

Abstract
Background
Tumor necrosis factor (TNF) superfamily ligands that provoke a dilated cardiac phenotype signal through a common scaffolding protein termed TNF receptor associated factor 2 (TRAF2); however, virtually nothing is known with regard to TRAF2 signaling in the adult mammalian heart.
Methods and Results
We generated multiple founder lines of mice with cardiac restricted overexpression of TRAF2 and characterized the phenotype of mice with higher expression levels of TRAF2 (MHC-TRAF2HC). MHC-TRAF2HC transgenic mice developed a time-dependent increase in cardiac hypertrophy, LV dilation and adverse LV remodeling, and a significant decrease in LV +dP/dt and −dP/dt when compared to littermate (LM) controls (p < 0.05 compared to LM). During the early phases of LV remodeling there was a significant increase in total matrix metalloproteinase (MMP) activity that corresponded with a decrease in total myocardial fibrillar collagen content. As the MHC-TRAF2HC mice aged, there was a significant decrease in total MMP activity accompanied by an increase in total fibrillar collagen content and an increase in myocardial tissue inhibitor of metalloproteinase-1 levels. There was a significant increase in NF-κB activation at 4 – 12 weeks and JNK activation at 4 weeks in the MHCs TRAF2HC mice. Transciptional profiling revealed that > 95% of the hypertrophic/dilated cardiomyopathy-related genes that were significantly upregulated genes in the MHC-TRAF2HC hearts contained κB elements in their promoters.
Conclusions
These results show for the first time that targeted overexpression of TRAF2 is sufficient to mediate adverse cardiac remodeling in the heart.
Keywords: tumor necrosis factor superfamily, dilated cardiomyopathy, inflammation, TNF receptor associated factor 2
Sustained inflammatory signaling in the heart leads to the development of a cardiomyopathy that is characterized by left ventricular (LV) dilation, LV dysfunction and myocardial fibrosis. If the inciting cause for the inflammation is resolved (e.g. viral myocarditis or sepsis), the cardiomyopathy is often fully reversible, even though the extent of LV dysfunction may have been quite severe. If, on the other hand, inflammation is sustained, and/or the inciting agent is persistent (e.g. Chagas disease), sustained inflammatory signaling frequently leads to the development of an irreversible cardiomyopathy. Although a number or pro-inflammatory cytokines have been implicated in this process, the cytokine that has been characterized most extensively thus far is tumor necrosis factor (TNF [TNFSF2]). Indeed, mice with cardiac-restricted overexpression of TNF in the heart demonstrate consistent morphological and structural changes in the myocardium including left ventricular (LV) wall thinning, LV dilatation, LV dysfunction, and increased myocardial fibrillar collagen content that overlaps the phenotype observed in inflammation-induced dilated cardiomyopathy. 1–3
TNF is the prototypical member of the TNF superfamily (TNFSF), which consists of 19 well-characterized ligands and 34 TNF superfamily receptors (TNFRSF). Members of the TNF superfamily of ligands and receptors are expressed in a broad variety of cell types, including myocardial cells.4 Without exception, all members of the TNF superfamily exhibit pro-inflammatory activity. Of note, recent studies have identified a potential role for TNF superfamily ligands/receptors in terms of mediating inflammatory responses in the heart, including FasL/Fas (TNFSF6/TNFRSF6), TWEAK (tumor necrosis factor-like weak inducer of apoptosis)/TWEAKR (TNFSF12/TNFRSF12),5 and RANKL (Receptor activator of NF-kB ligand)/RANK (TNFSF11/TNFRSF11A).6 Although cardiac-restricted overexpression of FasL does not lead to a dilated cardiomyopathy,7 overexpression of TNF, TWEAK and RANKL are each sufficient to provoke a dilated cardiac phenotype. In contrast to FasL, TNF, TWEAK and RANKL signal through TNFSF receptors that engage a common scaffolding protein termed TNF receptor associated factor 2 (TRAF2), that is recruited to the TNFSF receptors following engagement of their cognate ligands. Importantly, TRAF2 engages downstream signal transduction pathways that have been implicated in the development of a dilated cardiac phenotype, including mitogen activated protein kinases, NF-κB and c-jun N-terminal kinases (JNK).8 Taken together, these observations suggested the interesting possibility that TRAF2 may coordinate the signaling events that contribute to the changes in LV structure and function that occur during inflammation-induced cardiomyopathy. To address this question, we generated lines of transgenic mice with cardiac-restricted overexpression of TRAF2. Here we show for the first time that cardiac-restricted overexpression of TRAF2 is sufficient to mediate adverse cardiac remodeling in the heart, and thus phenocopies many aspects of the deleterious effects of TNF superfamily ligand signaling.
Methods
Transgenic Mice
We generated founder lines of transgenic mice with cardiac-restricted expression of murine tumor necrosis factor receptor associated factor 2 (TRAF2), using the alpha-myosin heavy chain (MHC) promoter (a gift from Jeff Robbins) to target TRAF2 to the cardiac myocyte, as described.9 Mice expressing the highest copy number of TRAF2 (referred to here as MHC-TRAF2HC) were selected for further study. The MHC-TRAF2HC lines of mice were generated and maintained on an FVB background. Age matched littermate mice (LM) that lack the transgene were used as controls in these experiments.
All experiments were approved by the Institutional Animal Care and Use Committees at the Baylor College of Medicine and Washington University School of Medicine and were conducted in accordance with the guidelines of the Baylor College of Medicine and Washington University School of Medicine, Animal Care and Research Advisory Committee and the rules governing animal use, as published by the NIH.
Characterization of MHC-TRAF2HC Mice
MHC-TRAF2HC and LM control mice were evaluated at 4, 8 and 12 weeks age using standard morphometric and histological analyses.10 Cardiac hypertrophy was evaluated by determining the heart-weight to body-weight ratio, as described.11 Histologic assessment of cardiac myocyte dimensions was performed using lectin staining. Briefly, myocardial samples were deparaffinized, hydrated, and incubated with Triticum vulgaris Lectin-TRITC conjugate.11 Nuclei were stained with DAPI (Vector Labs, Burlingame, CA,USA). Myocardial sections (endocardial, mid-myocardial, and epicardial) were examined under 400x magnification (Nikon Eclipse E800, Nikon Inc.), and the outline of cardiac myocytes was traced using Metavue software (v6.1, Universal Imaging Corp.). We measured the length and diameter (at the level of the nucleus) of cardiac myocytes that had clearly identified intercalated disks. A minimum of 100 myocytes was examined at the endocardial, mid-myocardial, and epicardial levels for each treatment group, and the results averaged. Myocardial ultrastructure was examined in the hearts of 12 week LM and MHC-TRAF2HC mouse hearts by transmission electron microscopy, as described.1
LV Structure and Function
LV structure and function were assessed using 2D directed M-mode echocardiography as described previously (see Supplemental Material for details).12
Extracellular Matrix
Deparaffinized sections of perfusion fixed hearts from 4, 8 and 12 week old MHC-TRAF2HC and littermate control mice were stained using the picrosirius red technique, as described (see Supplemental Material for details).1 MMP activity from 4, 8 and 12 week old MHC-TRAF2HC and LM control mouse hearts were obtained using gelatin zymography, as previously described (see Supplemental Material for details).13 The intensity of the zymographic bands was quantified by image analysis software (Image-J, NIH). Levels of TIMP-1 were measured in myocardial extracts from MHC-TRAF2HC mice and littermate controls at 4, 8 and 12 weeks of by ELISA (Amersham RPN 2611), according to manufacturer’s recommendations.1
Cell Death
Cardiac myocyte apoptosis was assessed in the hearts of 4, 8 and 12 week LM and MHC-TRAF2HC mice by TUNEL, using a commercially available kit (In-Situ Cell-Death Detection Kit; Roche Applied Science, Mannheim, Germany), according to the manufacturer’s suggestions. Cardiac myocytes were distinguished from non-myocyte cell types within the myocardium, as described.14 Sections were counterstained with the nucleic acid binding dye, 4′,6′-diamidino-2-phenylindole hydrochloride ([DAPI]; Vector Labs) to visualize the total number of myocyte nuclei in each myocardial section. The number of TUNEL positive nuclei per high-power field (400 x) was determined in 8 randomly selected fields by an investigator blinded to the experimental group being studied. We also examined for the presence or absence of myocyte cell necrosis at 4 weeks by Evans Blue dye (EBD) staining. Briefly, mice were injected intraperitoneally with 1% EBD (1% w/v [Sigma #E-2129, St. Louis, MO]) in PBS (filter sterilized) and sacrificed after 18 hours.15 Hearts were excised, cannulated and rinsed with 10ml PBS (1ml/min) followed by 10ml Z-fix fixative. Hearts were then paraffin-embedded and sectioned. Fluorescence microscopy (200x) was then performed using a filter set with an excitation of 510 – 560 nm and an emission of 590 nm.
NF-κB activation and JNK activity
Activation of NF-κB in nuclear extracts obtained from 4 –12 MHC-TRAF2HC and LM control mice was determined by electrophoretic mobility-shift assays (EMSAs), using an NF-κB oligonucleotide consensus sequence (5′-AGT TGA GGG GAC TTT CCC AGG C-3′ [Santa Cruz Biotechnology, Santa Cruz, CA]), as described.9 The specificity of binding was determined by competition with a 20X molar excess of the respective unlabeled oligonucleotide. NF-κB activation was also assessed at 12 weeks in nuclear extracts obtained from 12 week MHC-TRAF2HC, MHC-TRAF2LC, and LM control hearts using a NF-κB factor family (p65 [RelA], p50, p52, RelB) ELISA (Active Motif, Carlsbad, CA), which was performed exactly according to manufacturer’s instructions. Data represent the average of duplicate samples from each heart. JNK activity was determined in littermate control and TRAF2 hearts at 4, 8 and 12 weeks of age, using the SAPK/JNK Assay kit (Cat # 9810 from Cell Signaling Technology, Inc., Danvers, MA), according to the manufacturer’s instructions.
Gene Expression Profiling in MHC-TRAF2HC Hearts
To identify potential molecular pathways responsible for the phenotype of the MHC-TRAF2HC mice, gene expression profiling was performed. Briefly, Total RNA was extracted from the 4 LM controls hearts and 4 MHC-TRAF2HC hearts using TRIzol reagent (Invitrogen, Carlsbad, CA) per the manufacturer’s instructions. RNA was further processes and hybridized to a Mouse Ref-8 Illumina BeadChip by the Genome Technology Access Center (GTAC) at Washington University School of Medicine and scanned with the BeadStation system from Illumina, Inc. (San Diego, CA). Quality standards for hybridization, labeling, staining, background signal, and basal level of housekeeping gene expression for each chip were verified. After scanning the probe array, the resulting image was analyzed using the GenomeStudio software (Illumina, Inc., San Diego, CA). The background was subtracted, and log transformation and quantile normalization were performed. Differentially expressed genes between MHC-TRAF2HC and LM control mice were determined using ANOVA testing with contrasts using Partek GS (Partek, St. Louis, MO), using an unadjusted p value < 0.05 and a fold change of 1.2 or greater. Changes in gene expression were analyzed by the SAM (Statistical Analysis of Microarray) program and plotted.16 Expected differentially expressed genes are reported on the x axis, whereas observed genes that were differentially expressed are displayed in the y axis. A false discovery rate (FDR) less than 5% was used for the SAM plots. Functional analysis and pathway analysis was performed using Database for Annotation Visualization and Integrated Discovery (DAVID).17 Lists of genes that were significantly different in the MHC-TRAF2HC mouse hearts were analyzed by KEGG (Kyoto Encyclopedia of Genes and Genomes) functional pathway analysis. Identification of potential κB sites within significantly dysregulated genes was performed using UCSC_TFBS option in the DAVID functional annotation tool.
Statistical Analysis
Data are expressed as a mean ± SEM. Two-way repeated analysis of variance (ANOVA) was used to test for differences between MHC-TRAF2HC mice and littermate controls at 4, 8 and 12 weeks of age. Post-hoc ANOVA testing was performed between groups at individual time points, where appropriate, using the Tukey test. Differences in LV ventricular dimension, +dP/dt and -+dP/dt, and necropsy data between the MHC-TRAF2HC mice and their LM controls were performed using a non-paired Student’s t-test. All data analyses were performed using commercially available statistical software (STATVIEW version 5.0, SAS institute Inc., Cary, North Carolina). Significant differences were said to exist at p < 0.05.
Results
Characterization of MHC-TRAF2HC Mice
The generation and characterization of the transgenic founder lines with targeted overexpression of murine TRAF2 is reported in the Supplemental Material. Founder lines that expressed ≥ 10 copies of the transgene developed a dilated phenotype by 12 weeks of age that was similar to the phenotype observed in lines of mice with targeted overexpression of TNF.1,2 For the studies reported herein we selected mice that expressed 24 copies of the TRAF2 transgene, referred to herein as MHC-TRAF2HC mice.
Morphology and ultrastructure
Figure 1 depicts the characterization of the MHC-TRAF2HC mouse hearts in comparison to LM controls at 12 weeks of age. As shown, the MHC-TRAF2HC mice develop a dilated cardiac phenotype (Figure 1A) characterized by an increased heart-weight-to-body ratio (Figure 1C). The increased heart-weight-to-body ratio was secondary to a significant increase in heart weight (mg) in the MHC-TRAF2HC mice at 12 weeks(130.5±5.5 vs. 112.9±5.4, p < 0.05), insofar as the body weight (gm) was not different in the MHC-TRAF2HC and LM control mice, respectively (25.5±0.8 vs. 24.1±0.8; p > 0.05), at 12 weeks. Light microscopic evaluation of the 12 week hearts from MHC-TRAF2HC mice revealed that the histological appearance was similar to LM controls. Notably, there was no discernible inflammatory infiltrate (Figure 1B) by hematoxylin and eosin staining. Transmission electron microscopic evaluation of the 12 week LM hearts (Figure 1D and 1E) revealed a characteristic linear array of sarcomeres and myofibrils. In contrast, the myofibrils in the 12 week old MHC-TRAFHC mice were less organized, with loss of sarcomere registration, effacement of the Z-line, loss of the M-line, and accumulation of protein aggregates, consistent with the ultrastructural changes previously reported in MHCsTNF mice.1,18
Figure 1.

Characterization of MHC-TRAF2HC transgenic mice. (A) Photographs of whole hearts, coronal and sagittal sections of both LM and MHC-TRAF2HC mouse hearts (12 weeks). (B) Representative hematoxylin-eosin stained cross sections at the level of the papillary muscles (400x). (C) Heart-weight-to-body-weight ratio (mg/g) of LM and MHC-TRAF2HC at 8 and 12 weeks (n= 6–8 mice/group/time point) (*= p< 0.05 vs. LM at the indicated time point) (D–G) Representative transmission electron micrographs from 12 week MHC-TRAF2HC transgenic mice and LM at low (x7500, D,F) and high (x 60,000, E,G) magnification. Protein aggregates are enclosed by the circles.
LV Structure and Function
As shown in Figure 2A, there was progressive increase inLV end-diastolic dimension (LVEDD) in the MHC-TRAF2HC mice that was significantly (p < 0.05) different from LM controls by 8 weeks and 12 weeks. There was also a corresponding significant (p<0.05) increase in the r/h ratio in the MHC-TRAF2HC mouse hearts at 12 weeks of age when compared to LM, consistent with adverse cardiac remodeling (Figure 2B). Importantly, the increase in r/h ratio was not secondary to decreased wall thickness in the MHC-TRAF2HC mice, insofar as the LV wall thickness was similar in the LM and MHC-TRAF2HC mice at 12 weeks of age (1.19 + 0.08 vs. 1.16 + 0.07, respectively, p > 0.05). The increase in heart-weight-to-body ratio in the MHC-TRAF2HC mice at 12 weeks was accompanied by a ~ 24% increase in myocyte length (276.2 ± 3.5 μm vs. 222.5 ± 3.4 μm, p<0.001) and a corresponding ~ 11% decrease in myocyte width (50.9 ± 1.1 vs. 56.9 ± 1.8, p<0.01) compared to age matched LMs, consistent with cardiac myocyte elongation observed in eccentric remodeling of the heart. In order to determine whether targeted expression of TRAF2 resulted in LV dysfunction, as we have observed in lines of mice with targeted overexpression of TNF,19 we measured LV +dP/dt and −dP/dt ex vivo in a buffer perfused Langendorff apparatus. As shown in Figures 2C and 2D, there was a significant (p <0.01) decrease in peak +dP/dt and peak −dP/dt in 12 week MHC-TRAF2HC mice when compared to LM controls.
Figure 2.

LV structure and function. (A) 2-D echocardiographic assessment of left-ventricular end diastolic dimension (LVEDD) and (B) r/h (radius/wall thickness) ratio of 12 week LM and MHC-TRAF2HC at 4, 8 and 12 weeks (n= 6–8 mice/group/time point). (*= p< 0.05 vs. LM at the indicated time point) (C) LV +dP/dt and (D) LV −dP/dt assessed in 12 week in LM and MHC-TRAF2HC (n= 6 mice/group) (*= p< 0.05 vs. LM at 12 weeks).
Extracellular Matrix
To characterize the alterations in the extracellular matrix associated with LV remodeling in the MHC-TRAF2HC mice, we evaluated time-dependent changes in myocardial fibrillar collagen content using picrosirius red staining. Figure 3A shows representative histological sections of myocardium at 12 weeks, and Figure 3B summarizes the result of group data. As shown, LV myocardial fibrillar collagen content was similar (p>0.05) in MHC-TRAF2HC and LM control hearts at 4 weeks. However, as the mice aged there was a significant (2.1 fold) increase in collagen content in the MHC-TRAF2HC mice at 8 weeks (p=0.002) and a significant (4.4 fold) increase at 12 weeks (p=0.019) compared to LM controls, consistent with our prior observations in mice with targeted overexpression of TNF.1
Figure 3.

Myocardial fibrillar collagen content, MMP activity and TIMP levels. (A) Representative picrosirius red staining (400x) for myocardial fibrillar collagen content in LM and MHC-TRAF2HC mice. (B) Group data for % collagen volume for LM control and MHC-TRAF2HC mice at 4, 8 and 12 weeks (n=5 mice/group/time point). (*= p< 0.05 vs. LM at the indicated time point) (C) Group data for total MMP zymographic activity (arbitrary densitometric units) in LM control and MHC-TRAF2HC at 4, 8 and 12 weeks of age weeks (n=5–6 mice/group/time point). (*= p< 0.05 vs. LM at the indicated time point) (D) TIMP-1 levels in LM control and MHC-TRAF2HC control mice at 4, 8 and 12 weeks of age. (n= 6–8 mice/group/time point). (*= p< 0.05 vs. LM at the indicated time point)
To determine whether the increased fibrillar collagen content in the MHC-TRAF2HC mice was secondary to changes in matrix metalloproteinases (MMP) activity and/or myocardial levels of tissue inhibitors of metalloproteinase (TIMP) activity, we examined MMP activity and TIMP levels. Figures 3C and 3D show two important findings. First, there was a ~ 40% increase in MMP zymographic activity in the MHC-TRAF2HC mice at 4 weeks of age compared to LM controls (p<0.05). However, at 8 and 12 weeks of age, the MMP activity in the MHC-TRAF2HC mice declined significantly and was not significantly different (p > 0.05) in MHC-TRAF2HC and LM hearts at 8 and 12 weeks. The decrease in MMP activity was accompanied by an increase in TIMP-1 levels, which were significantly greater (p < 0.05) in the MHC-TRAF2HC mice by 12 weeks of age. Taken together, the decrease in MMP activity and increase in TIMP levels seen at 12 weeks of age is consistent with the increase in fibrosis observed in the MHC-TRAF2HC mice at 12 weeks of age.
Cell Death
Previously, we have reported that cardiac remodeling in lines of transgenic mice with targeted overexpression of TNF develops progressive cardiac myocyte apoptosis that is secondary to activation of the intrinsic and extrinsic death pathways.10,20 Figure 4A illustrates representative fluorescent micrographs of TUNEL staining in MHC-TRAF2HC at 12 weeks, and Figure 4B summarizes the results of group data at 4, 8 and 12 weeks of age. As shown, the number of TUNEL positive nuclei was significantly increased (p < 0.05) in the MHC-TRAF2HC hearts when compared to LM controls, whereas the prevalence of TUNEL positive nuclei was similar (p > 0.05) in MHC-TRAF2HC and LM hearts at 8 and 12 weeks of age, suggesting that progressive cardiac myocyte apoptosis was unlikely to contribute to the progressive cardiac dilation observed in the MHC-TRAF2HC mice at 8 and 12 weeks of age (Figure 1D). There was no evidence of myocyte necrosis in the MHC-TRAF2LC or LM control hearts (see Supplemental Figure 3), consistent with our observations in the MHCsTNF mice.14
Figure 4.
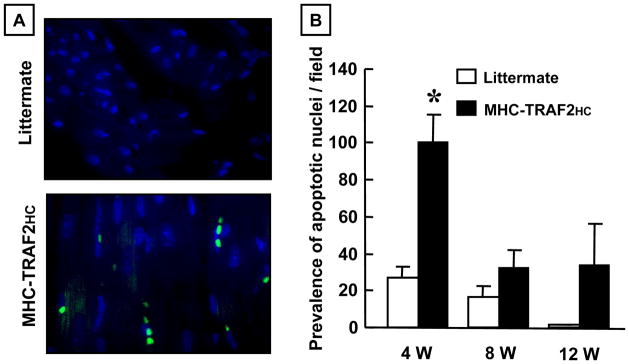
Prevalence of apoptosis. (A) Fluorescence microscopic (400x) images of TUNEL staining in LM control and MHC-TRAF2HC mice at 4 weeks of age. TUNEL stained apoptotic nuclei appear green. DAPI staining (blue) was used to determine the total nuclei per high power field. (B) Group data for prevalence of myocyte apoptosis in LM and MHC-TRAF2HC mice at 4, 8 and 12 weeks (n=5 mice/group/time point). (* p < 0.05 vs. LM at 4 weeks)
NF-κB activation and JNK activity
Previous studies suggest that the effects of TRAF2 in non-myocytes are mediated by activation of NF-κB and/or JNK.21 To determine whether these pathways were activated in the MHC-TRAF2HC transgenic mouse hearts, we assessed NF-κB activation by EMSA and ELISA, as well and JNK activity. Figure 5A shows a representative EMSA at 12 weeks of age, whereas Figure 5B summarizes the results of group data at 4 – 12 weeks. The salient finding shown by Figure 5B is that NF-κB activation was significantly increased at 4, 8 and 12 weeks in the MHC-TRAF2HC mice compared to LM control mice. The specificity of the DNA-protein interaction was determined by cold-chase experiments, which showed that the labeled NF-κB-DNA complexes were disrupted by a 20- fold excess of unlabeled oligonucleotide. As shown in Supplemental Figure 3, there was a significant increase in p50, RelB and p52 subunits in the MHC-TRAF2HC hearts when compared to LM controls. Moreover, there was a significant increase in p50, RelB and p52 subunits in the MHC-TRAF2HC hearts when compared to MHC-TRAF2LC hearts, consistent with a gene dosage effect of TRAF2 on activation of canonical and non-canonical signaling in the MHC-TRAF2HC mice. The important finding shown by Figure 5C is that although JNK activity was increased in the hearts of the MHC-TRAF2HC mice at 4 weeks compared to LM, there was no significant difference in JNK activity at 8 and 12 weeks, consistent with our findings in the MHC-TRAF2LC mouse hearts.9
Figure 5.

NF-κB activation and JNK activity. (A) Representative electromobility shift assay of NF-κB binding in nuclear extracts in 12 week littermate control (LM) and MHC-TRAF2HC (TG) mouse hearts. To determine the specificity of DNA-protein binding, nuclear extracts from MHC-TRAF2HC hearts were treated with a 20x excess of unlabeled oligonucleotide, and by supershift assays using polyclonal antibody directed against the p50 component of NF-κB (B) Densitometric quantification of NF-κB activity in 4, 8 and 12 week old LM and MHC-TRAF2HC mice. (n=3–5 mice per group per time point) (* p<0.05 vs. LM controls). (*= p< 0.05 vs. LM at 4,8 and 12 weeks) (C) Densitometric quantification of phospho-JNK activity using an in gel kinase assay in LM and MHC-TRAF2HC mice at 4, 8 and 12 weeks of age. (n=4 mice per group per time point) (* p < 0.05 vs. LM controls at 4 weeks).
Transcriptional Profiling in MHC-TRAF2HC Mice
To determine the mechanisms responsible for the dilated cardiac phenotype in the MHC-TRAF2HC mouse hearts we performed transcriptional profiling at 12 weeks of age. As shown by the SAM plot in Supplemental Figure 4A, 1136 genes were significantly upregulated and 823 genes were significantly downregulated in the 12 week MHC-TRAF2HC mouse hearts compared to LM controls. KEGG functional analysis (Supplemental Figure 4B) identified significant changes in gene expression in pathways implicated in both hypertrophic (p < 0.005) and dilated cardiomyopathy (p < 0.03) in the MHC-TRAF2HC compared to LM control hearts. Figure 6 displays a diagram of the cardiac hypertrophy/dilated cardiomyopathy related cardiac myocyte genes that were either upregulated or downregulated in the MHC-TRAF2HC, hearts compared to LM controls. Notably, genes that were involved with altered cytoskeletal linkage (α7 integrin subunit, cytosolic actin, desmin, α- and γ-sarcoglycan), decreased muscle contraction (α- and β-myosin heavy chain, troponin I), impaired excitation contraction coupling (β-1,4 subunits of the dihyropyridine receptor, ryanodine receptor, and SERCA2A) and increased myocardial fibrosis (TGF-β1) were altered in the transgenic mouse hearts (see Supplemental Table 2). Of the 37 cardiac hypertrophy/dilated cardiomyopathy pathway genes identified in the KEGG analysis, 35 genes (94.6%) had predicted κB sites when analyzed by the UCSC_TFBS option in DAVID, consistent with the significant increase in NF-κB activity observed in the MHC-TRAF2HC hearts.
Figure 6.

Transcriptional profiling. KEGG functional analysis identified significant changes in gene expression in pathways implicated in both hypertrophic and dilated cardiomyopathy in the MHC-TRAF2HC compared to LM control hearts. The diagram illustrates the specific changes in cardiac hypertrophy/dilated cardiomyopathy-related genes in relation to their anatomic location within the cardiac myocyte in MHC-TRAF2HC hearts vs. LM controls at 12 weeks. Identification of potential κB sites within significantly dysregulated genes was performed using UCSC_TFBS option in the DAVID functional annotation tool. (Key: red triangles denote increased gene expression; green triangles denote decreased gene expression; * denotes genes containing predicted κB sites in the promoter)
Discussion
The results of this study, in which we generated and characterized lines of transgenic mice with cardiac restricted overexpression of TRAF2 (MHC-TRAF2HC), demonstrate that TRAF2-mediated signaling leads to a dilated cardiac phenotype that overlaps the phenotype observed in transgenic mice overexpressing TNF superfamily ligands.1–3,5 The following lines of evidence support this statement. First, MHC-TRAF2HC transgenic mice develop progressive left ventricular dilation (Figure 2A), cardiac hypertrophy (Figure 1C), and adverse cardiac remodeling (increased r/h ratio [Figure 2B]). Second, the 12 week MHC-TRAF2HC mouse hearts had significantly decreased LV +dP/dt and −dP/dt when compared to LM controls (Figure 2C and 2D). Analysis of myocardial ultrastructure in 12 week old MHC-TRAF2HC disclosed sarcomere disarray, with effacement of the Z-line and loss of the M-line, thus providing a potential ultrastructural explanation for the observed decline in LV systolic and diastolic function (Figure 1F and 1G). Third, LV dilation in the MHC-TRAF2HC was accompanied by increased MMP activation at 4 weeks, followed by increased TIMP expression and increased myocardial fibrosis at 8 and 12 weeks, consistent with prior observations in the MHCsTNF transgenic mice(Figure 3A, 3B).22 Fourth, although we did observe increased myocyte apoptosis in the MHC-TRAF2HC mice at 4 weeks (Figure 4B), the prevalence of cardiac myocyte apoptosis did not increase progressively over time, as observed in the MHCsTNF transgenic mice.10,14 The reasons for the differences in the prevalence of myocyte apoptosis in the two models are not clear, but may be related to the complexity of TRAF2-mediated signaling, which can transmit pro-death or pro-life signals depending on the context of TRAF2 activation.23 Finally, we observed increased NF-κB activation from 4–12 weeks (Figure 5B) and increased JNK activity at 4 weeks of age (Figure 5C) in the MHC-TRAF2HC mice, consistent with the known signaling pathways downstream from TRAF2.21
Although this study was not designed to delineate the specific signaling pathways that are responsible for heart failure phenotype in the MHC-TRAF2HC mice, our results suggests an important role for NF-κB signaling. As shown in Figure 6, the transcriptional profiling studies show that TRAF2 signaling leads to alterations in cardiac myocyte gene expression that have been associated with the development of heart failure, including alterations in the cytoskeleton genes, activation of “fetal” gene programs, changes in excitation contraction coupling, as well as alterations in genes that that regulate remodeling of the extracellular matrix. The observation that NF-κB activation persisted from 4–12 weeks and occurred pari passu with the development and progression of cardiac remodeling, coupled with the observation that ~ 95% of the genes indentified in the KEGG cardiac hypertrophy/dilated cardiomyopathy functional pathway analysis (Figure 6 and Supplemental Table 2) contain predicted κB binding sites, suggests an important mechanistic role for TRAF2-mediated activation of NF-κB. Moreover, this point of view is consistent with a prior report which demonstrated that backcrossing TNF transgenic mice (TNF 1.6) with a transgenic mice expressing a cardiac-restricted dominant negative IκB transgene (“3M”) resulted in improved fractional shortening, decreased cardiac hypertrophy, and improved survival.24 Data from the present study do not support a critical role for TRAF2-mediated JNK activation in the development of a dilated cardiac phenotype, insofar as JNK activation was not significant beyond 4 weeks of age in the MHC-TRAF2HC mice. The mechanism for the decrease in JNK signaling in the MHC-TRAF2HC from 8–12 weeks is not known, but is consistent with the known effects of NF-κB mediated dampening of JNK activation through GADD45β.25
TRAF Mediated Signaling in the Heart
TNF and its superfamily of ligands and receptors represent a double-edged sword for the cardiovascular system. That is, members of the TNF superfamily play an important role in mediating homeostatic response in the heart, as well as deleterious effects when activated either inappropriately and/or in a sustained manner.26 Relevant to this discussion, many of the TNF receptor superfamily members (e.g. type 1 TNF receptor [TNFR1, TNFRSF1A] and FAS) contain death domains that initiate signal transduction pathways that lead to caspase activation and apoptotic cell death, consistent with the known deleterious effects of TNFSF/TNFRSF signaling. Recently, a family of cytoplasmic proteins has been identified that is capable of both negatively and positively regulating apoptotic pathways, as well as inducing the expression of genes that promote cell survival. Members of this family of signal transduction molecules were first described because of their ability to bind to the type 2 TNF receptor (TNFR2, TNFRSF1B) and, therefore, were given the name TNF receptor-associated factors (TRAFs). Subsequent studies have demonstrated that TRAFs serve as adapter proteins for a wide variety of innate immune cell surface receptors, and play important roles in regulating not only apoptosis, but also in mediating stress responses. TRAF proteins are cytoplasmic adapter proteins that can interact directly with the intracellular domains of cell surface receptors (e.g.TNFR2), or can be recruited indirectly though adaptor proteins that bind directly to the cytoplasmic tail of the TNFRSF member (e.g. TRADD binding TNFR1). Recruitment of TRAF2 to the cytoplasmic domains of receptors leads to the assembly of signaling complexes that activate mitogen activated kinase kinase kinases that converge on JNK and NF-κB signaling.21 To date, seven mammalian TRAFs (TRAF1-7) have been identified, of which TRAF 2, 3 and 6 are known to be expressed in the heart.21 With the exception of TRAF2, which is capable of upregulating cytoprotective pathways, and is cardioprotective when expressed at low levels,9 virtually nothing is known with regard to the role of TRAFs in the mammalian heart. Here we show that overexpression of TRAF2 in the heart phenocopies the dilated cardiomyopathic phenotype observed in TNF and sTWEAK transgenic mice. 1–3,5 While the precise reasons for the beneficial and deleterious cardiac phenotypes in mice that express, respectively, low and high levels of TRAF2 is not known, our data suggest that these differences are related to a “gene dosage” effect, insofar as there was a significantly greater degree of activation of canonical and non-canonical NF-κB subunits in the MHC-TRAF2HC mouse hearts when compared to MHC-TRAF2LC mouse hearts (Supplemental Figure 3). Nonetheless, we cannot exclude the formal possibility that the effects observed in the MHC-TRAF2HC mice were non-specific, and were secondary to high levels of expression of a the transgene, as has been reported for inert proteins that have been overexpressed in the heart.
Conclusions
This study shows for the first time that TRAF2- mediated signaling is sufficient to confer a heart failure phenotype in the adult mammalian heart. Given that TRAF2 associates directly or indirectly with the majority of TNF superfamily receptor members expressed in the heart (TNFR1, TNFR2, RANK and TWEAKR), and that the respective cognate ligands for these receptors, including TNF, RANKL, and TWEAK provoke a heart failure phenotype,6 the results of this study raise the interesting possibility that TRAF2 may serve as a nodal convergence point that orchestrates inflammatory responses in the heart following cardiac injury. Given that TRAF2 mediated signaling can be targeted through activation of GPR120 with omega-3 fatty acids,27 which have been shown to be beneficial in heart failure,28 or possibly through disruption of E3 ligases which are essential for TRAF2 signaling,29 the current observations may have therapeutic importance as well.
Supplementary Material
Sustained inflammatory signaling in the heart leads to the development of a cardiomyopathy that is characterized by left ventricular (LV) dilation, LV dysfunction and myocardial fibrosis. Although a number or pro-inflammatory cytokines have been implicated in this process, the cytokine that has been characterized most extensively thus far is tumor necrosis factor (TNF). All TNF superfamily ligands that provoke a dilated cardiac phenotype signal through a common scaffolding protein termed TNF receptor associated factor 2 (TRAF2); however, virtually nothing is known with regard to TRAF2 signaling in the adult mammalian heart. We generated transgenic mice with cardiac restricted overexpression of TRAF2 and characterized the phenotype of mice with high levels of TRAF2 (MHC-TRAF2HC) expression. MHC-TRAF2HC transgenic mice developed a time-dependent increase in cardiac hypertrophy, LV dilation and adverse LV remodeling with myocardial fibrosis, and a significant decrease in LV +dP/dt and −dP/dt when compared to littermate controls. These changes were accompanied by a significant increase in NF-βB activation at 4 – 12 weeks and JNK activation at 4 weeks in the MHCs TRAF2HC mice. Transciptional profiling revealed that > 95% of the hypertrophic/dilated cardiomyopathy-related genes that were significantly upregulated genes in the MHC-TRAF2HC hearts contained βB elements in their promoters. Viewed together, these results suggest that TRAF2 is sufficient to mediate adverse cardiac remodeling in the heart, and raise the interesting possibility that TRAF2 may serve as a nodal convergence point that orchestrates inflammatory responses in the heart following cardiac injury.
Acknowledgments
The authors would like to acknowledge the technical assistance of Dorellyn Lee and Lora Staloch.
Sources of Funding
This research was supported by research funds from the N.I.H. (RO1 HL89543 R01 HL58081, RO1 111094, T32 HL 007706, T32 HL007081).
Footnotes
Disclosures
None.
References
- 1.Sivasubramanian N, Coker ML, Kurrelmeyer K, DeMayo F, Spinale FG, Mann DL. Left ventricular remodeling in transgenic mice with cardiac restricted overexpression of tumor necrosis factor. Circulation. 2001;2001:826–831. doi: 10.1161/hc3401.093154. [DOI] [PubMed] [Google Scholar]
- 2.Kubota T, McTiernan CF, Frye CS, Slawson SE, Koretsky AP, Demetris AJ, Feldman AM. Dilated cardiomyopathy in transgenic mice with cardiac specific overexpression of tumor necrosis factor-alpha. Circ Res. 1997;81:627–635. doi: 10.1161/01.res.81.4.627. [DOI] [PubMed] [Google Scholar]
- 3.Bryant D, Becker L, Richardson J, Shelton J, Franco F, Pechock RM, Thompson M, Giroir BP. Cardiac Failure in transgenic mice with myocardial expression of tumor necrosis factor-a (TNF) Circulation. 1998;97:1375–1381. doi: 10.1161/01.cir.97.14.1375. [DOI] [PubMed] [Google Scholar]
- 4.Ueland T, Aukrust P, Damas JK, Gullestad L, Yndestad A. The tumor necrosis factor superfamily in heart failure. Future Cardiol. 2006;2:101–111. doi: 10.2217/14796678.2.1.101. [DOI] [PubMed] [Google Scholar]
- 5.Jain M, Jakubowski A, Cui L, Shi J, Su L, Bauer M, Guan J, Lim CC, Naito Y, Thompson JS, Sam F, Ambrose C, Parr M, Crowell T, Lincecum JM, Wang MZ, Hsu YM, Zheng TS, Michaelson JS, Liao R, Burkly LC. A novel role for tumor necrosis factor-like weak inducer of apoptosis (TWEAK) in the development of cardiac dysfunction and failure. Circulation. 2009;119:2058–2068. doi: 10.1161/CIRCULATIONAHA.108.837286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ueland T, Yndestad A, Oie E, Florholmen G, Halvorsen B, Froland SS, Simonsen S, Christensen G, Gullestad L, Aukrust P. Dysregulated osteoprotegerin/RANK ligand/RANK axis in clinical and experimental heart failure. Circulation. 2005;111:2461–2468. doi: 10.1161/01.CIR.0000165119.62099.14. [DOI] [PubMed] [Google Scholar]
- 7.Nelson DP, Setser E, Hall DG, Schwartz SM, Hewitt T, Klevitsky R, Osinska H, Bellgrau D, Duke RC, Robbins J. Proinflammatory consequences of transgenic fas ligand expression in the heart. J Clin Invest. 2000;105:1199–1208. doi: 10.1172/JCI8212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gupta S, Sen S. Role of the NF-kappaB signaling cascade and NF-kappaB-targeted genes in failing human hearts. J Mol Med (Berl) 2005;83:993–1004. doi: 10.1007/s00109-005-0691-z. [DOI] [PubMed] [Google Scholar]
- 9.Burchfield JS, Dong JW, Sakata Y, Gao F, Tzeng HP, Topkara VK, Entman ML, Sivasubramanian N, Mann DL. The Cytoprotective Effects of Tumor Necrosis Factor are Conveyed Through Tumor Necrosis Factor Receptor Associated Factor 2 in the Heart. Circ Heart Fail. 2010;3:157–164. doi: 10.1161/CIRCHEARTFAILURE.109.899732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Haudek SB, Taffet GE, Schneider MD, Mann DL. TNF provokes cardiomyocyte apoptosis and cardiac remodeling through activation of multiple cell death pathways. J Clin Invest. 2007;117:2692–2701. doi: 10.1172/JCI29134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sakata Y, Chancey AL, Divakaran VG, Sekiguchi K, Sivasubramanian N, Mann DL. Transforming growth factor-beta receptor antagonism attenuates myocardial fibrosis in mice with cardiac-restricted overexpression of tumor necrosis factor. Basic Res Cardiol. 2007;103:60–68. doi: 10.1007/s00395-007-0689-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nemoto S, Vallejo JG, Knuefermann P, Misra A, DeFreitas G, Carabello BA, Mann DL. Escheria coli lipolysaccharide-induced left ventricular dysfunction: the role of toll-like receptor-4 in the adult mammalian heart. Am J Physiol. 2002;282:H2316–H2323. doi: 10.1152/ajpheart.00763.2001. [DOI] [PubMed] [Google Scholar]
- 13.Diwan A, Dibbs Z, Nemoto S, DeFreitas G, Carabello BA, Sivasubramanian N, Wilson EM, Spinale FG, Mann DL. Targeted overexpression of noncleavable and secreted forms of tumor necrosis factor provokes disparate cardiac phenotypes. Circulation. 2004;109:262–268. doi: 10.1161/01.CIR.0000109642.27985.FA. [DOI] [PubMed] [Google Scholar]
- 14.Engel D, Peshock R, Armstong RC, Sivasubramanian N, Mann DL. Cardiac myocyte apoptosis provokes adverse cardiac remodeling in transgenic mice with targeted TNF overexpression. Am J Physiol Heart Circ Physiol. 2004;287:H1303–H1311. doi: 10.1152/ajpheart.00053.2004. [DOI] [PubMed] [Google Scholar]
- 15.Hamer PW, McGeachie JM, Davies MJ, Grounds MD. Evans Blue Dye as an in vivo marker of myofibre damage: optimising parameters for detecting initial myofibre membrane permeability. J Anat. 2002;200:69–79. doi: 10.1046/j.0021-8782.2001.00008.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Esteller M. Non-coding RNAs in human disease. Nat Rev Genet. 2011;12:861–874. doi: 10.1038/nrg3074. [DOI] [PubMed] [Google Scholar]
- 17.Huang dW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 18.Panagopoulou P, Davos CH, Milner DJ, Varela E, Cameron J, Mann DL, Capetanaki Y. Desmin mediates TNF-alpha-induced aggregate formation and intercalated disk reorganization in heart failure. J Cell Biol. 2008;181:761–775. doi: 10.1083/jcb.200710049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mann DL. Cytokines as mediators of disease progression in the failing heart. In: Hosenpud JD, Greenberg BH, editors. Congestive Heart Failure. Philadelphia: Lippincott Williams & Wilkins; 2006. pp. 205–225. [Google Scholar]
- 20.Broqvist M, Dahlstrom U, Karlberg BE, Matklund T. Neuroendocrine response in acute heart failure and the influence of treatment. Eur Heart J. 1989;10:1075–1083. doi: 10.1093/oxfordjournals.eurheartj.a059429. [DOI] [PubMed] [Google Scholar]
- 21.Arch RH, Gedrich RW, Thompson CB. Tumor necrosis factor receptor-associated factors (TRAFs)--a family of adapter proteins that regulates life and death. Genes Dev. 1998;12:2821–2830. doi: 10.1101/gad.12.18.2821. [DOI] [PubMed] [Google Scholar]
- 22.Hodgson DM, Zingman LV, Kane GC, Perez-Terzic C, Bienengraeber M, Ozcan C, Gumina RJ, Pucar D, O’Coclain F, Mann DL, Alekseev AE, Terzic A. Cellular remodeling in heart failure disrupts K(ATP) channel-dependent stress tolerance. EMBO J. 2003;22:1732–1742. doi: 10.1093/emboj/cdg192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xia ZP, Chen ZJ. TRAF2: a double-edged sword? Sci STKE. 2005;2005:e7. doi: 10.1126/stke.2722005pe7. [DOI] [PubMed] [Google Scholar]
- 24.Higuchi Y, Chan TO, Brown MA, Zhang J, DeGeorge BR, Jr, Funakoshi H, Gibson G, McTiernan CF, Kubota T, Jones WK, Feldman AM. Cardioprotection afforded by NF-kappaB ablation is associated with activation of Akt in mice overexpressing TNF-alpha. Am J Physiol Heart Circ Physiol. 2006;290:H590–H598. doi: 10.1152/ajpheart.00379.2005. [DOI] [PubMed] [Google Scholar]
- 25.De Smaele E, Zazzeroni F, Papa S, Nguyen DU, Jin R, Jones J, Cong R, Franzoso G. Induction of gadd45beta by NF-kappaB downregulates pro-apoptotic JNK signalling. Nature. 2001;414:308–313. doi: 10.1038/35104560. [DOI] [PubMed] [Google Scholar]
- 26.Mann DL. Inflammatory Mediators and the Failing Heart: Past, Present, and the Foreseeable Future. Circ Res. 2002;91:988–998. doi: 10.1161/01.res.0000043825.01705.1b. [DOI] [PubMed] [Google Scholar]
- 27.Oh DY, Talukdar S, Bae EJ, Imamura T, Morinaga H, Fan W, Li P, Lu WJ, Watkins SM, Olefsky JM. GPR120 is an omega-3 fatty acid receptor mediating potent anti-inflammatory and insulin-sensitizing effects. Cell. 2010;142:687–698. doi: 10.1016/j.cell.2010.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gissi-HF Investigators. Effect of n-3 polyunsaturated fatty acids in patients with chronic heart failure (the GISSI-HF trial): a randomised, double-blind, placebo-controlled trial. Lancet. 2008;372:1223–1230. doi: 10.1016/S0140-6736(08)61239-8. [DOI] [PubMed] [Google Scholar]
- 29.Jia L, Sun Y. SCF E3 ubiquitin ligases as anticancer targets. Curr Cancer Drug Targets. 2011;11:347–356. doi: 10.2174/156800911794519734. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.