

Abstract
Autoantibodies may be found years before an autoimmune disease becomes clinically apparent. For systemic lupus erythematosus (SLE), those to RNA-binding proteins, to phospholipids, and to double-stranded DNA, in particular, have been found in sera of SLE patients years before the diagnosis was made. New data now show in an unbiased way that, in patients with early SLE, no single antibody class or specificity is associated with progression to SLE. Rather, an increasing number of autoantibody specificities, such as to thyroid antigens, was observed in patients progressing. This points to more generalized B cell autoreactivity during progression to SLE, underlying lupus disease manifestations.
In a previous issue of Arthritis Research & Therapy, Nancy Olsen and colleagues from the Penn State Hershey Medical Center and the University of Texas Southwestern Medical Center present an analysis of a wide range of autoantibodies in patients with 'incomplete' SLE [1]. Within the Dallas Regional Autoimmune Disease registry (DRADR) cohort, they have followed patients who, with one exception, did not fulfill American College of Rheumatology criteria for systemic lupus erythematosus (SLE) at baseline using an autoantigen array.
Out of 22 patients, only 3 developed additional SLE criteria during a follow-up of 3.8 ± 0.6 years. These three patients showed some possible differences in their serology. At baseline, they had higher titers of antibodies to hemocyanin and PL-7 (threonyl-tRNA synthetase), and somewhat higher levels of antibodies to thyroglobulin, thyroid peroxidase, proliferating cell nuclear antigen, β2-microglobulin, and C1q, not all of which are typically associated with SLE. On follow-up, these patients showed a significant increase in anti-La/SS-B and LC1 (liver cytosol type 1) antibodies, and a trend towards increasing anti-Ro/SS-A antibodies. In addition, they were more likely to develop new antibody specificities, in contrast to the remaining patients, where this was seen in less than one-third.
The authors mainly present their work as an attempt to finding predictors for developing SLE. While the approach is of interest, the sample size, the limited follow-up time, and some technical issues preclude firm conclusions as of yet. Only three patients progressed to SLE - the others maintained a stable phenotype. This may indicate that the majority of patients had already reached a stable phenotype of 'undifferentiated connective tissue disease' at enrollment in the study. However, the few patients who progressed clinically may hint at early immune events that define SLE.
In a variety of autoimmune diseases, specific autoantibodies clearly precede disease. In autoimmune blistering diseases of the skin, there is evidence that antibodies determine both clinical phenotype and disease onset. Desmoglein-1 antibodies are associated with pemphigus foliaceus, while desmoglein-3 antibodies occur in pemphigus vulgaris. Blistering occurs at the sites where the targets of these antibodies occur naturally [2]. Intramolecular epitope spreading, to particular epitopes of desmoglein-1, apparently explains the onset of clinical features of disease, which reverses in disease remission [3].
In rheumatoid arthritis, antibodies to citrullinated peptides, which are quite specific for the disease, may be present for years in individuals with no joint symptoms [4]. In contrast to pemphigus, however, no individual epitope of anti-citrullinated protein antibodies has been associated with the onset of clinical disease. Rather, the range of specificities, and the titres of antibodies, increase as patients approach disease onset [5].
In SLE, likewise, as has been known for some time, autoantibodies are present for years before the diagnosis [6], and anti-double-stranded DNA, anti-Ro, anti-La, and anti-phospholipid antibodies, in particular. These antibodies also predict development of SLE in patients with undifferentiated connective tissue disease and patients fulfilling criteria of mixed connective tissue disease [7]. However, the potential spectrum of antibodies in SLE is much broader than in the above-mentioned diseases [8].
These authors now demonstrate in an unbiased longitudinal analysis that these less typical SLE autoantibodies may have prognostic significance. Antibodies to hemocyanin, PL-7, thyroglobulin, thyroid peroxidase, or proliferating cell nuclear antigen are not among those usually provoking a search for an underlying diagnosis of SLE. Indeed, some of them are associated with other autoimmune diseases, such as anti-thyroid peroxidase and anti-thyroglobulin with autoimmune thyroid disease, or anti-PL-7 with polymyositis (Figure 1). In their work, the number of antigens targeted by autoantibodies appears more important than any given specificity, and a rapid increase in the number of antigens targeted by autoantibodies accompanied transformation into SLE.
Figure 1.
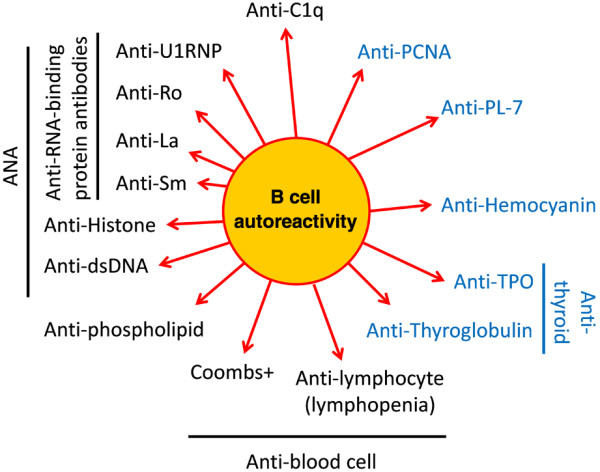
Autoantibodies found in early systemic lupus erythematosus. Typical systemic lupus erythematosus-associated autoantibodies are in black, those that are usually associated with other diseases are in blue letters. Overall, the wide antibody variety points to more generalized B-cell autoreactivity underlying systemic lupus erythematosus. dsDNA, double-stranded DNA; PCNA, proliferating cell nuclear antigen; TPO, thyroid peroxidase.
The findings in the manuscript by Olsen and colleagues thus paint a picture of increasing B cell autoreactivity, manifest as multiple autoantibodies during the progression of clinical SLE. There is undoubtedly a propensity to developing antibodies to specific nuclear antigens in SLE. Mechanistically this has been linked to defects in DNA processing, apoptotic clearance and Toll-like receptor sensing of nucleic antigens [9]. Most of the patients in this series were ANA positive at inclusion. However, broadening of the antibody spectrum, with much less specificity, may constitute a second step toward SLE. On a cellular level, the same concept is reflected by a marked increase in plasmablasts in active SLE, of which only a subset make anti-double-stranded DNA antibodies [10].
A range of known mechanisms may underlie this broader reactivity. Among others, polymorphisms in BLK, PTPN22 and Src family kinases have associations with SLE, and may lead to generalized B- and T-cell hyper-responsiveness [11]. Interferon-α production has been strongly associated with SLE. Excess production by plasmacytoid dendritic cells of type 1 interferons may, in response to Toll-like receptor stimulation by nuclear antigens and immune complexes, provoke a more generalized activation of humoral immunity. In other words, B cells may be intrinsically hyper-responsive and autoreactive, or they may simply be responding appropriately to inappropriate signals from a defective innate immune system (or both).
Similarly, the reasons why the majority of patients did not progress, despite autoantibodies at baseline, are also unclear, but may be of importance to understanding pathogenesis. For example, preservation of certain normal tolerance mechanisms in these patients may have been sufficient to keep B-cell autoreactivity in check. One might conclude, therefore, that autoantibodies are necessary, but not sufficient, to cause clinical SLE.
The results of this unique cohort therefore indicate that no one antibody target or class of autoantibody mark the transition to SLE, but rather a generalized broadening of the antibody repertoire. Recognizing this may be relevant to both better understand SLE diagnosis and classification and help target studies on abnormalities underlying lupus.
Abbreviations
SLE: systemic lupus erythematosus.
Competing interests
The authors declare that they have no competing interests.
See related research by Olsen et al., http://arthritis-research.com/content/14/4/R174
Contributor Information
Martin Aringer, Email: martin.aringer@uniklinikum-dresden.de.
Edward Vital, Email: E.M.J.Vital@leeds.ac.uk.
References
- Olsen NJ, Li Q-Z, Quan J, Wang L, Mutwally A, Karp DR. Autoantibody profiling to follow evolution of lupus syndromes. Arthritis Res Ther. 2012;14:R174. doi: 10.1186/ar3927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amagai M, Tsunoda K, Zillikens D, Nagai T, Nishikawa T. The clinical phenotype of pemphigus is defined by the anti-desmoglein autoantibody profile. J Am Acad Dermatol. 1999;40:167–170. doi: 10.1016/S0190-9622(99)70183-0. [DOI] [PubMed] [Google Scholar]
- Li N, Aoki V, Hans-Filho G, Rivitti EA, Diaz LA. The role of intramolecular epitope spreading in the pathogenesis of endemic pemphigus foliaceus (fogo selvagem) J Exp Med. 2003;197:1501–1510. doi: 10.1084/jem.20022031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rantapaa-Dahlqvist S, de Jong BA, Berglin E, Hallmans G, Wadell G, Stenlund H, Sundin U, van Venrooij WJ. Antibodies against cyclic citrullinated peptide and IgA rheumatoid factor predict the development of rheumatoid arthritis. Arthritis Rheum. 2003;48:2741–2749. doi: 10.1002/art.11223. [DOI] [PubMed] [Google Scholar]
- Bos WH, Wolbink GJ, Boers M, Tijhuis GJ, de Vries N, van der Horst-Bruinsma IE, Tak PP, van de Stadt RJ, van der Laken CJ, Dijkmans BA, van Schaardenburg D. Arthritis development in patients with arthralgia is strongly associated with anti-citrullinated protein antibody status: a prospective cohort study. Ann Rheum Dis. 2010;69:490–494. doi: 10.1136/ard.2008.105759. [DOI] [PubMed] [Google Scholar]
- Arbuckle MR, McClain MT, Rubertone MV, Scofield RH, Dennis GJ, James JA, Harley JB. Development of autoantibodies before the clinical onset of systemic lupus erythematosus. N Engl J Med. 2003;349:1526–1533. doi: 10.1056/NEJMoa021933. [DOI] [PubMed] [Google Scholar]
- Cappelli S, Bellando RS, Martinovic D, Tamas MM, Pasalic K, Allanore Y, Mosca M, Talarico R, Opris D, Kiss CG, Tausche AK, Cardarelli S, Riccieri V, Koneva O, Cuomo G, Becker MO, Sulli A, Guiducci S, Radic M, Bombardieri S, Aringer M, Cozzi F, Valesini G, Ananyeva L, Valentini G, Riemekasten G, Cutolo M, Ionescu R, Czirjak L, Damjanov N. et al. "To be or not to be," ten years after: evidence for mixed connective tissue disease as a distinct entity. Semin Arthritis Rheum. 2011;41:589–598. doi: 10.1016/j.semarthrit.2011.07.010. [DOI] [PubMed] [Google Scholar]
- Li QZ, Zhou J, Wandstrat AE, Carr-Johnson F, Branch V, Karp DR, Mohan C, Wakeland EK, Olsen NJ. Protein array autoantibody profiles for insights into systemic lupus erythematosus and incomplete lupus syndromes. Clin Exp Immunol. 2007;147:60–70. doi: 10.1111/j.1365-2249.2006.03251.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman A, Isenberg DA. Systemic lupus erythematosus. N Engl J Med. 2008;358:929–939. doi: 10.1056/NEJMra071297. [DOI] [PubMed] [Google Scholar]
- Jacobi AM, Mei H, Hoyer BF, Mumtaz IM, Thiele K, Radbruch A, Burmester GR, Hiepe F, Dorner T. HLA-DRhigh/CD27high plasmablasts indicate active disease in patients with systemic lupus erythematosus. Ann Rheum Dis. 2010;69:305–308. doi: 10.1136/ard.2008.096495. [DOI] [PubMed] [Google Scholar]
- Sestak AL, Furnrohr BG, Harley JB, Merrill JT, Namjou B. The genetics of systemic lupus erythematosus and implications for targeted therapy. Ann Rheum Dis. 2011;70(Suppl 1):i37–i43. doi: 10.1136/ard.2010.138057. [DOI] [PubMed] [Google Scholar]