethanol: role of nuclear factor-kappa B, interleukin-6, and granulocyte colony-stimulating factor. Shock. 2004;21:519–525. doi: 10.1097/01.shk.0000126905.75237.07. [DOI] [PubMed] [Google Scholar]
- 99.Zakaria el R, Campbell JE, Peyton JC, et al. Postresuscitation tissue neutrophil infiltration is time-dependent and organ-specific. J Surg Res. 2007;143:119–125. doi: 10.1016/j.jss.2007.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Akhtar S, Li X, Kovacs EJ, et al. Interleukin-18 delays neutrophil apoptosis following alcohol intoxication and burn injury. Mol Med. 2011;17:88–94. doi: 10.2119/molmed.2010.00080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Horie Y, Yamagishi Y, Kato S, et al. Role of ICAM-1 in chronic ethanol consumption-enhanced liver injury after gut ischemia-reperfusion in rats. Am J Physiol Gastrointest Liver Physiol. 2002;283:G537–G543. doi: 10.1152/ajpgi.00098.2002. [DOI] [PubMed] [Google Scholar]
- 102.Shi HP, Deitch EA, Da Xu Z, et al. Hypertonic saline improves intestinal mucosa barrier function and lung injury after trauma-hemorrhagic shock. Shock. 2002;17:496–501. doi: 10.1097/00024382-200206000-00010. [DOI] [PubMed] [Google Scholar]
- 103.Kuebler JF, Jarrar D, Toth B, et al. Estradiol administration improves splanchnic perfusion following trauma-hemorrhage and sepsis. Arch Surg. 2002;137:74–79. doi: 10.1001/archsurg.137.1.74. [DOI] [PubMed] [Google Scholar]
- 104.Sheth SU, Palange D, Xu DZ, et al. Testosterone depletion or blockade in male rats protects against trauma hemorrhagic shock-induced distant organ injury by limiting gut injury and subsequent production of biologically active mesenteric lymph. J Trauma. 2011;71:1652–1658. doi: 10.1097/TA.0b013e31823a06ea. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Mattox KL, Maningas PA, Moore EE, et al. Prehospital hypertonic saline/dextran infusion for post-traumatic hypotension. The U.S.A. Multicenter Trial. Ann Surg. 1991;213:482–491. doi: 10.1097/00000658-199105000-00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Angle N, Cabello-Passini R, Hoyt DB, et al. Hypertonic saline infusion: can it regulate human neutrophil function? Shock. 2000;14:503–508. [PubMed] [Google Scholar]