

Abstract
Introduction
An NZB-derived genetic locus (Sle2c2) that suppresses autoantibody production in a mouse model of induced systemic lupus erythematosus contains a polymorphism in the gene encoding the G-CSF receptor. This study was designed to test the hypothesis that the Sle2c2 suppression is associated with an impaired G-CSF receptor function that can be overcome by exogenous G-CSF.
Methods
Leukocytes from B6.Sle2c2 and B6 congenic mice, which carry a different allele of the G-CSF receptor, were compared for their responses to G-CSF. Autoantibody production was induced with the chronic graft-versus-host-disease (cGVHD) model by adoptive transfer of B6.bm12 splenocytes. Different treatment regimens varying the amount and frequency of G-CSF (Neulasta®) or carrier control were tested on cGVHD outcomes. Autoantibody production, immune cell activation, and reactive oxygen species (ROS) production were compared between the two strains with the various treatments. In addition, the effect of G-CSF treatment was examined on the production autoantibodies in the B6.Sle1.Sle2.Sle3 (B6.TC) spontaneous model of lupus.
Results
B6.Sle2c2 and B6 leukocytes responded differently to G-CSF. G-CSF binding by B6.Sle2c2 leukocytes was reduced as compared to B6, which was associated with a reduced expansion in response to in vivo G-CSF treatment. G-CSF in vivo treatment also failed to mobilize bone-marrow B6.Sle2c2 neutrophils as it did for B6 neutrophils. In contrast, the expression of G-CSF responsive genes indicated a higher G-CSF receptor signaling in B6.Sle2c2 cells. G-CSF treatment restored the ability of B6.Sle2c2 mice to produce autoantibodies in a dose-dependent manner upon cGVHD induction, which correlated with restored CD4+ T cells activation, as well as dendritic cell and granulocyte expansion. Steady-state ROS production was higher in B6.Sle2c2 than in B6 mice. cGVHD induction resulted in a larger increase in ROS production in B6 than in B6.Sle2c2 mice, and this difference was eliminated with G-CSF treatment. Finally, a low dose G-CSF treatment accelerated the production of anti-dsDNA IgG in young B6.TC mice.
Conclusion
The different in vivo and in vitro responses of B6.Sle2c2 leukocytes are consistent with the mutation in the G-CSFR having functional consequences. The elimination of Sle2c2 suppression of autoantibody production by exogenous G-CSF indicates that Sle2c2 corresponds to a loss of function of G-CSF receptor. This result was corroborated by the increased anti-dsDNA IgG production in G-CSF-treated B6.TC mice, which also carry the Sle2c2 locus. Overall, these results suggest that the G-CSF pathway regulates the production of autoantibodies in murine models of lupus.
Introduction
Systemic lupus erythematosus (SLE) is an autoimmune disease with a complex etiology in which the production of pathogenic autoantibodies (autoAbs) results in cellular and tissue damage. Aside from B cells, which produce these autoAbs, and CD4+ T cells, which provide B cell help for the generation of class-switched, affinity maturated autoAbs, essentially every other immune cell subset has been implicated in SLE pathogenesis. The strong genetic basis of SLE is sustained by a large number of polymorphisms that have been identified in recent years through association studies in large cohorts of patients and controls [1]. Mouse models of SLE have been used extensively to study both the cellular and genetic basis of SLE, and overall, the results obtained from these models have largely been validated in SLE patients. In particular, murine models have revealed a large number of SLE susceptibility genes, which are organized in the same three broad pathways: apoptosis and processing of apoptotic debris, toll-like receptor (TLR) signaling and type I IFN pathways, and lymphocyte activation in both SLE patients and SLE-prone mice [2,3]. The genetic analysis of the NZM2410 mouse model has also shown the existence of both SLE-resistance and suppressor genes. Consequently, the SLE-resistant strain C57BL/6 (B6) carries susceptibility genes that were revealed when combined with either other susceptibility genes provided by the NZM2410 lupus-prone genome, or when subjected to a strong immune stimulation [4,5].
The bm12- chronic graft vs host disease (cGVHD) model is a well-defined model of induced lupus in which B6.C-H2bm12 lymphocytes are transferred into H-2b B6 hosts. Within 3 weeks of transfer, mice develop lupus-like phenotypes including lymphocyte activation and anti-nuclear autoAbs, which are dependent on interactions between donor CD4+ T cells and host autoreactive B cells [6]. We have shown that B6.Sle2c2 mice, which are B6 mice carrying an NZM2410 (NZB)-derived genomic region on the telomeric potion of the Sle2 locus, are profoundly resistant to bm12-cGVHD induction as compared to their B6 congenic controls [5]. Using mixed bone-marrow (BM) chimeras and functional assays, we have shown that Sle2c2 suppression is mediated by BM-derived cells, but not by T cells, B cells, or dendritic cells (DCs). We mapped Sle2c2 resistance to a short genomic interval that contains Csf3r, the gene encoding for the granulocyte-colony stimulation factor (G-CSF) receptor (G-CSFR) [5]. The NZM2410 allele of Csf3r (Csf3rN as opposed to the B6 allele Csf3rB) carries a mutation in exon 10 (rs13477964) that results in a S378N substitution in its extracellular domain.
G-CSF is an essential factor in the recruitment and function of neutrophils [7]. G-CSFR expression regulates GVHD in mice [8]. Moreover, neutrophils have recently been implicated at multiple levels in the pathology of SLE or related systemic autoimmune diseases [9]. Neutrophils inflict direct damage to the vasculature [10], the kidneys [11] and the skin [12] of lupus patients. They also contribute to SLE pathogenesis through their direct production of type I IFN [13], or through the highly immunogenic neutrophil extracellular traps (NETs) amplifying the production of either type I IFN [14] or IL-17 [15], although these later findings have been refuted in the MRL/lpr lupus-prone mouse model [16]. G-CSF also controls the number of myeloid derived suppressor cells (MDSCs) [17], which are potent suppressors of alloreactive T cell responses in a GVHD model [17]. In MRL/lpr mice, the percentage of MDSCs increases as disease progresses [18], corroborating our findings of an expanded GR1lo MDSC population in B6 mice with established cGVHD [5].
These studies lead us to hypothesize that the N378S mutation in the Csf3r gene impairs G-CSFR function. This mutation is located in the solvent-exposed part of the receptor in a putative fibronectin III domain outside the solved structure of the protein [5]. It is therefore impossible at this point to predict its functional significance, although it has the potential to affect ligand binding. We hypothesized that the N378S mutation is responsible for the differential bm12-cGVHD response between B6 and B6.Sle2c2 mice, which we tested by comparing their response to exogenous G-CSF. We reasoned that if bm12-cGVHD resistance in B6.Sle2c2 mice was mediated by a gain of function in suppressive cells such as MDSCs, G-CSF treatment would induce bm12-cGVHD resistance in B6 mice. In contrast, if bm12-cGVHD resistance in B6.Sle2c2 mice was due to a loss of function or recruitment in inflammatory neutrophils, then G-CSF treatment would alleviate bm12-cGVHD resistance in B6.Sle2c2 mice, which would develop autoAbs and lymphocyte activation comparable to that of B6. Here we demonstrate a weaker binding of mouse G-CSF to B6.Sle2c2 leukocytes as compared to B6, as well as a defective expansion of B6.Sle2c2 myeloid cells and neutrophils in response to in-vivo G-CSF treatment. Furthermore, exogenous G-CSF restored bm12-cGVHD responses in B6.Sle2c2 mice. Finally, the B6.Sle1.Sle2.Sle3 congenic mice that display a full-blown spontaneous lupus as its NZM2410 parental strain carry the Sle2c2 locus [19]. It was therefore predicted that G-CSF treatment would accelerate their autoAb production, which we showed in this study with a low-dose treatment. Overall these results suggest that Sle2c2 prevents the induction of systemic autoimmunity by impairing the production of inflammatory neutrophils due to a decreased ability of the G-CSFR to bind its ligand.
Materials and methods
Mice
The B6.Sle2c2 mice carrying an NZM2410 (NZB)-derived interval located between D4Mit11 and D4Mit72 have previously been described [5]. B6 and B6.C-H2bm12/KhEg (bm12) were originally purchased from the Jackson Laboratory (Bar Harbor, ME, USA). Cohorts of mice within an experiment were sex- and age-matched, but the reported phenotypes were neither affected by sex or age. The NZM2410-derived triple congenic mice B6.Sle1.Sle2.Sle3 (B6.TC) have been previously described [19]. All animal protocols were approved by the Institutional Animal Care and Use Committee at the University of Florida.
cGVHD induction and G-CSF treatment
bm12-cGVHD was induced according to an established protocol [6]. Briefly, 50 to 80 × 106 bm12 splenocytes were injected intraperitoneally into 2 to 4 month-old B6 or B6.Sle2c2 mice. Serum was collected for autoAb detection by ELISA as previously described [6] at day (d) 0, 7, 14 and 21 after induction. Antinuclear antibody (ANA) stains were conducted on slides containing fixed Hep-2 cells (Inova Diagnotics, San Diego, CA, USA) with mouse sera diluted 1:40, and revealed with fluorescein isothiocyanate (FITC)-conjugated anti-mouse IgG (Southern Biotech, Birmingham, AL, USA) at 1:50 dilution. Staining intensity was calculated with morphometry software (Metamorph, Molecular Devices, Sunnyvale, CA, USA) for a standardized level of green fluorescence averaged on 10 to 20 randomly selected Hep-2 cells per sample. Samples with an average staining intensity > 1 were considered positive. At d21, mice were sacrificed and splenocytes assessed by flow cytometry.
bm12-cGVHD-induced mice were treated with subcutaneous injections of 1.2, 12.0, or 120.0 ug pegylated human G-CSF (Neulasta®, huG-CSF, Amgen, Thousand Oaks, CA, USA), all starting at d1 of induction. Some huG-CSF injections were repeated every 7 d resulting in 4 different treatment regimens: one injection of 120 ug (120 × 1), two or three injections of 12 ug (12 × 2 and 12 × 3), and three injections of 1.2 ug (1.2 × 3). Control mice were injected with 5% dextrose as vehicle control. Each experimental group contained at least five recipient mice per treatment per strain, and control groups were performed side by side for each treatment group. Groups of four un-manipulated mice per strain were injected with huG-CSF and blood was collected every 2 to 3 d to compare the dose-response effect of G-CSF of peripheral blood leukocytes (PBLs) between strains. Serum mouse (m) G-CSF was measured by ELISA with reagents from Peprotech (Rocky Hills, NJ, USA) in sera diluted 1:50. B6.TC and B6 female mice were treated with six weekly injections of 1 ug of Neulasta or 5% dextrose starting at 2 months of age and were sacrificed at week 7. Serum was collected every two weeks and anti-dsDNA IgG, anti-chromatin IgG and total IgG were measured as previously described in sera diluted at 1:100 and 1:5000, respectively [6].
Flow cytometry
Splenocytes or PBLs were blocked with anti-CD16/CD32 (2.4G2), then stained with pre-titrated amounts of the following FITC-, phycoerythrin (PE)-, allophycocyanin-, or biotin-conjugated Abs: CD4 (RM4-5), CD69 (H1.2F3), CD44 (IM7), CD62L (MEL-14) B220 (RA3-6B2), CD86 (GL1), CD80 (16-10A1), CD22.2 (Cy34.1), I-ab (AF6-120.1), CD11b (M1/70), CD11c (HL3), Ly6C/G (GR-1/RB6-8C5) or isotype controls, all from BD Biosciences (San Diego, CA, USA). Biotinylated Abs were revealed by streptavidin-PercP-Cy5a. p-signal transducer and activator of transcription 3 (Stat3) (Y705) staining was performed after fixation and permeabilization (BD Cytofix/Cytoperm kit). G-CSF binding was performed by incubating splenocytes for 1 h with 25 ng of mG-CSF (ProSpec, East Brunswick, NJ, USA) biotinylated with a Biotin XX Microscale protein labeling kit (Molecular Probes, Eugene, OR, USA), detected by flow cytometry with PE-streptavidin. As negative controls, cells were incubated only with PE-streptavidin. ROS production was detected with 1 μM dihydrorhodamine 123 (DHR, Molecular Probes). Cell staining was analyzed using a FACScalibur or LSRFortessa (Becton Dickinson, Franklin Lakes, NJ, USA). At least 30,000 (PBLs) and 50,000 (splenocytes) events were acquired per sample, and dead cells were excluded based on scatter characteristics.
Gene expression analysis
Splenocytes and BM cells from B6 and B6.Sle2c2 mice (three each) were cultured in complete medium at a concentration of 106 cells/ml with 0.0, 0.1, or 1.0 ug/ml of Neulasta for 8 h. RNA was extracted and cDNA was synthesized as previously described [20]. Expression of Itgam and Mpo relative to Gapdh was quantified by quantitative PCR measuring the incorporation of Sybergreen (Molecular Probes). The primer sequences were: Itgam 5'-CCC CAA TTA CGT AGC GAA TG-3'and 5'- TGC TGC GAA GAT CCT AGT TG -3'; Mpo 5'- TGC TCT CGA ACA AAG AGG GT-3' and 5'- CTT TGA CAG CCT GCA CGA-3'; Gapdh 5'- AGC TTG TCA TCA ACG GGA AG -3' and 5'- GTG GTT CAC ACC CAT CAC AA -3'. Results were calculated with the ΔΔ cycle threshold (Ct) method and normalized to the average values for untreated B6 samples.
Statistics
Statistical analyses were performed with GraphPad Prism5, using the two-tailed unpaired t-test or Bonferroni multiple comparison test when multiple groups were compared. Non-parametric statistics were performed when data were not normally distributed. Graphs show mean and standard error of the mean (SEM), and statistical significance is represented as *P < 0.05, **P < 0.01, and ***P < 0.001.
Results
B6.Sle2c2 leukocytes bind less G-CSF and expand less to exogenous G-CSF than B6
To determine whether the S378N mutation in the extracellular domain of the G-CSFR-affected ligand binding, we compared in vitro binding of mG-CSF between B6 and B6.Sle2c2 splenocytes. As expected, the highest binding was found on GR1hi CD11b+ neutrophils, whereas intermediate levels were found on GR1lo CD11b+ and B cells (Figure 1A). However, every B6.Sle2c2 cell subset showed a significantly lower binding of mG-CSF, either as percentage of G-CSF+ cells (Figure 1A), or as the mean fluorescence intensity (data not shown). This was not due to different endogenous levels of G-CSF, since the two strains presented similar serum G-CSF levels (B6, 1.10 ± 0.21 ng/ml; B6.Sle2c2, 1.44 ± 0.39 ng/ml, n = 20 per strain). To test whether the decreased G-CSF binding by B6.Sle2c2 leukocytes provides functional consequences, we compared the effect of in vivo huG-CSF treatment on the expansion of myeloid cells between the two strains (Figure 1B). Although human and mouse G-CSF are not identical, a large number of studies, including one by our group [21], have shown that huG-CSF is fully functional in mice. The pegylated form of G-CSF in Neulasta has an extended half-life and a time course analysis showed that the maximum response occurs 4 d after treatment (data not shown). At that time point, a dose-dependent expansion of CD11b+, GR1hi CD11b+ and GR1lo CD11b+ PBLs was observed in both strains but a greater amount of hu-G-CSF was required for B6.Sle2c2 cells to achieve the level of B6 cell expansion (Figure 1B).
Figure 1.
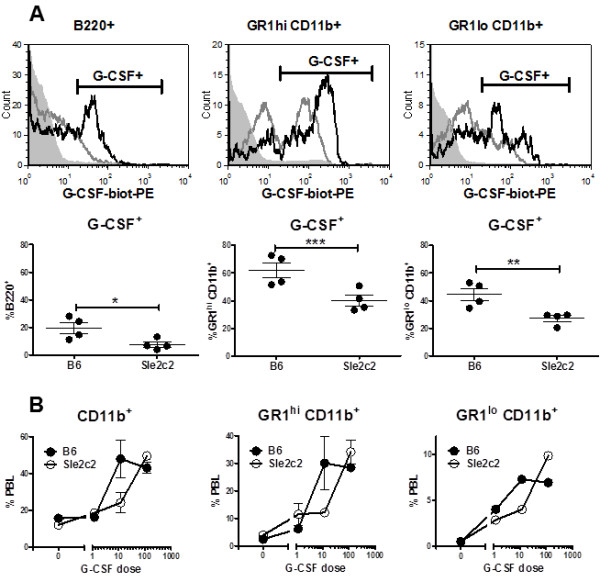
Differential response of B6.Sle2c2 and B6 leukocytes to granulocyte-colony stimulation factor (G-CSF). (A) Mouse (m)G-CSF binding by B6 and B6.Sle2c2 leukocytes measured with biotinylated G-CSF and steptavidin-PE (G-CSF-biot-PE). The top row shows representative overlays of the B6 (black) and Sle2c2 (gray) histograms, with the gray-filled histograms showing the controls without added mG-CSF for the indicated gated cell populations. (B) Total CD11b+, GR1hi CD11b+ and GR1lo CD11b+ peripheral blood leukocytes (PBLs) in untreated B6 and B6.Sle2c2 mice (n = 6 per strain), and 4 d after injections of 1.2, 12.0 or 120.0 ug human (hu)G-CSF (n = 2 per dose per strain). The graphs show mean and standard error of the mean with the significance value from the t-test (*P < 0.05; **P < 0.01; ***P < 0.001).
One of the functions of G-CSF is to mobilize neutrophils from BM to the periphery [7]. We therefore compared the numbers of neutrophils in the BM and the spleen of B6 and B6.Sle2c2 mice 4 d after treatment with G-CSF or dextrose control (Figure 2). As expected, the percentage of neutrophils decreased in the BM of G-CSF-treated B6 mice (Figure 2A and 2C) and increased in their spleens (Figure 2B and 2C). In contrast, the percentage of neutrophils in the BM of G-CSF-treated B6.Sle2c2 mice was higher than in the BM of dextrose-treated mice (Figure 2A and 2C), which corresponded to a lack of mobilization of the B6.Sle2c2 neutrophils to the spleen (Figure 2B and 2C). The percentages of neutrophils in the blood of these G-GCF-treated mice reflected this difference in mobilization, with an increased percentage of circulating B6 neutrophils but a decreased percentage of circulating B6.Sle2c2 neutrophils (Figure 2D).
Figure 2.

Differential neutrophil mobilization in response to human granulocyte-colony stimulation factor (huG-CSF) in B6 and B6.Sle2c2 mice. Percentage of Ly6G+ CD11b+ neutrophils present in the bone marrow (BM) (A) and spleen (B) in B6 and B6.Sle2c2 mice 4 d after an injection with 5% dextrose (Dex) or 1 ug hu-G-CSF (GCSF). The graphs show mean and standard error of the mean with the significance of the Bonferroni multiple comparison test performed on three mice per group. The experiment was repeated and the same results were obtained, although with different absolute values (*P < 0.05; **P < 0.01; ***P < 0.001).
To determine whether G-CSF signaling differed between B6.Sle2c2 and B6 leukocytes, we used three well-known targets, CD11b and myeloperoxidase (MPO) expression and STAT3 phosphorylation [22]. Ex vivo, CD11b expression on splenocytes was higher in B6.Sle2c2 than in B6 mice (Figure 3A). Treatment with 120 ug of hu-G-CSF significantly increased CD11b expression on CD11b+ PBLs in both strains, but it remained at a higher level on B6.Sle2c2 cells. This effect of exogenous G-CSF was abolished by d9 after injection. Similar results were obtained with the expression of CD16/32 (another G-CSF-responsive gene) on CD11b+ PBLs (data not shown). We next compared CD11b/Itgam and Mpo message expression in BM and spleen cells obtained from B6 and B6.Sle2c2 mice 4 d after treatment with 1 ug hu-G-CSF, then treated for 8 h with hu-G-CSF in vitro (Figure 3B). In both strains, we observed that hu-G-CSF decreased both Itgam and Mpo expression. The response pattern was however different between the two strains, with B6.Sle2c2 cells being more responsive than B6 at equal doses. In addition, we observed, as for CD11b protein expression after the in vivo treatment (Figure 3A), an overall higher expression of Itgam and Mpo in B6.Sle2c2 than in B6 cells. Ex vivo STAT3 phosphorylation was also significantly higher in splenocytes from untreated B6.Sle2c2 mice; either when considering the percentage of CD11b+, GR1hi CD11b+ and GR1lo CD11b+ cells expressing pSTAT3 (Figure 3C), or pSTAT3 mean fluorescence intensity (data not shown). Although factors other than G-CSF signaling may contribute to the differential CD11b expression and STAT3 phosphorylation, the two strains only differ by the Sle2c2 interval (< 15 Mb) [5]. While these results demonstrate a lower G-CSF binding and expansion to exogenous G-CSF by leukocytes expressing the Sle2c2 Cfs3r allele, evidence of greater G-CSFR signaling in leukocytes expressing the Sle2c2 Cfs3r allele was also observed. These apparently paradoxical results should be put in perspective that very little is known about G-CSFR signaling and G-CSFR-responsive genes in primary myeloid cells, as all the studies have been performed with myeloid cell lines (reviewed in [22]). Our results however clearly showed that B6 and B6.Sle2c2 myeloid cells respond differently to G-GSF both in vivo and in vitro by multiple processes, including myeloid/granulocyte expansion, mobilization, and G-CSFR-responsive gene expression.
Figure 3.
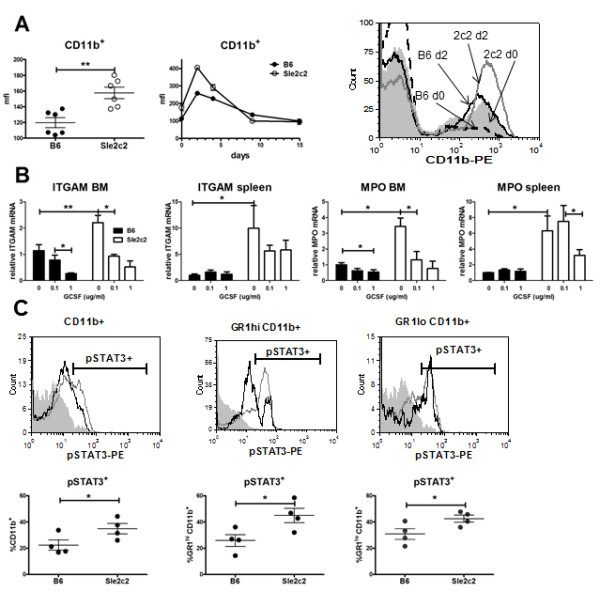
Differential response of the granulocyte-colony stimulation factor receptor (G-CSFR) target genes in response to G-CSF in B6.Sle2c2 and B6 leukocytes. (A) CD11b is upregulated in B6.Sle2c2 peripheral blood leukocytes (PBLs) compared to B6 (left panel). Human (hu)G-CSF (120 ug) injected on day (d)1 (arrow) induced a greater CD11b upregulation in B6.Sle2c2 than in B6 PBLs (middle panel, n = 4 per strain). The overlay histograms on the right show representative CD11b expression in B6 splenocytes at d0 (dash) and d2 (black), and in B6.Sle2c2 splenocytes at d0 (filled gray) and d2 (open gray). (B) Itgam and Mpo relative gene expression in bone marrow (BM) and spleen cells cultured for 8 h in medium alone and with 0.1 or 1.0 ug/ml hu-G-CSF. Filled bars represent B6 and open bars represent B6.Sle2c2 (n = 3 per group). In each graph, the data were normalized to the average of untreated B6 values. (C) Intracellular pSTAT3 staining in total CD11b+, GR1hi CD11b+ and GR1lo CD11b+ splenocytes (four mice per strain). The top row shows representative overlays of the B6 (open black) and Sle2c2 (open gray) histograms, with the filled silver histograms showing the isotype control. The graphs show mean and standard error of the mean with the significance value from the t-test (A and C) or the Bonferroni multiple comparison test (B) (*P < 0.05; **P < 0.01; ***P < 0.001).
Exogenous G-CSF restores cGVHD responses in B6.Sle2c2 mice
If the S378N mutation in the G-CSFR is responsible for the cGVHD resistance in B6.Sle2c2 mice, we postulated that huG-CSF treatment should eliminate the difference in cGVHD response between B6 and B6.Sle2c2 mice. If resistance in B6.Sle2c2 mice is due to a G-CSFR loss of function, huG-CSF treatment should restore cGVHD response in B6.Sle2c2 mice. On the other hand, if resistance is due to a G-CSFR gain of function, cGVHD resistance would be expected in huG-CSF-treated B6 mice. Based on our time course analysis showing a maximal effect at d4 after injection that was eliminated at d7, we tested a combination of treatment protocols in which the dose (1.2, 12.0, or 120.0 ug) and the frequency (one-, two-, or three-weekly injections starting at cGVHD induction) varied. Two treatment protocols (120 × 1 and 12 × 2) raised the production of anti-dsDNA IgG in B6.Sle2c2 mice significantly above the dextrose-treated controls (Figure 4A left), and to a level similar to the B6 controls, especially in the 12 × 2 cohort. For the 120 × 1 cohort, a drop in the third week was observed, possibly because the pharmacological effect of the single huG-CSF treatment was diminishing. With these two treatment regimens combined, the amount of anti-dsDNA and anti-chromatin IgG at d21 was significantly higher in the B6.Sle2c2 mice treated with huG-SCF than in controls (Figure 4B). Moreover, the autoAbs produced by treated B6.Sle2c2 mice reached equivalent levels as in treated B6 mice. cGVHD induced autoAbs in B6 mice with strong Hep-2 staining that combined cytoplasmic and nuclear patterns (Figure 4C). The 12 × 2 treatment induced Hep-2-staining in B6.Sle2c2 mice similar in intensity and pattern as in B6 mice (Figure 4C). Similar results were obtained with the 120 × 1 treatment, although with a more variable intensity (data not shown). The 1.2 × 3 treatment had no effect, indicating that the dose was too low. The 12 × 3 treatment had an intermediate effect in B6.Sle2c2 mice, and interestingly was the only regimen that induced a significant decrease in the B6 anti-dsDNA IgG response (Figure 4A right). The other treatments showed a trend in lowering autoAbs produced by B6 mice, but the difference was not significant. Overall, these results demonstrate that exogenous G-CSF eliminates the resistance to cGVHD induction in B6.Sle2c2 mice in a dose-dependent manner, with no significant effect in B6 mice at the same dose. This suggests that an impaired G-CSFR response mediates the cGVHD resistance in the B6.Sle2c2 strain, which can be compensated by an excess G-CSF to drive equilibrium binding or signaling to the receptor.
Figure 4.
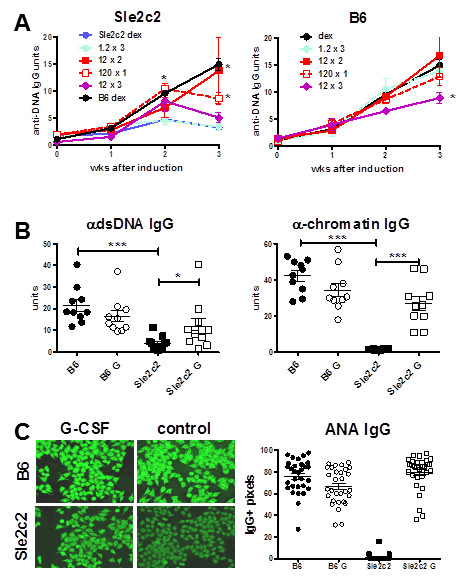
Exogenous human granulocyte-colony stimulation factor (huG-CSF) restores autoantibody (autoAb) production in B6.Sle2c2 mice after induction of chronic graft vs host disease (cGVHD). (A) huG-CSF-treatment induced anti-dsDNA IgG in B6.Sle2c2 mice in a dose-dependent manner (left), but had little effect in B6 mice (right). Five mice per strain per treatment were used. (B) Anti-dsDNA and anti-chromatin IgG found at d21 in the combined 120 × 1 and 12 × 2 cohorts. (C) Representative ANA IgG staining of Hep-2 cells incubated with sera collected at 21 d in each of the four treatment groups in the 120 × 1 and 12 × 2 cohorts and average staining intensity of 10 to 20 randomly selected cells per sample. Means and standard error of the mean with the significance value from the t-test for comparison of treated mice and controls (dex) are shown for each strain for a given time point in A, and from Dunnett's multiple comparison test between groups as indicated in B and C (*P < 0.05; **P < 0.01; ***P < 0.001).
The restoration of cGVHD response by G-CSF increases CD4+ T cell activation and expands the DC and neutrophil populations
cGVHD increases the percentage of blasts and the expression of activation markers such as class II MHC, CD22 and CD69 on B6 but not on B6.Sle2c2 B cells [5]. Treatment with huG-CSF has minimal effects on these B cell parameters (data not shown). It decreased CD22 expression in a dose-dependent fashion on B6 but not in B6.Sle2c2 B cells. However, these changes in B cell activation did not correspond to the observed changes in autoAb production. This suggested that huG-CSF did not restore the cGVHD response through B cells. Host CD4+ T cells are also activated by cGVHD in B6 but not in B6.Sle2c2 mice [5]. B6.Sle2c2 mice have a significantly greater percentage of splenic CD4+ T cells 3 weeks after cGVHD induction (21.58 ± 0.66% vs 16.35 ± 0.48%, P < 0.001). All huG-CSF treatments reduced the size of the CD4+ T cell compartment in both strains, and the 12 × 2 treatment, which was the most successful in restoring a cGVHD response in B6.Sle2c2 mice, resulted in an equivalent percentage of CD4+ T cells between strains (17.19 ± 0.55% vs 16.18 ± 0.11%, P > 0.05). The 12 × 2 treatment also significantly increased the percentage of CD4+ T cell blasts in B6.Sle2c2 mice (controls: B6.Sle2c2: 8.54 ± 0.54% vs B6: 16.57 ± 0.75%, P > 0.0001; 12 × 2: 14.50 ± 1.26% vs 15.64 ± 1.89%, P > 0.05), increased CD69 expression, and reduced the naïve and expanded the effector memory CD4+ T cells in B6.Sle2c2 mice (Figure 5A). The similar values in 12 × 2 treated B6 and B6.Sle2c2 mice correlated with restored autoAb production in the B6.Sle2c2 mice (Figure 4). The percentage of naïve CD4+ T cells was still higher in treated B6.Sle2c2 than in treated B6 spleens, yet the difference was smaller than in untreated mice (1.43- vs 2.15-fold). The huG-CSF treatments (12 × 3 and 1.2 × 3) that did not restore autoAbs production in B6.Sle2c2 mice had no effect on CD4+ T cell activation (data not shown).
Figure 5.
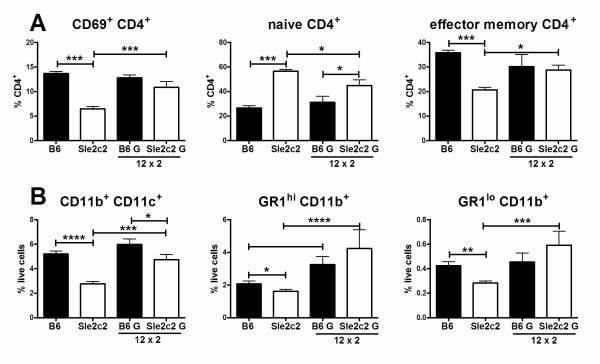
Restoration of chronic graft vs host disease (Cgvhd)-induced autoantibody (autoAb) by human granulocyte-colony stimulation factor (huG-CSF) correlates with CD4+ T cell activation, as well as DC and GR-1+ cell expansion. (A) huG-CSF 12 × 2 treatment increased CD69 expression, reduced the percentage of naïve CD62L+ CD44- CD4+ T cells and expanded the percentage of the CD62L- CD44+ effector memory CD4+ T cells in B6.Sle2c2 mice compared to dextrose-treated controls. (B) huG-CSF 12 × 2 treatment increased the percentage of CD11b+ CD11c+, GR1hi CD11b+ and GR1lo CD11b+ cells in B6.Sle2c2 mice compared to dextrose-treated controls. Means and standard error of the mean are shown with the significance value from Dunnett's multiple comparison tests between groups with five mice per group (*P < 0.05; **P < 0.01; ***P < 0.001).
The percentage of CD11b+ CD11c- GR1- macrophages was equivalent between B6 and Sle2c2 mice 3 weeks after cGVHD induction. huG-CSF treatments increased the percentage of macrophages in a dose-dependent manner similarly between these two strains without any correlation with autoAb production (data not shown). In contrast, the percentage of CD11b+ CD11c+ myeloid dendritic cells (DCs) was significantly lower in B6.Sle2c2 than in B6 mice, and the 12 × 2 treatment significantly expanded this cell population in the spleen of B6.Sle2c2 mice to a level similar to that of treated B6 mice (Figure 5B). The percentage of CD11b+ GR1hi neutrophils was significantly higher in un-manipulated B6.Sle2c2 than in B6 mice (3.97 ± 0.55% vs 2.50 ± 0.16%, P = 0.03). Three weeks after cGVHD induction, this difference was inverted, with B6.Sle2c2 values being significantly lower than B6 (Figure 5B). As in the blood of non-induced mice (Figure 1B), 12 × 2 treatment expanded the neutrophil population in the spleen and resulted in equivalent percentages in both strains. Finally, the percentage of CD11b+ GR1lo cells was similar in untreated and unmanipulated B6.Sle2c2 and B6 mice (0.49 ± 0.04 vs 0.50 ± 0.02%, P > 0.05), but three weeks after cGVHD induction, this population was significantly depleted in B6.Sle2c2 mice (Figure 5B). Also as in the blood of non-induced mice (Figure 1B), the 12 × 2 expanded the CD11b+ GR1lo population in B6.Sle2c2 mice three weeks after cGVHD induction and resulted in equivalent percentages in both strains (Figure 5B). Overall, these results showed that restoration of the cGVHD response in B6.Sle2c2 mice by huG-CSF treatment correlated with an increase in CD4+ T cell activation, and an expansion of the DC, neutrophils and CD11b+ GR1lo cell populations.
Reactive oxygen species (ROS) production is differentially up-regulated in the cGVHD response between B6.Sle2c2 and B6 mice
B6.Sle2c2 spleens contain significantly more ROS+ granulocytes cells (Figure 6A and 7B) and myeloid (Figure 6B and 7A) than B6, although no difference was observed for the intensity of ROS expression (mean fluorescence intensity, MFI) on ROS+ cells (data not shown). The same result was obtained with PBL (data not shown). Bm12-cGVHD induction expanded by over 5-fold the population of ROS+ granulocytes in both strains, and eliminated the difference between strains (Figure 6A and 7D). The percentage of ROS+ myeloid splenocytes was also similar between strains after cGVHD induction (Figure 6B and 7C). The ROS MFI was, however, significantly higher on B6 than B6.Sle2c2 granulocytes after cGVHD induction (Figure 6C). A time course analysis of ROS expression in B6 and B6.Sle2c2 PBLs at d3, 10 and 21 after cGVDH induction demonstrated a robust ROS induction in both strains, but that reached significantly higher levels in B6 than in B6.Sle2c2 mice at d21 (Figure 6D). The 12 × 2 treatment induced a higher ROS production in B6.Sle2c2 PBLs eliminating the difference with B6 mice, either treated or controls (Figure 6D). Therefore, steady state levels of ROS are higher in B6.Sle2c2 than in B6 mice. cGVHD induction greatly upregulates ROS production to a higher level in B6 than in B6.Sle2c2 mice, which is corrected by huG-CSF treatment. Taken together, these results suggest that variations in ROS production regulate the cGVHD response and that G-CSF signaling contributes to these variations.
Figure 6.

Reactive oxygen species (ROS) production is differentially upregulated between B6.Sle2c2 and B6 mice. B6.Sle2c2 mice present a higher percentage of GR1+ (A) and CD11b+ (B) splenocytes than B6. Bm12-chronic graft vs host disease (cGVHD) expands the GR1+ ROS+ in both strains, but eliminates the differences between strains. (C) At day (d)21 after cGVHD induction the ROS mean fluorescence intensity (MFI) was significantly higher in B6 GR1+ splenocytes than in B6.Sle2c2. (D) Time course analysis of ROS MFI in total peripheral blood leukocytes (PBLs) during cGVDH induction in mice treated with 12 × 2 human granulocyte-colony stimulation factor (huG-CSF). Means and standard error of the mean for five mice per group, with the significance value from the t-test are shown (*P < 0.05; ** P < 0.01). These data are representative of two sets of experiments.
Figure 7.
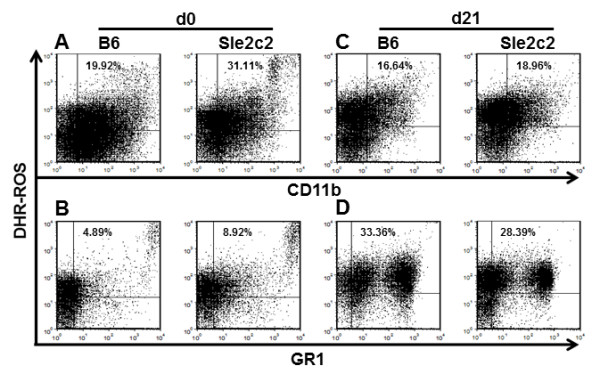
Representative fluorescence-activated cell sorting (FACS) plots of reactive oxygen species (ROS) production in B6 and B6.Sle2c2 splenocytes. Splenocytes were co-stained for CD11b (A and C) or GR1 (B and D) in un-induced mice (d0) and at d21 after bm12-chronic graft vs host disease (cGVHD) induction. The positions of the quadrants were determined with anti-CD11 and anti-GR1 isotype controls, and with cells stained with surface makers in the absence of the Dihydrorhodamine 123 (DHR) dye. The percentages correspond to double-positive ROS+ CD11b+ or GR1+ live splenocytes.
Exogenous G-CSF accelerates anti-dsDNA IgG in lupus-prone B6.TC mice
B6.TC mice first produce anti-chromatin IgG between 2 to 3 months of age then anti-dsDNA IgG between 4 and 5 months of age [19]. The presence of Sle2c2 in the B6.TC genome predicts that G-CSF treatment would accelerate their autoAb production as it did in the induced lupus model. We treated 2-month-old B6.TC and B6 female mice with six weekly injections of either 1ug hu-G-CSF or dextrose. It has been shown that neutralizing anti-hu G-CSF antibodies develop with a greater number of injections [23]. G-CSF treated B6.TC mice developed anti-dsDNA IgG significantly faster than control mice (Figure 8A). The G-CSF treatment did not induce anti-dsDNA IgG in B6 mice (Figure 8B). G-CSF, however, had no effect on anti-chromatin IgG, which increased at a similar rate in both treated and control B6.TC mice, and stayed at background levels in treated and control B6 mice (Figure 8C). G-CSF treatment did not affect total IgG levels (Figure 8D) or IgM (data not shown) in either strain, indicating that the effect of G-GSF was specific for anti-dsDNA IgG. This was confirmed when the production of anti-dsDNA IgG was normalized to total IgG (Figure 8E). It was not the case for anti-chromatin IgG (Figure 8F), which may be related to the fact that the anti-chromatin response was already ongoing when the treatment was initiated. Nevertheless, these results show that exogenous G-CSF increased production of anti-dsDNA IgG in mice carrying the Sle2c2 locus in a spontaneous model of lupus.
Figure 8.

Human granulocyte-colony stimulation factor (huG-CSF) treatment accelerates anti-dsDNA IgG production in B6.TC mice. Anti-dsDNA IgG production in B6.TC mice (A) and B6 controls (B) that were treated weekly with 1 ug huG-CSF (GCSF) or 5% dextrose (dextrose) starting at 2 months of age (n = 3 per group). Results for B6.TC mice were normalized to their individual anti-dsDNA IgG levels before treatment (week 0) set as 1. Results for B6 were normalized to the average units of all B6.TC mice before treatment to account for the large difference between B6.TC and B6 anti-dsDNA IgG production. (C) Anti-chromatin IgG production in the four cohorts. Unit values are shown because minimal variation was observed before treatment between and within cohorts. The regression lines between the huG-CSF-treated B6.TC and B6 cohorts have significantly different slopes (P = 0.005) but not between dextrose-treated cohorts (P = 0.17). (D) Total serum IgG in the four cohorts. B6.TC mice had significantly more IgG than B6 before treatment (152 vs 95 ug/ml, P = 0.001), but that difference disappeared with time. The huG-CSF treatment did not change total IgG production in neither strain. Anti-dsDNA (E) and chromatin (F) IgG in B6.TC mice normalized to their total IgG production. The autoantibody (autoAb) unit/total IgG ratios were normalized to the individual values before treatment set as 1. The graphs show mean and standard error of the mean at the indicated time points after treatment. For A and E, the P-values correspond to an F-test comparing the slopes for the two linear regression lines corresponding to the two treatments.
Discussion
This study was performed to test the hypothesis that the S378N mutation in the Csf3r gene results in defective G-CSFR signaling and is responsible for the differential autoimmune response between B6 and B6.Sle2c2 mice. In our previous study, we speculated that bm12-cGVHD resistance in B6.Sle2c2 mice was mediated by enhanced MDSC function or number. However, G-CSF treatment did not induce bm12-cGVHD resistance in B6 mice, but instead eliminated bm12-cGVHD resistance in B6.Sle2c2 mice. We therefore conclude that B6.Sle2c2 mice are resistant to autoimmunity due to their incapacity to mobilize pro-inflammatory G-CSF-responsive BM-derived cells, most likely neutrophils. Indeed, we report here that B6.Sle2c2 leukocytes bind less G-CSF and expand less in response to in vivo G-CSF treatment. On the other hand, G-CSFR-responsive genes are expressed at higher levels in B6.Sle2c2 leukocytes, which present a greater STAT3 phosphorylation, indicating a higher level of G-CSFR signaling. Although the lower cell expansion to G-CSF and the higher G-CSFR signaling suggest opposing responses, it is possible that the latter is a steady-state compensatory response to the former. This hypothesis will require extensive comparison of G-CSFR signaling between the two alleles, including in cell lines transfected with G-CSFR constructs differing only at S378N.
The lower binding of G-CSF and impaired cellular expansion in response to exogenous G-CSF lead us to hypothesize that exogenous G-CSF would compensate for a loss of function phenotype. To this end, we indeed found that two huG-CSF treatment regimens restored the production of autoAbs to a level similar to that found in B6. These treatments also restored CD4+ T cell activation as well as DC and granulocyte expansion in B6.Sle2c2 mice to the same levels as in B6. This suggested that these cell populations are effectively modified by higher G-CSF availability and they are involved in the production of autoAbs in this model.
We also found that B6.Sle2c2 myeloid cells and granulocytes produced more ROS at steady-state than B6 cells. A recent study found a strong association between a coding mutation in the human NCF2 gene with SLE susceptibility, and determined that the disease associated allele was associated with a lower ROS production [24]. This is consistent with decreased levels of Ncf1, a gene encoding for another unit of the nicotinamide adenine dinucleotide phosphate-oxidase (NADPH) oxidase complex, being associated with collagen-induced arthritis [25] and experimental allergic encephalomyelitis [26]. Upon bm12-cGVHD induction, ROS production is upregulated to a greater level in the B6 than B6.Sle2c2 granulocytes. huG-CSF treatment increased ROS production in B6.Sle2c2 mice, which was concomitant with their production of autoAbs. These results suggest that the NZM2410 allele of Csf3r results in a higher ROS production at steady state levels of G-CSF that maintains CD4+ T cell tolerance, as it has been shown in other models [27]. We showed here that bm12-cGVHD induction recruits inflammatory neutrophils, as is the case in human SLE [28], of which B6.Sle2c2 mice cannot achieve the same expansion as B6 mice unless exogenous G-CSF is provided. Signaling through the G-CSFR is critical for the development and function of inflammatory granulocytes, whereas G-CSFR deficiency is protective in collagen-induced arthritis [29]. We therefore propose that impaired G-CSF binding and G-CSFR signaling is protective again systemic autoimmunity in two phases, first by producing higher levels of ROS, which maintains T cell tolerance, and second by failing to support the expansion of inflammatory neutrophils, which can amplify the autoimmune response either through the type I IFN or Th17 pathways [9].
We have previously reported that there is no difference in Csf3r expression between B6 and B6.Sle2c2 mice [5]. Genomic sequencing of the Csf3r locus has identified a large number of intronic polymorphisms and four coding synonymous mutations, but S378N was the only coding non-synonymous mutation identified between the NZM2410 and B6 genomes (unpublished observations). None of the genes located within Sle2c2 locus have known functions in the G-CSF pathway. This implies that the differences that we report here between B6 and B6.Sle2c2 mice in their response to G-CSF are most likely due to the S378N polymorphism. This will, however, need to be formally addressed by stringent structure/function studies.
We have used the bm12-cGVHD model to test the hypothesis that Sle2c2 suppression involves the G-CSF pathway, as it is a very robust model that leads to a rapid synchronized production of autoAbs with very little inter-individual variation. However, we have evidence that Sle2c2 also suppresses spontaneous autoimmune responses in two NZM2410-derived models [30,31] and we have now shown that a low-dose treatment with G-CSF accelerates the spontaneous production of anti-dsDNA IgG in B6.TC mice, which includes Sle2c2 [19].
An association between G-CSF treatment and disease exacerbation is supported by clinical findings, where G-CSF treatment of neutropenic SLE patients can be associated with severe flares [32]. Moreover, depending on the dose, G-CSF treatment can either prevent or accelerate disease in the MRL/lpr mouse model of SLE [33]. While these two reports did not explore the mechanism of disease exacerbation, we propose that it overlaps with the resistance or susceptibility to induced autoAb production mediated by the S378N G-CSFR polymorphism, and that the B6.Sle2c2 and B6.TC strains offer an excellent model to identify the cellular and molecular bases on how G-CSF modulates SLE pathogenesis.
Contrary to the results reported here, G-CSF treatment has been reported to promote reversal [21] or prevention [34] of type 1 diabetes in the NOD mouse. In this model, NADPH oxidase impairment through Ncf1-deficiency was associated with disease protection [35], contrary to the results obtained in SLE patients with NCF2-impaired function [24]. These results suggest an inverse correlation between the protective effect of exogenous G-CSF and the protective effect of the NADPH oxidase products in autoimmune diseases. This has significant implications for devising strategies targeting the G-CSF pathways in opposite direction in diseases such as SLE and type I diabetes.
Conclusions
Exogenous G-CSF increases the production of autoAb in B6.Sle2c2 and B6.TC mice and both of these strains carry the same mutation in the G-CSFR. Furthermore, we have shown a defective mobilization of BM neutrophils in response to G-CSF treatment of B6.Sle2c2 mice as well as a different expression of G-CSF-responsive genes between B6 and B6.Sle2c2 mice. Overall, these data suggest that mice carrying the G-CSFR mutation are defective in mobilizing and/or activating inflammatory neutrophils, which inhibits the production of autoAbs. Further experiments will be required to demonstrate a direct role of neutrophils in this process.
Abbreviations
ANA: antinuclear antibodies; autoAb: autoantibody; B6: C57BL/6; BM: bone-marrow; bm12: B6.C-H2bm12; cGVHD: chronic graft vs host disease; ct: cycle threshold; DC: dendritic cell; ELISA: enzyme-linked immunosorbent assay; FACS: representative fluorescence-activated cell sorting; FITC: fluorescein isothiocyanate; G-CSF: granulocyte-colony stimulation factor; G-CSFR: granulocyte-colony stimulation factor receptor; huG-CSF: human G-CSF; IFN: interferon; Ig: immunoglobulin; IL: interleukin; MDSC: myeloid-derived suppressor cell; MFI: mean fluorescence intensity; mG-CSF: mouse G-CSF; MPO: myeloperoxidase; NADPH: nicotinamide adenine dinucleotide phosphate-oxidase; NET: neutrophil extracellular trap; PBL: peripheral blood leukocyte; PCR: polymerase chain reaction; PE: phycoerythrin; ROS: reactive oxygen species; SEM: standard error of the mean; SLE: systemic lupus erythematosus; STAT3: signal transducer and activator of transcription 3; TLR: toll-like receptor.
Competing interests
The authors declare that they have no conflict of interest
Authors' contributions
ML performed the flow cytometry analysis and the in vitro assays. RS performed the neutrophil mobilization assays, the expression assays in G-CSF-responsive genes and the treatment study with lupus-prone mice. LZ performed the cGVHD induction and G-CSF treatments of these mice, antibody measurements and flow cytometry. CW assisted with the conception and implementation of the in vivo G-CSF treatment studies. YYZ assisted with the neutrophil mobilization assay. MA participated in the study design and coordination, as well as in data interpretation. LM conceived the study, overviewed data analysis and interpretation, and wrote the paper. All authors read and approved the final manuscript.
Contributor Information
Margareta Lantow, Email: Margareta.Lantow@klinik.uni-regensburg.de.
Ramya Sivakumar, Email: rsivakumar09@ufl.edu.
Leilani Zeumer, Email: zeumer@pathology.ufl.edu.
Clive Wasserfall, Email: clive@ufl.edu.
Ying-Yi Zheng, Email: yyzheng2@ufl.edu.
Mark A Atkinson, Email: atkinson@ufl.edu.
Laurence Morel, Email: morel@ufl.edu.
Acknowledgements
The authors are grateful to Dr Clayton Mathews for stimulating discussions and to Nathalie Kanda for excellent animal care. Supported by the National Institutes of Health (RO1 AI068965 to LM and PO1 AI42288 to MAA).
References
- Tsokos GC. Systemic lupus erythematosus. N Engl J Med. 2011;365:2110–2121. doi: 10.1056/NEJMra1100359. [DOI] [PubMed] [Google Scholar]
- Morel L. Genetics of SLE: evidence from mouse models. Nat Rev Rheumatol. 2010;6:348–357. doi: 10.1038/nrrheum.2010.63. [DOI] [PubMed] [Google Scholar]
- Harley IT, Kaufman KM, Langefeld CD, Harley JB, Kelly JA. Genetic susceptibility to SLE: new insights from fine mapping and genome-wide association studies. Nat Rev Genet. 2009;10:285–290. doi: 10.1038/nrg2571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morel L, Tian XH, Croker BP, Wakeland EK. Epistatic modifiers of autoimmunity in a murine model of lupus nephritis. Immunity. 1999;11:131–139. doi: 10.1016/S1074-7613(00)80088-6. [DOI] [PubMed] [Google Scholar]
- Xu Z, Vallurupalli A, Fuhrman C, Ostrov D, Morel L. An NZB-derived locus suppresses chronic graft versus host disease and autoantibody production through non-lymphoid bone-marrow derived cells. J Immunol. 2011;186:4130–4139. doi: 10.4049/jimmunol.1003512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris SC, Cheek RL, Cohen PL, Eisenberg RA. Autoantibodies in chronic graft versus host result from cognate T-B interactions. J Exp Med. 1990;17:503–517. doi: 10.1084/jem.171.2.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christopher MJ, Link DC. Regulation of neutrophil homeostasis. Curr Opin Hematol. 2007;14:3–8. doi: 10.1097/00062752-200701000-00003. [DOI] [PubMed] [Google Scholar]
- Morris ES, MacDonald KPA, Kuns RD, Morris HM, Banovic T, Don ALJ, Rowe V, Wilson YA, Raffelt NC, Engwerda CR, Burman AC, Markey KA, Godfrey DI, Smyth MJ, Hill GR. Induction of natural killer T cell-dependent alloreactivity by administration of granulocyte colony-stimulating factor after bone marrow transplantation. Nat Med. 2009;15:436–441. doi: 10.1038/nm.1948. [DOI] [PubMed] [Google Scholar]
- Kaplan MJ. Neutrophils in the pathogenesis and manifestations of SLE. Nat Rev Rheumatol. 2011;7:691–9. doi: 10.1038/nrrheum.2011.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villanueva E, Yalavarthi S, Berthier CC, Hodgin JB, Khandpur R, Lin AM, Rubin CJ, Zhao W, Olsen SH, Klinker M, Shelay D, Denny MF, Plumas J, Chaperot L, Kretzler M, Bruce AT, Kaplam MJ. Netting neutrophils induce endothelial damage, infiltrate tissues, and expose immunostimulatory molecules in systemic lupus erythematosus. J Immunol. 2011;187:538–552. doi: 10.4049/jimmunol.1100450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakkim A, Furnrohr BG, Amann K, Laube B, Abed UA, Brinkmann V, Herrmann M, Voll RE, Zychlinsky A. Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc Natl Acad Sci USA. 2010;107:9813–9818. doi: 10.1073/pnas.0909927107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obermoser G, Sontheimer RD, Zelger B. Overview of common, rare and atypical manifestations of cutaneous lupus erythematosus and histopathological correlates. Lupus. 2010;19:1050–1070. doi: 10.1177/0961203310370048. [DOI] [PubMed] [Google Scholar]
- Denny MF, Yalavarthi S, Zhao W, Thacker SG, Anderson M, Sandy AR, McCune WJ, Kaplan MJ. A distinct subset of proinflammatory neutrophils isolated from patients with systemic lupus erythematosus induces vascular damage and synthesizes type I IFNs. J Immunol. 2010;184:3284–3297. doi: 10.4049/jimmunol.0902199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Romo GS, Caielli S, Vega B, Connolly J, Allantaz F, Xu Z, Punaro M, Baisch J, Guiducci C, Coffman RL, Barrat FJ, Banchereau J, Pascual V. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci Transl Med. 2011;3:73ra20–73ra20. doi: 10.1126/scitranslmed.3001201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin AM, Rubin CJ, Khandpur R, Wang JY, Riblett M, Yalavarthi S, Villanueva EC, Shah P, Kaplan MJ, Bruce AT. Mast cells and neutrophils release IL-17 through extracellular trap formation in psoriasis. J Immunol. 2011;187:490–500. doi: 10.4049/jimmunol.1100123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell AM, Kashgarian M, Shlomchik MJ. NADPH oxidase inhibits the pathogenesis of systemic lupus erythematosus. Sci Transl Med. 2012;4:157ra141. doi: 10.1126/scitranslmed.3004801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Highfill SL, Rodriguez PC, Zhou Q, Goetz CA, Koehn BH, Veenstra R, Taylor PA, Panoskaltsis-Mortari A, Serody JS, Munn DH, Tolar J, Ochoa AC, Blazar BR. Bone marrow myeloid-derived suppressor cells (MDSCs) inhibit graft-versus-host disease (GVHD) via an arginase-1-dependent mechanism that is up-regulated by interleukin-13. Blood. 2010;116:5738–5747. doi: 10.1182/blood-2010-06-287839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwata Y, Furuichi K, Kitagawa K, Hara A, Okumura T, Kokubo S, Shimizu K, Sakai N, Sagara A, Kurokawa Y, Ueha S, Matsushima K, Kaneto S, Wada T. Involvement of CD11b+ GR-1low cells in autoimmune disorder in MRL-Faslpr mouse. Clin Exp Nephrol. 2010;14:411–417. doi: 10.1007/s10157-010-0309-9. [DOI] [PubMed] [Google Scholar]
- Morel L, Croker BP, Blenman KR, Mohan C, Huang G, Gilkeson G, Wakeland EK. Genetic reconstitution of systemic lupus erythematosus immunopathology with polycongenic murine strains. Proc Natl Acad Sci USA. 2000;97:6670–6675. doi: 10.1073/pnas.97.12.6670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potula H, Xu Z, Croker PP, Morel L. Cyclin-dependent kinase inhibitor Cdkn2c deficiency promotes B1a cell expansion and autoimmunity in a mouse model of lupus. J Immunol. 2012;189:2931–40. doi: 10.4049/jimmunol.1200556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker MJ, Xue S, Alexander JJ, Wasserfall CH, Campbell-Thompson ML, Battaglia M, Gregori S, Mathews CE, Song S, Troutt M, Eisenbeis S, Williams J, Shatz DA, Haller MJ, Atkinson MA. Immune depletion with cellular mobilization imparts immunoregulation and reverses autoimmune diabetes in nonobese diabetic mice. Diabetes. 2009;58:2277–2284. doi: 10.2337/db09-0557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panopoulos AD, Watowich SS. Granulocyte colony-stimulating factor: molecular mechanisms of action during steady state and 'emergency' hematopoiesis. Cytokine. 2008;42:277–288. doi: 10.1016/j.cyto.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Kruijf E-JFM, van Pel M, Hagoort H, Kruysdijk D, Molineux G, Willemze R, Fibbe WE. Repeated hematopoietic stem and progenitor cell mobilization without depletion of the bone marrow stem and progenitor cell pool in mice after repeated administration of recombinant murine G-CSF. Human Immunol. 2007;68:368–374. doi: 10.1016/j.humimm.2007.01.017. [DOI] [PubMed] [Google Scholar]
- Jacob CO, Eisenstein M, Dinauer MC, Ming W, Liu Q, John S, Quismorio FP Jr, Reiff A, Myones BL, Kaufman KM, McCurdy D, Harley JB, Silverman E, Kimberly RP, Vyse TJ, Gaffney PM, Moser KL, Klein-Gitelman M, Wagner-Weiner L, Langefeld CD, Armstrong DL, Zidovetzki R. Lupus-associated causal mutation in neutrophil cytosolic factor 2 (NCF2) brings unique insights to the structure and function of NADPH oxidase. Proc Natl Acad Sci USA. 2012;109:E59–E67. doi: 10.1073/pnas.1113251108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olofsson P, Holmberg J, Tordsson J, Lu S, Akerstrom B, Holmdahl R. Positional identification of Ncf1 as a gene that regulates arthritis severity in rats. Nat Genet. 2003;33:25–32. doi: 10.1038/ng1058. [DOI] [PubMed] [Google Scholar]
- Hultqvist M, Olofsson P, Holmberg J, Backstrom BT, Tordsson J, Holmdahl R. Enhanced autoimmunity, arthritis, and encephalomyelitis in mice with a reduced oxidative burst due to a mutation in the Ncf1 gene. Proc Natl Acad Sci USA. 2004;101:12646–12651. doi: 10.1073/pnas.0403831101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hultqvist M, Olsson LM, Gelderman KA, Holmdahl R. The protective role of ROS in autoimmune disease. Trends Immunol. 2009;30:201–208. doi: 10.1016/j.it.2009.03.004. [DOI] [PubMed] [Google Scholar]
- Bosch X. Systemic lupus erythematosus and the neutrophil. N Engl J Med. 2011;365:758–760. doi: 10.1056/NEJMcibr1107085. [DOI] [PubMed] [Google Scholar]
- Eyles JL, Hickey MJ, Norman MU, Croker BA, Roberts AW, Drake SF, James WG, Metcalf D, Campbell IK, Wicks IP. A key role for G-CSF-induced neutrophil production and trafficking during inflammatory arthritis. Blood. 2008;112:5193–5201. doi: 10.1182/blood-2008-02-139535. [DOI] [PubMed] [Google Scholar]
- Xu Z, Duan B, Croker BP, Wakeland EK, Morel L. Genetic dissection of the murine lupus susceptibility locus Sle2: contributions to increased peritoneal B-1a cells and lupus nephritis map to different loci. J Immunol. 2005;175:936–943. doi: 10.4049/jimmunol.175.2.936. [DOI] [PubMed] [Google Scholar]
- Xu Z, Cuda CM, Croker BP, Morel L. The NZM2410-derived lupus susceptibility locus Sle2c1 increases TH17 polarization and induces nephritis in Fas-deficient mice. Arthritis Rheum. 2011;63:764–774. doi: 10.1002/art.30146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasiliu IM, Petri MA, Baer AN. Therapy with granulocyte colony-stimulating factor in systemic lupus erythematosus may be associated with severe flares. J Rheumatol. 2006;33:1878–1880. [PubMed] [Google Scholar]
- Zavala F, Masson A, Hadaya K, Ezine S, Schneider E, Babin O, Bach JF. Granulocyte-colony stimulating factor treatment of lupus autoimmune disease in MRL-lpr/lpr mice. J Immunol. 1999;163:5125–5132. [PubMed] [Google Scholar]
- Kared H, Masson A, dle-Biassette H, Bach JF, Chatenoud L, Zavala F. Treatment with granulocyte colony-stimulating factor prevents diabetes in NOD mice by recruiting plasmacytoid dendritic cells and functional CD4(+)CD25(+) regulatory T-cells. Diabetes. 2005;54:78–84. doi: 10.2337/diabetes.54.1.78. [DOI] [PubMed] [Google Scholar]
- Tse HM, Thayer TC, Steele C, Cuda CM, Morel L, Piganelli JD, Mathews CE. NADPH oxidase deficiency regulates th lineage commitment and modulates autoimmunity. J Immunol. 2010;185:5247–5258. doi: 10.4049/jimmunol.1001472. [DOI] [PMC free article] [PubMed] [Google Scholar]