

Abstract
Glycogen storage disease type IX (GSD IX) is described as a benign condition that often does not require treatment. Most patients with the disease are thought to outgrow the childhood manifestations, which include hepatomegaly, poor growth, and ketosis with or without hypoglycemia. Long term complications including fibrosis and cirrhosis have seldom been reported in the most common subtype, GSD IXα. We present two cases of children with GSD IXα who had fibrosis at the time of diagnosis in addition to the commonly reported disease manifestations. Structured therapy with frequent doses of uncooked cornstarch and protein supplementation was initiated, and both children responded with improved growth velocity, increased energy, decreased hepatomegaly and improved well-being. Additionally, radiographic features of fibrosis improved. We propose that GSD IXα is not a benign condition. Even in patients with a less severe presentation, consideration of a structured treatment regimen to improve quality of life appears warranted.
Keywords: Glycogen storage disease type IX, Cirrhosis, Treatment
Introduction
Glycogen storage disease type IX is likely the most common type of glycogen storage disease, but there has been minimal research on the natural history and treatment of this condition. The first case of hepatic phosphorylase b kinase (PhK) deficiency appeared in the literature in 19661. By mixing homogenates of the patient[‘s liver and of normal liver, the low phosphorylase activity was determined to result from deficiency of phosphorylase kinase and not phosphorylase itself. Phosphorylase activity was then restored to normal values by the addition of phosphorylase b kinase from rabbit muscle, further substantiating this conclusion. The first patient described with what is now called glycogen storage disease type IX (GSD IX) was a girl, and the disease was thought to be inherited in an autosomal recessive pattern. Shortly thereafter, other pedigrees were more compatible with X-linked inheritance.2 The X-linked form of the disease was initially classified as GSD VIII, but both inheritance patterns are now classified as GSD IX since they are caused by deficiency of the same enzyme complex. The classification GSD VIII no longer exists.3
It is now known that liver PhK is comprised of four subunits (α, β, γ, and δ), and subtypes of liver PhK deficiency have been classified by the subunit where mutations have been found. The most common subtype is GSD IXα, accounting for about 75% of all GSD IX.3 It is caused by a mutation in the PHKA2 gene on the X-chromosome, and as such has also been called X-linked glycogenosis (XLG). The other two subtypes are inherited in an autosomal-recessive manner, with males and females equally affected. Mutations in the PHKB gene result in GSD IXβ, with PhK deficiency both in liver and muscle. However, the muscle symptoms can be mild or absent; thus, this subtype may be clinically indistinguishable from the liver PhK deficiencies caused by other mutations. The gamma subunit, encoded by the PHKG2 gene, contains the catalytic site of the enzyme. Mutations in this gene are known to be associated with a more severe phenotype that can present with cirrhosis in childhood.4 Mutations in gene that code for the delta subunit of the enzyme have not been described to date.
GSD IX is characterized by childhood onset of hepatomegaly, growth retardation and fasting ketosis. Hypoglycemia is not always pronounced because fatty acid oxidation and gluconeogenesis are intact, and normal blood glucose concentrations may be maintained. The symptoms and biochemical abnormalities are thought to improve with age, and progression to cirrhosis has been deemed rare except in the small subset of patients with PhK deficiency caused by mutations in the PHKG2 gene. Treatment of disease manifestations has traditionally been based upon symptoms, and it is widely believed that some individuals require no treatment at all. In this case series, we report 2 patients with mutations in the PHKA2 gene that presented with cirrhosis at the time of diagnosis. While minimal hypoglycemia was occurring, prominent ketosis was present. With aggressive therapy with protein and cornstarch, all biochemical and laboratory abnormities were ameliorated, and clinical improvement has occurred.
Case Reports
Patient 1
Patient 1 is a former 9 pound 5 ounce male delivered at term following an uncomplicated pregnancy. No hypoglycemia was documented in the perinatal period, and he had no difficulty with the postnatal transition. Abdominal distension was noted at one year of age, but no abnormalities were detected on abdominal ultrasound. Throughout childhood, frequent nausea and vomiting occurred in the morning. However, he was thriving otherwise, and developmental milestones were achieved appropriately. At 6 years of age, hepatic transaminases were found to be elevated (ALT 308 U/L, AST 336 U/L), and abdominal ultrasound revealed marked hepatomegaly. The patient underwent a liver biopsy, and gross pathology revealed diffuse enlargement of the hepatocytes with focal macrovesicular steatosis. Abundant glycogen was present on PAS staining, and cirrhosis was present with portal to portal fibrosis. Based upon the findings, GSD type IV was suspected, and the patient was referred for evaluation for a liver transplant. However, amylopectin inclusions were not demonstrated, and sequencing of the GBE1 gene was normal. A repeat biopsy was performed, and again diffuse ballooning of the hepatocytes with glycogen and portal to portal fibrosis were noted. Focal regenerative nodules were present, and enzymatic studies were inconclusive.
Due to the suspicion of glycogen storage disease, frequent feeds were initiated, but marked transaminase elevation persisted. Liver transplantation was recommended, but the patient was referred to our program at 7 years of age for a second opinion before the procedure occurred. Even though minimal hypoglycemia was being documented, monitoring revealed profound morning ketosis. At 7 years 6 months of age, the patient was admitted to our metabolic unit for initiation of a formal treatment regimen. Metabolic monitoring revealed both day and night ketosis with post-prandial hyperlactatemia. Therapy with uncooked cornstarch (dosed 3 times per day) and protein (2.5 g/kg/day) was commenced based upon the results, and a dramatic improvement in energy occurred with resolution of his morning nausea and vomiting. Subsequent mutation analysis confirmed X-linked glycogenosis with a hemizygous sequence change in the PHKA2 gene with c.883C>T in exon 9, altering the arginine codon at position 295 to a cysteine codon (p.Arg295Cys). This mutation has been previously reported in the XLG2 subtype of GSD IX5.
With the initiation of formal therapy, a dramatic increase in growth occurred (Figure 1), and metabolic derangements ameliorated (Table 1). Suboptimal growth was a concern throughout childhood; however, with treatment, growth velocity exceeded the 99th percentile for age, and a height increase of more than 1.4 standard deviations was seen in less than 2 years (Figure 1, red). Serum concentrations of hepatic transaminases, triglycerides, and lactate were persistently elevated before treatment. Following initiation of a structured treatment regimen, all laboratory studies markedly improved. Anemia that was present before treatment resolved, and hepatic transaminase concentrations normalized. The liver size decreased by 6.2 cm in the course of one year. Serial abdominal ultrasonography demonstrated resolution of his irregular hepatic contour, suggesting an improvement in fibrosis.
Figure 1.
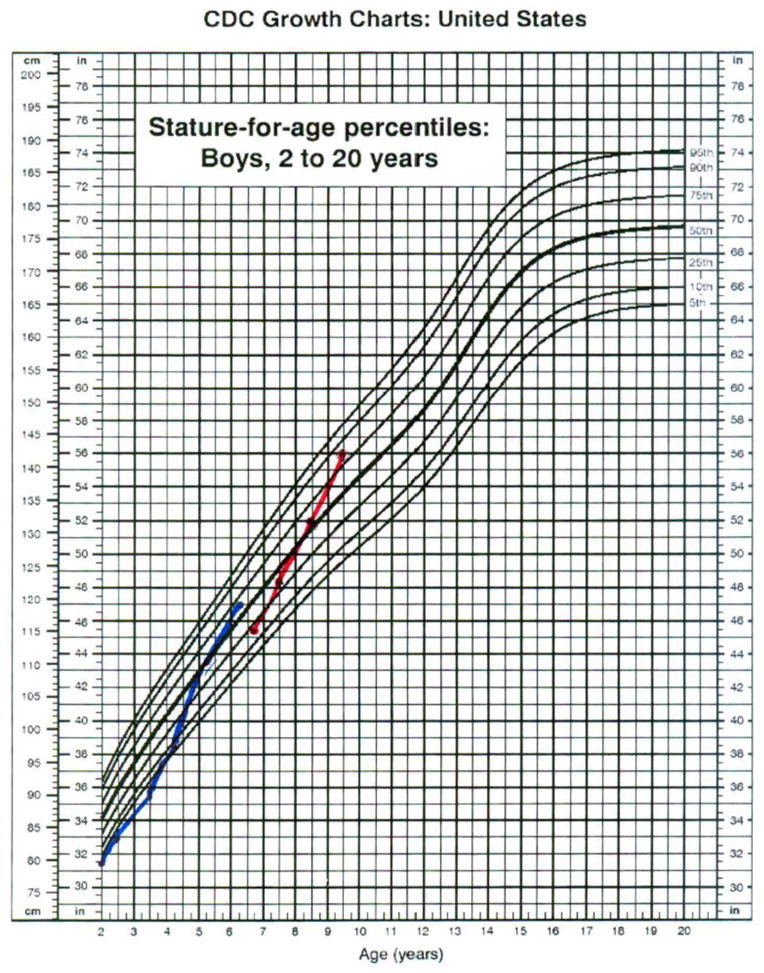
SOURCE: Developed by the National Center for Health Statistics in collaboration with the National Center for Chronic Disease Prevention and Health Promotion (2000).
Table 1.
Markers of Metabolic Control for Patient 1
| Age (years) | AST (U/L) | ALT (U/L) | Triglycerides (mg/dL) |
|---|---|---|---|
| 6–6/12 | 788 | 523 | |
| 7–3/12 | 424 | 328 | 326 |
| 7–9/12 | 172 | 163 | 222 |
| 8–0/12 | 174 | 166 | 349 |
| 8–6/12 | 61 | 102 | 105 |
| 9–0/12 | 100 | 140 | 129 |
| 9–3/12 | 73 | 136 | 174 |
| 9–6/12 | 64 | 91 | 168 |
Patient 2
This patient is a 5 pound, 13 ounce male product of a term gestation complicated by poor maternal weight gain. No hypoglycemia was noted during the perinatal transition. Throughout infancy, the patient was constantly hungry, and he breast fed on demand every 2 hours around the clock. Severe reflux was problematic, and he had a history of projectile vomiting that most frequently occurred in the morning. During the second year of life, his growth velocity dramatically decreased, and screening tests revealed iron deficiency anemia in the setting of chronic diarrhea. Further evaluation revealed massive hepatomegaly and marked hepatic transaminase elevation associated with an anion gap acidosis (AST 681 U/L, ALT 410 U/L, TCO2 15, anion gap 20). A percutaneous liver biopsy revealed swollen hepatocytes and portal to portal fibrosis with bridging, consistent with early cirrhosis. Glycogen storage disease was suspected although excessive glycogen could not be demonstrated.
A second liver biopsy was performed at 3 years of age to establish a diagnosis. Elevated glycogen was found in his liver, and enzymatic testing was consistent with type IX glycogen storage disease. The diagnosis of phosphorylase kinase deficiency was confirmed genetically when a mutation was discovered in the PHKA2 gene, c.133C>T, altering the arginine codon at position 45 to a tryptophan codon (p.Arg45Trp [R45W]). Therapy was initiated with a regimen of frequent feeds throughout the day, but concern about suboptimal energy and abnormal laboratory studies persisted. Home monitoring of blood ketones revealed marked ketosis after an overnight fast, and he became profoundly ketotic with beta-OH-butyrate concentrations greater than 3 mmol/L during a gastrointestinal illness.
The patient was referred to our program at 3 years 10 months of age, and, and supplementation with uncooked cornstarch and protein was initiated. With a formal therapeutic regimen, dramatic clinical and biochemical improvement was noted. Energy level and stamina markedly increased, and growth acceleration occurred. A height increase of 2.73 standard deviations was seen within 3 years of initiating therapy (Figure 1, blue). Hepatic transaminase elevation and hyperlipidemia normalized on therapy (Table 2). Less than two years after initiation of therapy, he had no evidence of hepatic fibrosis on ultrasound exam.
Table 2.
Markers of Metabolic Control for Patient 2
| Age (years) | AST (U/L) | ALT (U/L) | Triglycerides (mg/dL) |
|---|---|---|---|
| 2–0/12 years | 1423 | 778 | 561 |
| 2–6/12 years | 707 | 659 | 372 |
| 2–9/12 years | 947 | 885 | 493 |
| 3–0/12 years | 59 | 71 | 353 |
| 3–6/12 years | 42 | 60 | 325 |
| 4–0/12 years | 40 | 53 | 151 |
| 5–0/12 years | 45 | 67 | 450 |
| 6–0/12 years | 34 | 50 | 394 |
| 6–3/12 years | 43 | 73 | 347 |
Discussion
Almost five decades after it was first described, GSD IX is widely regarded as a benign condition. Treatment is often deemed unnecessary because the childhood symptoms frequently improve with age. However, children who are untreated may experience untoward effects of the disease. Morning nausea and vomiting commonly occur, and school absence can lead to academic difficulties. Poor growth and pubertal delay may cause psychological distress even if catch-up growth occurs.6 Furthermore, there is evidence that peak bone mineral accretion is lower in adults with a history of delayed puberty, which may increase the risk of fracture later in life.7 All of these complications can be attributed to presence of chronic ketosis, and this metabolic abnormality has been under-appreciated. Severe ketosis (comparable to a diabetes patient having large ketones) can occur even in the setting of relative normoglycemia, and normalization of the ketone concentrations through treatment with cornstarch and protein normalizes other laboratory abnormalities and appears to decrease the risk of complications.
While mutations in the PHKG2 gene are known to be associated with a more severe phenotype, the XLG subtype of GSD IX due to mutations in PHKA2 has been regarded as a benign condition with minimal complications. A broad phenotypic spectrum has been reported in GSD IX caused by mutations in the PHKA2 gene, but only one case associated with fibrosis is reported in the literature until recently.8 We present two patients with PhK deficiency caused by a hemizygous mutation in the PHKA2 gene with hepatic fibrosis at the time of diagnosis, which is similar to another recent case report.9 In contrast to the aforementioned report, however, this series demonstrates the benefits of ketone monitoring and aggressive treatment for this condition. Both children in this series experienced a dramatic improvement in growth velocity, energy, biochemical abnormalities, hepatomegaly and overall well-being with the initiation of aggressive therapy.
There is mounting evidence that long term complications associated with other hepatic forms of GSD can be prevented by optimizing metabolic control. An association between elevated lactate concentrations and an increased frequency of long-term complications in GSD Ia patients was first reported in 2002.10 It has been shown that improved metabolic control was associated with a decreased risk of hepatocellular adenoma development in patients with GSD I.11 Similarly, optimal metabolic control has been reported to have a protective effect on the development of microalbuminuria and proteinuria in patients with GSD I.12 Normal bone density has also been associated with improved metabolic control in patients with GSD Ia.13 We report two patients with hepatic fibrosis that demonstrated improvement on serial ultrasound examination with improved metabolic control, and this adds to the increasing evidence that optimization of metabolic control should occur in GSD patients before pursuing liver transplantation.
Conclusion
We report two cases of patients with GSD IXα, a condition thought to be mild and often described as benign, who had fibrosis at the time of diagnosis. Both demonstrated considerable improvement in growth velocity, energy, biochemical abnormalities, hepatomegaly and overall well-being with the initiation of aggressive therapy. We propose that GSD IXα is not always a mild condition that improves with age that can be expected to be asymptomatic in adulthood, as is often reported in the literature. 3,14 Even in the absence of cirrhosis, aggressive therapy can improve quality of life.
HIghlights.
We describe two patients with a severe presentation of GSD IX alpha.
We treated with frequent doses of uncooked cornstarch and protein supplementation.
Structured frequent therapy resulted in the improvement of hepatic fibrosis on ultrasound exam.
Improved growth velocity and quality of life were seen with the initiation of a formal treatment regimen.
Patients with less severe disease may similarly experience improved growth velocity and quality of life on structured therapy.
Acknowledgments
Support for this project was provided in part by the National Institutes of Health (NIH) and National Center for Research Resources (NCRR) CTSA grant UL1 TR000064 (University of Florida). Philanthropic assistance was provided from Matthew’s GSD Type IX Fund and the Sturtz GSD Research Fund which are managed through the University of Florida Office of Development.
List of Abbreviations
- GSD
Glycogen storage disease
- PhK
Phosphorylase b kinase
- XLG
X-Linked glycogenosis
Footnotes
Funding: None of the authors has any financial interest in this work or conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hug G, Schubert WK, Chuck G. Phosphorylase kinase deficiency of the liver: deficiency in a girl with increased hepatic glycogen. Science. 1966 Sep 23;153(3743):1534–5. doi: 10.1126/science.153.3743.1534. [DOI] [PubMed] [Google Scholar]
- 2.Huijing F, Fernandes J. X-chromosomal inheritance of liver glycogenosis with phosphorylase kinase deficiency. Am J Hum Genet. 1969;21:267. [PMC free article] [PubMed] [Google Scholar]
- 3.Goldstein J, Austin S, Kishnani J, Bali D. Phosphorylase Kinase Deficiency. [Accessed November 26, 2012];GeneReviews at Gene Tests: Medical Genetics Information Resource. http://www.genetests.org.
- 4.Burwinkel B, Rootwelt T, Kvittingen EA, et al. Severe phenotype of phosphorylase kinase-deficient liver glycogenosis with mutations in the PHKG2 gene. Pediatr Res. 2003;54:834. doi: 10.1203/01.PDR.0000088069.09275.10. [DOI] [PubMed] [Google Scholar]
- 5.Ban K, Sugiyama K, Goto K, Mizutani F, Togari H. Detection of PHKA2 gene mutation in four Japanese patients with hepatic phosphorylase kinase deficiency. Tohoku J Exp. 2003;200:47–53. doi: 10.1620/tjem.200.47. [DOI] [PubMed] [Google Scholar]
- 6.Schippers HM, Smit GP, Rake JP, Visser G. Characteristic growth pattern in male X-linked phosphorylase-b kinase deficiency (GSD IX) J Inherit Metab Dis. 2003;26(1):43–7. doi: 10.1023/a:1024071328772. [DOI] [PubMed] [Google Scholar]
- 7.Finkelstein JS, Klibanski A, Neer RM. A longitudinal evaluation of bone mineral density in adult men with histories of delayed puberty. J Clin Endocrinol Metab. 1996 Mar;81(3):1152–5. doi: 10.1210/jcem.81.3.8772591. [DOI] [PubMed] [Google Scholar]
- 8.Beauchamp NJ, Dalton A, Ramaswami U, Niinikoski H, Mention K, Kenny P, Kolho KL, et al. Glycogen storage disease type IX: High variability in clinical phenotype. Mol Genet Metab. 2007;92:88–99. doi: 10.1016/j.ymgme.2007.06.007. [DOI] [PubMed] [Google Scholar]
- 9.Johnson AO, Goldstein JL, Bali D. Glycogen storage disease type IX: novel PHKA2 missense mutation and cirrhosis. J Pediatr Gastroenterol Nutr. 2012;55:90–2. doi: 10.1097/MPG.0b013e31823276ea. [DOI] [PubMed] [Google Scholar]
- 10.Weinstein DA, Wolfsdorf JI. Effect of continuous glucose therapy with uncooked cornstarch on the long-term clinical course of type 1a glycogen storage disease. Eur J Pediatr. 2002;161 (Suppl 1):S35–39. doi: 10.1007/s00431-002-1000-2. [DOI] [PubMed] [Google Scholar]
- 11.Wang DQ, Fiske LM, Carreras CT, Weinstein DA. Natural history of hepatocellular adenoma formation in glycogen storage disease type I. J Pediatr. 2011;159:442–6. doi: 10.1016/j.jpeds.2011.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Martens D, Rake JP, Navis G, Fidler V, Van Dael C, Smit GP. Renal function in glycogen storage disease type 1, natural course, and renopreservative effects of ACE inhibition. Clin J Am Soc Nephrol. 2009;4(11):1741–6. doi: 10.2215/CJN.00050109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Minarich LA, Kirpich A, Fiske LM, Weinstein DA. Bone mineral density in glycogen storage disease type Ia and Ib. Genet Med. 2012 doi: 10.1038/gim.2012.36. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Burwinkel B, Maichele AJ, Aagenaes O, Bakker HD, Lerner A, Shin YS, Strachan JA, et al. Autosomal glycogenosis of liver and muscle due to phosphorylase kinase deficiency is caused by mutations in the phosphorylase kinase β subunit (PHKB) Hum Mol Genet. 1997;6(7):1109–1115. doi: 10.1093/hmg/6.7.1109. [DOI] [PubMed] [Google Scholar]