

Abstract
MYOC mutations were originally identified in patients with juvenile open angle glaucoma (JOAG). Cell culture and mouse studies suggest that MYOC mutations cause glaucoma through a dominant-negative effect on myocilin protein secretion. We tested this hypothesis with patient samples in this study. Glaucoma and control patients underwent complete ocular examination. DNA samples from glaucoma patients, and unaffected relatives and controls were used for DNA sequencing of MYOC. Aqueous humor (AH) samples from glaucoma and control patients were obtained at the time of surgery. Myocilin protein in AH was detected by quantitative Western blot analysis. A de novo Val251Ala mutation of MYOC was found to segregate with disease in a family with autosomal dominant JOAG. Myocilin protein was detected in all control AH samples but not was nearly undetectable in AH samples from a patient heterozygous for the Val251Ala mutation. Our results using human patient samples are consistent with a dominant-negative effect of pathogenic MYOC mutations on myocilin secretion.
Keywords: aqueous humor, de novo mutation, glaucoma, myocilin
1. Introduction
Primary open angle glaucoma (POAG) is the leading cause of irreversible blindness due to optic nerve damage. An important risk factor for POAG is elevated intraocular pressure (IOP) which is determined by the balance of the flow of aqueous humor (AH) into and out of the anterior chamber of the eye [1]. In POAG, the rate of AH production is not affected and elevated IOP is caused by increased resistance of AH outflow through the trabecular meshwork (TM), a filtering structure composed of alternating layers of extracellular matrix and TM cells. The precise mechanism of increased resistance to AH outflow through the TM is not well understood [2].
Myocilin contains an olfactomedin-homology domain and is a secreted protein with unknown functions. The myocilin gene (MYOC) is located within the glaucoma locus GLC1A that was identified in a family with an early-onset form of glaucoma, juvenile open angle glaucoma (JOAG) inherited in an autosomal dominant manner [3]. Screening of candidate genes within GLC1A revealed mutations in MYOC segregating with disease in several JOAG families [4]. Although initially discovered in patients with JOAG, mutations in MYOC also account for 2–4% of POAG cases [4–6]. More than 70 disease-causing mutations in MYOC have been identified, the vast majority of which occur in exon 3 which encodes the olfactomedin-homology domain [7].
Disease-causing mutations in MYOC result in intracellular retention of the normally secreted protein, as shown in vitro with cell lines transfected with mutant MYOC [8, 9]. The non-secretion phenotype is a dominant-negative effect, since co-expression of mutant and normal wild-type myocilin in cell lines results in intracellular retention or reduced secretion of both forms [10]. The accumulation of myocilin in the endoplasmic reticulum (ER) has suggested that an ER stress response is likely the disease mechanism for MYOC mutations [11].
To date, there is little direct evidence that MYOC mutations lead to a non-secretion phenotype in the AH of human subjects only a few AH samples have been used to investigate possible effects of MYOC mutations [8], in part due to the difficulty of obtaining AH samples from the relatively rare patients with pathogenic MYOC mutations. In this study, we investigated the expression presence of myocilin in the AH of a JOAG patient with a disease-causing MYOC mutation to test the hypothesis that such mutation leads to non-secretion of myocilin into AH.
2. Materials and methods
This study adhered to the tenets of the Declaration of Helsinki and was approved by the Institutional Review Board of Vanderbilt Medical Center. Other than unavailable family members in the 3-generation pedigree (II1, II3 and III2, Fig. 1A), all patients underwent complete ocular examination, including 24 control subjects. Inclusion criteria for JOAG were absence of secondary causes, age of onset < 40 years, elevated IOP > 21 mm Hg, open iridocorneal angles and glaucomatous optic nerve damage with associated visual field defects.
Figure 1.

Pedigree of patients with juvenile open angle glaucoma (JOAG) caused by a de novo MYOC Val251Ala mutation. Autosomal dominant inheritance of disease is shown in the three-generation pedigree (A). Affected members are represented with filled symbols, with the proband indicated by an arrow. and DNA samples were unavailable from family members (II-1, II-3 and III-2) are indicated by diagonal lines. Two grandparents (I-1 and I-2) in the first generation were disease free, but their daughter (II-2) and three grandchildren (III-1, III-2 and III-3) were all affected. Sequencing of MYOC revealed that all affected individuals in the pedigree (II-2, III-1 and III-3) carry a heterozygous T>C substitution, resulting in an amino acid change Val251Ala (B). Two available normal individuals (I-1 and I-2) in the pedigree have homozygous allele TT in the location (C). Haplotype analysis of 4 polymorphic SNPs within MYOC revealed that the T>C substitution is a de novo mutation (asteriskarrow head) arising in II-2 and transmitted in an autosomal dominant fashion (A). Chromosomal location of the mutation and rs numbers for the genotyped SNPs are indicated (A).
Blood samples were obtained from patients after written consent forms were signed. DNA extractions from whole blood were performed on a Gentra Systems AutoPure robot using Puregene chemistry (Qiagen, Inc., Valencia, CA). PCR primers (Table 1) based on MYOC sequence (genomic, GRCh37/hg19 assembly; cDNA, NM_000261.1) were used to amplify exons 1 and 3, including proximal intronic sequence and partial 5′ promoter region. MYOC was sequenced in a total of 20 JOAG patients, including the proband. An additional 43 controls were screened for the Val 251Ala mutation. For haplotype analysis, PCR primers were designed to amplify regions within MYOC containing known SNPs (Table 1). PCR amplicons were sequenced using a 96-capillary ABI 3730xl DNA Analyzer (Life Technologies Corp., Carlsbad, CA). DNA sequence data was analyzed using Sequencher software, version 4.8 (Gene Codes Corp., Ann Arbor, MI). Novelty of identified MYOC variants was investigated by search of the variant databases NCBI dbSNP, Build 137 (Database of Single Nucleotide Polymorphisms, National Center for Biotechnology Information, National Library of Medicine), NHLBI Exome Variant Server (NHLBI GO Exome Sequencing Project, accessed September, 2012) and myocilin database, last updated on August 15, 2012.
Table 1. Primers used for PCR amplification and sequencing.
Primers for PCR amplification of MYOC gene and Sanger sequencing. One pair of primers was used for promoter region and Exon 1 including exon-intron junction, two pairs of primers were used for Exon 3 including exon-intron junctions. Four sets of primers used for haplotype analysis by genotyping SNPs are shown with respective SNP rs numbers. The Amplicon sizes of amplicons were are shown in base pairs (bp).
| Name | Sequence (5′ – 3′) | Target | Amplicon Size (bp) |
|---|---|---|---|
| M1.1F | GGTGCATAAATTGGGATGTTC | Promoter & Exon 1 | 999 |
| M1.1R | TTGTGCTAGCTGTGCAGTCTC | ||
| M3-3F | TGCGATAACTGAGGCGTAGAG | Exon 3 (Part 1) | 997 |
| M3-3R | GGAGGCTTTTCACATCTTGG | ||
| M3-2F | CATTGACTTGGCTGTGGATG | Exon 3 (Part 2) | 992 |
| M3.1R | CATCCTGCAATCACATCTCC | ||
| MS5F | CTCAATGAGTTTGCAGAGTG | rs235920 | 399 |
| MS5R | TCTGCTGTGCTGAGAGGTG | ||
| MS89F | TGAACCTTTGCTCAGATTG | rs235918 | 399 |
| MS89R | CCACTTTCACAAAAAGTGAC | ||
| MS4F | GTCCTGAACACCTGAGAATC | rs235868 | 396 |
| MS4R | GTAAGCAGGTTTAGGATTGG | ||
| MS2F | CTCAGGCCCAACTGTTATC | rs603930 | 295 |
| MS2R | AGAATTAGCTGGATGTGGTG |
AH samples were obtained from JOAG patients (n = 3) and controls (n=24) at the time of glaucoma or cataract surgery by a single surgeon (RWK) at the beginning of the procedures. Glaucoma was ruled out in control subjects undergoing cataract surgery. To obtain AH, a 30-gauge needle mounted on a tuberculin syringe was inserted through the clear cornea at the limbus into the central portion of the anterior chamber and 50 – 100 μl AH gently withdrawn. AH samples were frozen immediately after collection and stored at −80°C. AH was also obtained from 3 cadaver eyes within 24 h post-mortem (N1, N2 and N3 in Fig. 2) from donors with no documented history of glaucoma. The age at time of death for cadaver eyes was 36, 38 and 32 years for N1, N2 and N3, respectively.
Figure 2.
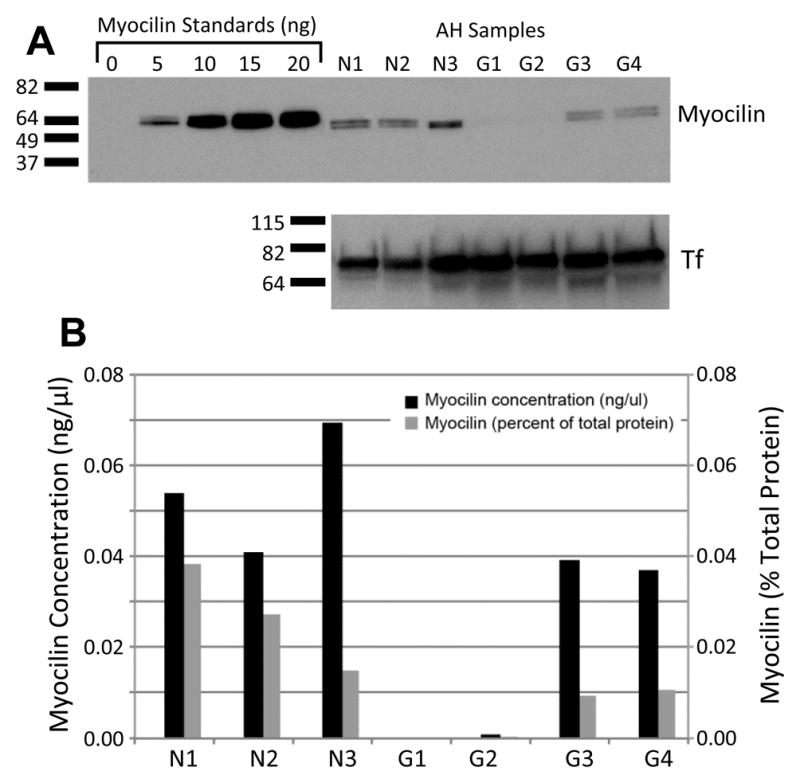
MYOC Val251Ala mutation causes non-secretion of protein into the aqueous humor. Nearly undetectable lelels of myocilin in AH of patient with Val251Ala MYOC mutation. Western analysis blot (A)of myocilin protein (upper panel) shows no detectable bands faint bands slightly above background in the AH samples of the patient with Val251Ala MYOC mutation (G1 and G2) in comparison with normal controls (N1, N2 and N3), and two JOAG patients (G3 and G4) that do not carry pathogenic MYOC mutations, but similar band intensities for transferrin as internal control (lower panel). G1 and G2 represent AH samples from left eye and right eye of the patient III-3 shown in Fig. 1A, obtained on separate surgeries. Recombinant myocilin standards for quantitation ranging from 0 ng to 20 ng are shown in the left-most 5 lanes. Quantitation of Western blot (B) shown in A is displayed as myocilin concentration (black bars) and amount of myocilin as a percentage of total AH proteins (light gray bars) in AH (B).
The amount of myocilin protein in AH samples from control and JOAG patients was determined by a quantitative Western blot method which includes recombinant myocilin standards in each gel, as previously described [12, 13], using a rabbit-polyclonal antibody that recognizes amino acids 108 to 131 of myocilin [14]. The intensity levels of myocilin were assessed with Image J software. For each gel, the amount of myocilin protein for each sample was calculated using a standard curve based on the band intensities of the myocilin standards (Fig. 3). Total protein concentration in AH was determined by Bradford assay.
Figure 3.
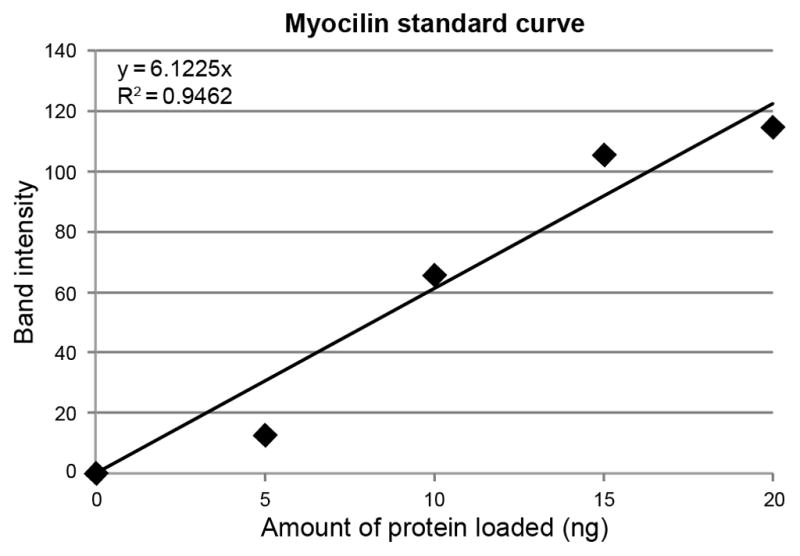
Representative graph of recombinant myocilin protein standard curve based on band intensity. A linear standard curve with R2 of 0.95 is shown.
3. Results
A 34 year-old non-Hispanic European Caucasian American male patient of European American ancestry presented to the glaucoma clinic with IOP of 40 mmHg, after having discontinued glaucoma medications. He had been previously diagnosed with JOAG at the age of 20 years at another institution and his diagnosis was confirmed in our clinic. According to the patient’s clinical history, both of his siblings and his mother were affected by JOAG. Diagnosis of JOAG was confirmed by clinical exam of the mother and available sibling of the initial patient (II-2 and III-1, Fig. 1A). As shown in Fig. 1A, the disease is transmitted in an autosomal dominant fashion in this family, consistent with most reports of familial cases of JOAG.
Sequencing of MYOC of the initial patient (III-3, Fig. 1) revealed that he is heterozygous for a T to C nucleotide substitution (NM_000261.1:c.752T>C) in exon 3, which changes the codon of amino acid 251 from GTA to GCA, resulting in a valine to an alanine substitution (NP_000252.1:p.Val251Ala) (Fig. 1B). The Val251Ala variant was not found in an additional 19 unrelated JOAG patients nor in 43 unrelated controls. In addition, the variant is not found in NCBI dbSNP or in data from the NHLBI Exome Variant Server which includes MYOC sequence data for 8,600 European American chromosomes, indicating that Val251Ala is a rare variant. Both the affected mother and affected sister are also heterozygous for the Val251Ala variant. The unaffected grandparents were homozygous for the normal wild-type allele (Fig. 1C). Haplotype analysis using 4 polymorphic markers within MYOC indicate that the Val251Ala variant is a de novo mutation originating in the mother (II-2, Fig. 1A). Segregation of the heterozygous Val251Ala mutation with disease is consistent with autosomal dominant inheritance of JOAG in this family.
To investigate the functional significance of the Val251Ala mutation, the amount of myocilin in the AH of the initial patient (III-3, Fig. 1A) was compared to controls by quantitative Western blot (Fig. 2). Doublet bands, caused by different myocilin glycosylation patterns, were detected at the expected molecular weight were detected for control AH samples from cadaver eyes from donors not affected by glaucoma (Lanes N1, N2 and N3, Fig. 2A). Myocilin protein was also detected in AH collected at the time of glaucoma surgery from 2 JOAG patients who did not carry disease-causing MYOC mutations (G3 and G4, Fig. 2). The first JOAG patient (G3, Fig. 2) carries two known benign polymorphisms, c.-83G>A and Arg76Lys [6]. No variants were detected in the second JOAG patient (G4, Fig. 2). Fig. 2B shows quantification of myocilin in the AH in terms of concentration (black bars) and percent of total protein (light gray bars). An additional set of 24 samples of AH from control patients unaffected by glaucoma were also investigated by quantitative Western, with myocilin protein detected in all samples. The average myocilin concentration of normal samples, including 24 samples obtained at the time of cataract surgery and 3 samples obtained from cadaver eyes, was 0.07±0.05 ng/μl. Myocilin protein from these 27 normal samples accounts for 0.038±0.024% of the total proteins in AH. For the initial patient with the Val251Ala mutation, AH humor samples were obtained from the left and right eyes at the time of surgery on separate occasions. In contrast to the other 27 control AH samples investigated, Myocilin protein was not detected by Western blot in either of the two both samples from the patient carrying the Val251Ala mutation were the only samples that showed only faint bands slightly above background (G1 and G2, Fig. 2), suggesting that this mutation results in non-secretion low levels of myocilin protein in into the AH.
4. Discussion
The Val251Ala mutation found in this study is likely pathogenic since it segregates with disease and is a rare allele not found in either dbSNP or within the Exome Variant Server data which includes MYOC sequence information for 8,600 chromosomes of the same similar ancestry as the study family. Similar to nearly all pathogenic MYOC mutations, the Val251Ala mutation is within the olfactomedin homology domain encoded by exon 3. A previous study reported a Val251Ala mutation, however, the nucleotide position of the mutation and the codon to amino acid conversion reported were erroneous [15]. Therefore, it is difficult to know if that variant was Val251Ala or Gly244Ala. The Val251Ala mutation reported in this study is a de novo mutation originating in the mother of the initially presenting patient.
Using AH samples from patients, we were able to test the hypothesis that pathogenic MYOC mutations result in non-secretion of myocilin in a dominant-negative fashion. The evidence for this hypothesis has been almost entirely based on cell culture and mouse models. Obtaining AH samples from patients with known pathogenic MYOC mutations is difficult since only 2–4% of POAG cases are attributable to MYOC mutations and because AH can only be obtained from patients at the time of surgery. One published study investigated myocilin expression in the AH of 3 patients heterozygous for a disease-associated truncation mutation, Q368X [8]. Although the truncated form of myocilin was not detected in the AH of patients with the Q368X mutation, the normal form was detected strongly in 2 of 3 patients and weakly in the other, a finding not entirely consistent with the dominant negative mechanism suggested by cell culture and transgenic mouse studies.
Our Western analysis of patient AH samples supports the hypothesis of a disease mechanism involving a dominant-negative effect on secretion of myocilin into the AH. The only AH samples with no detected almost undetectable myocilin were obtained from the patient heterozygous for the Val251Ala MYOC mutation. The highest level of myocilin in the Val251Ala patient was 0.001 ng/μl, 10-fold lower than the lowest concentration found in controls which was 0.015 ng/μl. Since the 2 samples of AH from this patient were obtained from his left and right eyes on separate surgical dates and gave the same result, the near lack of detection is unlikely due to differences in sample handling or storage. Myocilin protein was detected in all control AH samples obtained from patients unaffected by glaucoma and in samples from 2 patients affected by JOAG with no disease-causing MYOC mutations. One JOAG patient (G3, Fig. 2) with known benign Arg76Lys variant had detectable myocilin in the AH, consistent with the previous report that this variant does not affect myocilin protein secretion [9]. Our findings confirm are consistent with a dominant-negative effect of a disease-causing MYOC mutation on secretion of both normal wild-type and mutated forms of myocilin in patient AH. However, it should be noted that since wild-type homodimers can still form and be secreted [10], these results cannot distinguish between a dominant negative effect and haploinsufficiency. In addition, since direct measurement of intracellular myocilin protein is not possible for these patients, our results do not provide direct evidence of a specific effect on secretion.
Cell culture studies have shown that most pathogenic MYOC mutations investigated, including 3 close to the Val251Ala mutation reported here (C245Y, G246R and G252R), are not secreted, but rather accumulate in the ER [9, 16]. Intracellular accumulation of myocilin occurs because the mutated forms are unstable [11, 17] and aberrantly targeted to peroxisomes due to the uncovering of a cryptic peroxisomal targeting signal [18]. Accumulation of mis-folded myocilin in the ER results in an ER stress response which is likely the disease mechanism for MYOC mutations. Consistent with an ER stress mechanism, recently it has been shown that transgenic mice over-expressing a pathogenic Y437H MYOC mutation develop elevated IOP and glaucoma. The disease could be prevented by systemic or topical treatment with phenylbutyric acid [19, 20], a drug approved for use in treating urea cycle disorders and which has been shown to relieve ER stress [16]. Our finding that the glaucoma-causing Val251Ala mutation leads to non-secretion is associated with nearly undetectable levels of myocilin in human AH is consistent with the disease mechanisms suggested by cell culture and mouse studies and supports the possibility that patients with pathogenic MYOC mutations may benefit from treatments for ER stress such as phenylbutyric acid.
Acknowledgments
We are grateful to our patients and their family members for their willingness to participate in this study. We wish to acknowledge the following sources of support: National Institutes of Health R01 EY020894 (RWK) and R01 EY07065 (MFP); Research to Prevent Blindness (RWK is a recipient of a Career Development Award and MPF is a recipient of a Lew R. Wasserman Merit Award; Vanderbilt Eye Institute and Mayo Clinic Department of Ophthalmology are the recipients of unrestricted grants); School of Medicine, Vanderbilt University and Mayo Foundation. We also wish to thank Jessica Kunkel for assisting experiments and Jennifer Shaw for helping with illustrations.
Footnotes
Conflict of interest
All authors declare no conflict of interest relevant to this manuscript.
Web Resources
NCBI Database of Single Nucleotide Polymorphisms: http://www.ncbi.nlm.nih.gov/SNP/
NHLBI Exome Variant Server: http://evs/gs.washington.edu/EVS/
Myocilin database: http://www.myocilin.com
Image J software: http://rsb.info.nih.gov/ij/index.html
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kwon YH, Fingert JH, Kuehn MH, Alward WL. Primary open-angle glaucoma. N Engl J Med. 2009;360(11):1113–1124. doi: 10.1056/NEJMra0804630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Johnson M. What controls aqueous humour outflow resistance? Exp Eye Res. 2006;82(4):545–557. doi: 10.1016/j.exer.2005.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sheffield VC, Stone EM, Alward WL, Drack AV, Johnson AT, Streb LM, Nichols BE. Genetic linkage of familial open angle glaucoma to chromosome 1q21–q31. Nat Genet. 1993;4(1):47–50. doi: 10.1038/ng0593-47. [DOI] [PubMed] [Google Scholar]
- 4.Stone EM, Fingert JH, Alward WL, Nguyen TD, Polansky JR, Sunden SL, Nishimura D, Clark AF, Nystuen A, Nichols BE, Mackey DA, Ritch R, et al. Identification of a gene that causes primary open angle glaucoma. Science. 1997;275(5300):668–670. doi: 10.1126/science.275.5300.668. [DOI] [PubMed] [Google Scholar]
- 5.Alward WL, Fingert JH, Coote MA, Johnson AT, Lerner SF, Junqua D, Durcan FJ, McCartney PJ, Mackey DA, Sheffield VC, Stone EM. Clinical features associated with mutations in the chromosome 1 open-angle glaucoma gene (GLC1A) N Engl J Med. 1998;338(15):1022–1027. doi: 10.1056/NEJM199804093381503. [DOI] [PubMed] [Google Scholar]
- 6.Fingert JH, Heon E, Liebmann JM, Yamamoto T, Craig JE, Rait J, Kawase K, Hoh ST, Buys YM, Dickinson J, Hockey RR, Williams-Lyn D, et al. Analysis of myocilin mutations in 1703 glaucoma patients from five different populations. Hum Mol Genet. 1999;8(5):899–905. doi: 10.1093/hmg/8.5.899. [DOI] [PubMed] [Google Scholar]
- 7.Resch ZT, Fautsch MP. Glaucoma-associated myocilin: a better understanding but much more to learn. Exp Eye Res. 2009;88(4):704–712. doi: 10.1016/j.exer.2008.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jacobson N, Andrews M, Shepard AR, Nishimura D, Searby C, Fingert JH, Hageman G, Mullins R, Davidson BL, Kwon YH, Alward WL, Stone EM, et al. Non-secretion of mutant proteins of the glaucoma gene myocilin in cultured trabecular meshwork cells and in aqueous humor. Hum Mol Genet. 2001;10(2):117–125. doi: 10.1093/hmg/10.2.117. [DOI] [PubMed] [Google Scholar]
- 9.Gobeil S, Letartre L, Raymond V. Functional analysis of the glaucoma-causing TIGR/myocilin protein: integrity of amino-terminal coiled-coil regions and olfactomedin homology domain is essential for extracellular adhesion and secretion. Exp Eye Res. 2006;82(6):1017–1029. doi: 10.1016/j.exer.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 10.Gobeil S, Rodrigue MA, Moisan S, Nguyen TD, Polansky JR, Morissette J, Raymond V. Intracellular sequestration of hetero-oligomers formed by wild-type and glaucoma-causing myocilin mutants. Invest Ophthalmol Vis Sci. 2004;45(10):3560–3567. doi: 10.1167/iovs.04-0300. [DOI] [PubMed] [Google Scholar]
- 11.Liu Y, Vollrath D. Reversal of mutant myocilin non-secretion and cell killing: implications for glaucoma. Hum Mol Genet. 2004;13(11):1193–1204. doi: 10.1093/hmg/ddh128. [DOI] [PubMed] [Google Scholar]
- 12.Howell KG, Vrabel AM, Chowdhury UR, Stamer WD, Fautsch MP. Myocilin levels in primary open-angle glaucoma and pseudoexfoliation glaucoma human aqueous humor. J Glaucoma. 2010;19(9):569–575. doi: 10.1097/IJG.0b013e3181d13020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fautsch MP, Bahler CK, Vrabel AM, Howell KG, Loewen N, Teo WL, Poeschla EM, Johnson DH. Perfusion of his-tagged eukaryotic myocilin increases outflow resistance in human anterior segments in the presence of aqueous humor. Invest Ophthalmol Vis Sci. 2006;47(1):213–221. doi: 10.1167/iovs.05-0334. [DOI] [PubMed] [Google Scholar]
- 14.Fautsch MP, Bahler CK, Jewison DJ, Johnson DH. Recombinant TIGR/MYOC increases outflow resistance in the human anterior segment. Invest Ophthalmol Vis Sci. 2000;41(13):4163–4168. [PubMed] [Google Scholar]
- 15.Michels-Rautenstrauss K, Mardin C, Wakili N, Junemann AM, Villalobos L, Mejia C, Soley GC, Azofeifa J, Ozbey S, Naumann GO, Reis A, Rautenstrauss B. Novel mutations in the MYOC/GLC1A gene in a large group of glaucoma patients. Hum Mutat. 2002;20(6):479–480. doi: 10.1002/humu.9092. [DOI] [PubMed] [Google Scholar]
- 16.Yam GH, Gaplovska-Kysela K, Zuber C, Roth J. Sodium 4-phenylbutyrate acts as a chemical chaperone on misfolded myocilin to rescue cells from endoplasmic reticulum stress and apoptosis. Invest Ophthalmol Vis Sci. 2007;48(4):1683–1690. doi: 10.1167/iovs.06-0943. [DOI] [PubMed] [Google Scholar]
- 17.Burns JN, Turnage KC, Walker CA, Lieberman RL. The stability of myocilin olfactomedin domain variants provides new insight into glaucoma as a protein misfolding disorder. Biochemistry. 2011;50(26):5824–5833. doi: 10.1021/bi200231x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shepard AR, Jacobson N, Millar JC, Pang IH, Steely HT, Searby CC, Sheffield VC, Stone EM, Clark AF. Glaucoma-causing myocilin mutants require the Peroxisomal targeting signal-1 receptor (PTS1R) to elevate intraocular pressure. Hum Mol Genet. 2007;16(6):609–617. doi: 10.1093/hmg/ddm001. [DOI] [PubMed] [Google Scholar]
- 19.Zode GS, Kuehn MH, Nishimura DY, Searby CC, Mohan K, Grozdanic SD, Bugge K, Anderson MG, Clark AF, Stone EM, Sheffield VC. Reduction of ER stress via a chemical chaperone prevents disease phenotypes in a mouse model of primary open angle glaucoma. J Clin Invest. 2011;121(9):3542–3553. doi: 10.1172/JCI58183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zode GS, Bugge KE, Mohan K, Grozdanic SD, Peters JC, Koehn DR, Anderson MG, Kardon RH, Stone EM, Sheffield VC. Topical ocular sodium 4-phenylbutyrate rescues glaucoma in a myocilin mouse model of primary open-angle glaucoma. Invest Ophthalmol Vis Sci. 2012;53(3):1557–1565. doi: 10.1167/iovs.11-8837. [DOI] [PMC free article] [PubMed] [Google Scholar]