

Abstract
Resuscitation of Mtb is crucial to the etiology of Tuberculosis, because latent tuberculosis is estimated to affect one-third of the world population. The resuscitation-promoting factor RpfB is mainly responsible for Mtb resuscitation from dormancy. Given the impact of latent Tuberculosis, RpfB represents an interesting target for tuberculosis drug discovery. However, no molecular models of substrate binding and catalysis are hitherto available for this enzyme. Here, we identified key interactions involved in substrate binding to RpfB by combining x-ray diffraction studies and computational approaches. The crystal structure of RpfB catalytic domain in complex with N,N′,N″-triacetyl-chitotriose, as described here, provides the first, to our knowledge, atomic representation of ligand recognition by RpfB and demonstrates that the strongest interactions are established by the N-acetylglucosamine moiety in the central region of the enzyme binding cleft. Molecular dynamics analyses provided information on the dynamic behavior of protein-substrate interactions and on the role played by the solvent in RpfB function. These data combined with sequence conservation analysis suggest that Glu-292 is the sole residue crucial for catalysis, implying that RpfB acts via the formation of an oxocarbenium ion rather than a covalent intermediate. Present data represent a solid base for the design of effective drug inhibitors of RpfB. Moreover, homology models were generated for the catalytic domains of all members of the Mtb Rpf family (RpfA-E). The analysis of these models unveiled analogies and differences among the different members of the Rpf protein family.
Abbreviations used: Mtb, Mycobacterium tuberculosis; RpfB, Resuscitation promoting factor B; PDB, Protein Data Bank; RpfBc, catalytic domain of RpfB; NAG3, N,N',N"-triacetyl-chitotriose; NAG6, hexa-N- acetylchitohexaose; MD, molecular dynamics; RMSF, root mean-square fluctuation
Introduction
Tuberculosis (TB) is the world’s leading cause of death due to a single infectious agent, Mtb, with ∼2 million deaths and 10 million new cases per year. Current treatment of TB is not effective against multidrug-resistant, extensively drug-resistant, and totally drug-resistant strains, not responding to any standard treatment (1–3). Therefore, the development of novel pharmacological regimens having a significant impact on shortening the treatment of both drug-sensitive and drug-resistant TB patients is urgently required. Currently, one-third of the world’s population is estimated to be infected with the tubercle bacillus, the majority of which develop latent tuberculosis (4). Latently infected individuals do not display any signs of active TB. However, they have a finite risk of developing a clinically active disease; this risk is markedly increased in the presence of HIV coinfection (5). During dormancy, bacteria undergo a reversible state of low metabolic activity in which cells can persist for extended periods without division (6–9). Of importance, dormant mycobacteria show a low susceptibility to antibiotics; this makes the development of active compounds against latent TB an urgent task. As highlighted by the World Health Organization Global Plan to Stop TB 2011–2015, new drugs and drug combinations should be developed against both active and dormant TB (Global Plan to Stop TB, http://www.stoptb.org/global/plan/).
A key enzyme in Mtb resuscitation from dormancy is the resuscitation-promoting factor RpfB, one of the five Rpf encoded in Mtb genome (10,11). RpfB is the largest and most complex of the five Rpf proteins encoded by Mtb and the sole to produce delayed reactivation from chronic tuberculosis (12). This enzyme acts by hydrolyzing the major constituent of Mtb cell wall, peptidoglycan (PGN), which is formed by glycan chains of β (1–4)-linked N-acetyl glucosamine (NAG) and N-acetyl muramic acid, cross-linked by short peptide stems. In Mtb, these peptide stems are called DAP-type, as they contain meso-diaminopimelic acid (DAP) residues (13–15). Although structural features of a major portion of RpfB have been described (13), information on key interactions of RpfB with its substrate and key residues for catalysis is still lacking.
Despite a low sequence identity, RpfB catalytic domain exhibits structural similarity to lysozyme, an enzyme whose mechanism has been long debated (16–18). In hen egg white lysozyme (HEWL), two residues have been found to be critical to the enzyme activity, Glu-35 and Asp-52. Glu-35 acts as a proton donor to the glycosidic bond, cleaving the C-O bond in the substrate, whereas Asp-52 acts as a nucleophile to generate either ionic intermediate (16) or a covalent intermediate (17,18). In a second step, a water molecule is responsible for attacking either the glycosyl-enzyme intermediate (17,18) or the ionic intermediate (16). The role of the glutamic acid as a proton donor (Glu-292 in RpfB) is shared by all proposed mechanisms of action of lysozyme, whereas the type of intermediate formed and the following water addition step is still debated and likely depends on the lysozyme family.
Because no clues on RpfB catalytic mechanism are hitherto available, we have combined x-ray crystallography with molecular modeling and dynamics to obtain information on the interaction mode of this enzyme with its substrate. Here, we determined the structure of the complex of RpfB catalytic domain and its unliganded form in complex with the lysozyme inhibitor NAG3. This structure delivers the first, to our knowledge, atomic snapshot of the ligand recognition by RpfB. Crystallographic data were also integrated with MD simulations to investigate the dynamic behavior of protein-substrate interactions and to evaluate the role of protein solvation in RpfB action. Finally, the availability of a high resolution model for RpfB catalytic domain provided the opportunity to generate three-dimensional (3D)-models for the catalytic domains of the four Mtb homologs of RpfB, which share significant sequence similarities. These analyses provided clues into analogies and differences among different Mtb Rpfs.
Materials and Methods
Cloning, expression, and purification of proteins
The primers, Rv1009F 5′-CATGCCATGGGAATCGACGGAAGCATCTGGGA-3′, and Rv1009R 5′AGCCGGATCCTCAGCGCGCACCCGCT-3′, containing NcoI and HindIII restriction sites, were used to amplify the rpfB coding sequence starting at residue Ile-280 from H37Rv strain of Mtb. The polymerase chain reaction (∼260 bp) product was cloned into expression vector pETM-11, which gives a protein with a TEV-cleavable N-terminal poly-His tag, denominated RpfB280–362. The resulting positive plasmid was used to transform the Escherichia coli BL21(DE3) strain. The overnight culture was used to inculate 1 L of LB media containing 50 μg mL−1 kanamycin, the protein induction was performed when the OD600 value of 0.7 was reached, by the addition of 1 mM IPTG at 22°C. After ∼16 h the cells were harvested and the protein was isolated by sonicating cell pellets resuspended in 20 mL of binding buffer (5 mM imidazole, NaCl 300 mM, 50 mM Tris-HCl, 5% (v/v) glycerol, pH 8.0) in the presence of a protease-inhibitor cocktail (Roche Diagnostic). The lysate was cleared by centrifugation at 18,000 rpm, and the supernatant loaded on a 5 mL Ni-NTA column (Pharmacia), equilibrated with binding buffer. After washing with 10 volumes of binding buffer, a linear gradient of imidazole (5–300 mM) was applied to elute the protein. The fractions containing RpfB280–362 were pooled and dialyzed against 2 L of 50 mM Tris-HCl, 150 mM NaCl, 5% (v/v) glycerol, pH 8.0 with one exchange at 4°C. After removal of His-tag using TEV protease, the cleaved protein was purified by a second Ni-NTA affinity run. A final purification step was carried out using a Superdex 200 16/60 (Pharmacia) (50 mM TrisHCl, 150 mM NaCl, 10% (v/v) glycerol, pH 8.0). All purification steps were carried out at 20°C. The homogeneity of protein was tested by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). The calculated molecular mass by mass spectrometry is 9127 Da. Freshly concentrated protein, 8 mg/mL, was used for crystallization experiments.
Crystallization and data collection
Crystallization trials were performed at 293 K using the hanging drop vapor diffusion method. Preliminary crystallization conditions were setup using a robot station for high throughput crystallization screening (Hamilton STARlet NanoJet 8+1) and commercially available sparse-matrix kits (Crystal Screen kits I and II, Hampton Research, Index). After manual improvement of crystallization conditions by fine-tuning the protein and precipitant concentrations, only tiny crystals could be obtained. Optimization of the crystallization conditions was performed manually using the Hampton additive screen. Best crystals were obtained using a protein concentration of 6 mg mL−1, 0,5% (w/v) PEG8000, 1 M ammonium sulfate, 0.1 M Hepes, pH 7.5, and 120 mM benzamidine (add. 74, Hampton Research). Indeed, benzamidine significantly improved crystal morphology and x-ray diffraction properties. Crystals of RpfBcat apo-form were obtained after overnight retro soaking in a solution containing 1.2 M ammonium sulfate, 0,1 M Hepes, pH 7.5 to remove benzamidine from the catalytic site. To obtain crystals of complexes with NAG3, a further 3 h soaking was performed in the same solution and in the presence of 20 mM NAG3. NAG3 was purchased from Sigma-Aldrich. Diffraction data for Apo-RpfB and for RpfB-NAG3 complex were collected in-house at 100 K using a Rigaku Micromax 007 HF generator producing Cu Kα radiation and equipped with a Saturn944 charge-coupled device detector. Cryoprotection of the crystals was achieved by a fast soaking in paraffin oil. The data sets were scaled using the HKL2000 program package (19), Table 1.
Table1.
Data collection and refinement statistics
| RpfB-NAG3 | Apo RpfB | |
|---|---|---|
| A. Data collection | ||
| Space group | P21 | P21 |
| Cell parameters a, b, c (Å); | 42.45, 51.51, 66.71 | 42.37, 51.40, 66.66 |
| β (°) | 104.39 | 104.15 |
| Resolution range (Å) | 30–1.33 | 30–1.25 |
| N. of unique reflections | 64107 | 74037 |
| Average redundancy | 3.5 (3.0) | 5.0 (4.5) |
| Rmerge (%) | 6.0 (31.0) | 6.0 (12.6) |
| Completeness (%) | 98.2 (99.0) | 92.6 (75.5) |
| Mean I/σ(I) | 17.0 (3.0) | 51.0 (11.1) |
| B. Refinement | ||
| Rwork/Rwork (F > 4σ) | 16.4 | 17.7 |
| Rfree (%) | 20.5 | 21.9 |
| N. of residues | 320 | 320 |
| N. of waters | 570 | 600 |
| R.m.s. deviations | ||
| Bond lengths (Å) | 0.01 | 0.01 |
| Bond angles (°) | 1.5 | 1.6 |
Values in parentheses are for the highest resolution shells.
Structure refinement
The crystal structure of RpfBcat was solved by molecular replacement using the program Phaser and the structure of RpfBcat in complex with benzamidine (PDB code 4EMN) as a starting model. Crystallographic refinement was carried out against 95% of the measured data using the ccp4i program suite. The remaining 5% of the observed data, which were randomly selected, was used in Rfree calculations to monitor the progress of refinement. The program REFMAC was used for the refinement of the apo form of RpfBcat and its complex with NAG3. Noncrystallographic restraints were applied in REFMAC with medium restraints for main-chain atoms and loose restraints for side-chain atoms. Water molecules were incorporated into the structure in several rounds of successive refinement, using ARP/wARP followed by REFMAC runs (20,21). The pertinent refinement details along with the necessary statistics for the final protein model are given in Table 1. Atomic coordinates of unliganded and NAG3-bound forms have been deposited in the PDB with identification codes 4kl7 and 4kpm, respectively.
MD
MD simulations were performed both on ligand-free and on substrate-bound forms of RpfBcat. The crystallographic structure of the ligand-free form was used as the starting model for the former simulation. The structure of the complex between RpfBcat and NAG3 was used to generate an extended RpfBcat—NAG6 complex, with all six subsites of the protein saturated. This latter complex was used as the starting model in the substrate-bound simulation.
MD simulations were performed with the GROMACS package by using the all-atom AMBER99sb force field in combination with the TIP4P-ew explicit water model. NAG6 was parameterized by using GLYCAM06 (22). The latter was specifically optimized for MD simulations performed with the use of Ewald summation methods, and showed extremely good agreement with experimental data over a range of temperatures. To avoid any bias on the hydration status of the protein derived from the MD analyses, crystallographic water molecules were removed from the starting models. The simulations were carried out in the NPT ensemble with periodic boundary conditions at a constant temperature of 300 K. A rectangular box was used to accommodate the protein/peptide, water molecules, and ions. The systems considered for RpfBcat and RpfBcat—NAG6 included 4311 and 4706 water molecules, respectively.
Hydration analysis: solvent density map
The MD trajectories were analyzed to primary compute solvent density maps whose maxima are assumed to be the MD hydration sites. For each frame of the sampling, positions of water molecules are counted in a grid of 0.5Å after superimposing the current protein structure onto a reference one. To prevent sweeping effects due to backbone flexibility, frames were preliminary selected based on their Cα-root mean-square deviation (RMSD) with the reference structure. In particular, only structures with a Cα-RMSD value lower than 1Å were considered. The maps were stored in EDM format and drawn by means of curve levels.
Hydration analysis: residence time in the hydration sites
The time autocorrelation function P(τ) was adopted to retrieve the residence time of water molecules in the MD hydration sites, using a method previously adopted to unveil the role of collagen hydration in host-pathogen recognition (23–25). The expression is a function of the characteristic time τ and is given by
| (1) |
where the function delta, δ(Ξ(t), Ξ(t + τ)), assumes values of 0 or 1 depending on whether the indexes of the partners at times t and t + τ differ or not. The resulting function is then fitted with an exponential model.
Model building of the 3D-structure of catalytic domains of Mtb RpfB homologs
Model building was performed by using the program Modeller (http://modbase.compbio.ucsf.edu/ModWeb20-html/help.html) (26) and the structure of the free form of RpfBcat as a template. Taking into account the high sequence identity, standard parameters of the modeling procedure were used. The stereochemical quality of the models was evaluated by using the program Procheck (27).
Results
Crystallographic structure of RpfBcat in its ligand-free state
In all RpfB forms hitherto crystallized, the active site of the enzyme is occupied either by inhibitors or by residues belonging to other domains of symmetry-related copies (13,15). Moreover, despite extensive experimental trials using a variety of different conditions, attempts to crystallize the unliganded form of RpfB catalytic domain have been so far unsuccessful. These observations suggest that this domain is intrinsically endowed with a remarkable flexibility that may be important for catalysis. To gain detailed structural information into the active site of the enzyme in its ligand-free state, we designed a specific strategy based on the crystallization of adduct of the domain with benzamidine (15), followed by a retro soaking procedure to remove the ligand. This protocol preserved crystal quality, which diffracted at very high resolution (1.25 Å). Crystals of RpfBcat belong to the P21 space group, with four molecules in the asymmetric unit (Vm 1.96 Å3/Da, solvent content 37.2%). The vast majority of the residues of the resulting model, including those of the active site, are characterized by well-defined electron densities (Fig. 1 A).
Figure 1.
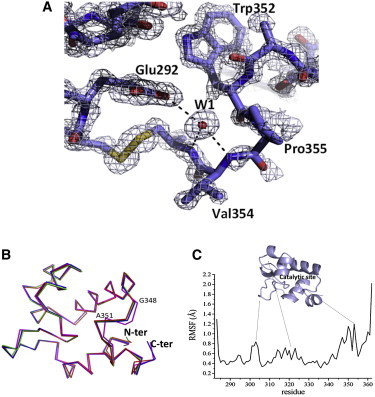
Structure and dynamics of ligand free RpfBcat (A) (2Fo-Fc) electron density map representing RpfB catalytic site. The water site W1 and its interactions with the catalytic site of RpfB are represented. (B) Superposition of the four molecules in the asymmetric unit of free RpfBcat. (C) RMSF values, calculated on backbone Cα atoms in the equilibrated region (10–100 ns) of the MD simulation of free RpfBcat. A cartoon representation of RpfBcat is reported to show the location of flexible loops on the domain structure.
The inspection of the maps corresponding to the active site clearly indicates that the retro soaking was successful and that the structure represents a ligand-free state of the domain (Fig. 1 A). Despite the absence of the ligand, the catalytic residue Glu-292 and the residues that compose the adjacent hydrophobic pocket (Ile-288, Trp-297, Val-309, Phe-311, Ile-337, and Trp-352) assume well-defined conformations. The quality of the model also provides a detailed description of the solvation of the active site. Indeed, a network of water molecules linked to the RpfBcat catalytic site has been identified. Of these, the strongest electron density and lowest B factors characterize the water site W1, which is hydrogen bonded to the side chain of Glu-292 and to the main-chain nitrogen atoms of Val-354 and Cys-355 (Fig. 1 A). Superposition of the four RpfBcat molecules in the asymmetric unit shows a highly conserved structure, with overall backbone RMSD values lower than 0.3 Å. However, a significant structural difference is observed in the D chain for the loop between Gly-348 and Pro-353, with a maximum displacement at Ala-351 (2.1 Å) (Fig. 1 B).
Dynamics of RpfBcat in its ligand-free state
To achieve a comprehensive characterization of the structural and dynamical properties of RpfBcat in a crystal-free context, extensive MD analyses of the domain were also carried out. In particular, an MD simulation of 100 ns was performed using the crystallographic structure of RpfBcat as a starting model. To assess the evolution of the RpfBcat structure in the simulation timescale (100 ns), a number of stereochemical parameters (gyration radius, secondary structure, and RMSD) were monitored along the trajectories. The trends of these parameters indicated that the simulation converged in ∼10 ns (data not shown). The RMSF profile shows that RpfBcat is a compact module that shows a significant flexibility (RMSF larger than 1.0 Å) in loop regions (Fig. 1 C). Consistent with the crystal structure, the loop Gly-348-Pro-353 close to the catalytic site also displays rather high RMSF values (Fig. 1 C), indicating that its flexibility is an intrinsic characteristic of RpfBcat structure and may modulate access to the substrate. In addition, this flexible region close to the catalytic site may be responsible for reluctance to crystallization of free RpfBcat.
The trajectory structures of the MD simulation were also used for hydration analysis aimed at identifying putative water molecules that may play a role in catalysis. The computed solvent density derived from the trajectory structures (see Methods for details) unveils the presence of a number of discrete peaks in the first hydration shell (Fig. 2), whose location is in close agreement with the position of crystallographic water molecules. The strength of water-protein interaction was assessed by using residence times, occupancy, and intensity of the peaks in the hydration map (Fig. 2). As a result, we identified a small set of five water sites with very high residence times (>10-fold the average) and correspond to well-defined crystallographic water molecules. Four of them likely play a structural role as they are either totally or nearly totally buried (Fig. S1 in the Supporting Material). Surprisingly, the fifth (W1) is solvent accessible and lines the active site pocket of the enzyme (Fig. 1 and Fig. S1). As frequently observed for water molecules tightly bound to proteins (28), W1 is strongly anchored to the N-terminus of an RpfBcat helix (the C-terminal one). Specifically, W1 is strongly h-bonded to the backbone nitrogen atoms of Val-354 and Cys-355 and to the side chain of Glu-292, an interaction that is strongly conserved in MD simulations. These considerations suggested to us that W1 may play a crucial role in the enzyme mechanism.
Figure 2.
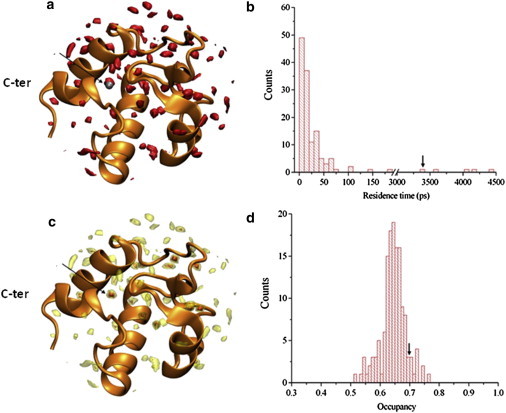
Hydration of RpfBcat studied by MD simulations. (A) Curve level of the solvent density map are shown in red (contour level set as two times the bulk values), the water W1 from x-ray structure is shown in silver. (C) Curve levels of the solvent density map are shown by using contour levels, with darkest curves set as 8 times the bulk solvent. (B) Distribution of the residence times in the first and second hydration layers. The residence time of the water W1 is indicated by an arrow. (D) Distribution of the occupancy factors in the first and second hydration layers. The occupancy of the water W1 is indicated by an arrow.
Crystal structure of RpfBcat in complex with NAG3
With the aim of understanding key interactions of NAG moieties with RpfB, we prepared crystals of RpfBcat in complex with NAG3, an effective competitive inhibitor of lysozyme. Crystals of the complex were generated by using crystals of the ligand-free form (see above) that were subsequently soaked with the sugar. This procedure yielded high resolution diffracting crystals for the complex (1.33 Å). Electron density was clearly visible only for the NAG3 molecule bound to the RpfBcat A chain of the asymmetric unit, whose catalytic cleft displays the largest solvent exposure. The electron density maps of the catalytic site of this chain allowed for a clear definition of NAG3 structure and its interactions with the enzyme (Fig. 3 A). Of the six potential sugar binding sites of RpfB, hypothesized by analogy with lysozyme and designated as −4 to +2 (formerly A, B, C, D, E, and F), NAG3 occupies sites −3, −2, and −1 (Fig. 3 B). Superposition of equivalent A chains of NAG3-bound RpfBcat and free RpfBcat clearly shows that a conformational change involves the loop between Gly-348 and Pro-353. Consistent with the flexibility observed for the loop Gly-348-Pro-353 in both the crystal structure of free Rpfcat and in MD results, this loop opens up the catalytic site upon NAG3 binding (Fig. 3 C).
Figure 3.
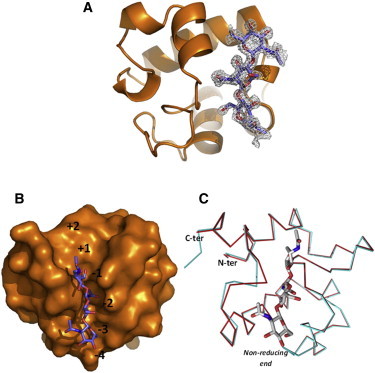
Crystal structure of the complex between RpfBcat and NAG3. (A) Omit Fo-Fc electron density map showing a NAG3 molecule in the enzyme binding site. (B) Location of NAG3 (stick representation) in the B, C, and D sites of RpfBcat binding cleft. RpfBcat is shown in a surface representation. The figure shows the deep cleft that allocates the acetyl moiety of NAG at the C site. (C) Superposition of A chains of NAG3-bound and free RpfBcat. Proteins are drawn in Cα trace, whereas NAG3 is draws in stick representation.
Analysis of interactions of NAG3 shows that the main anchor of the NAG3 molecule are sites −2 and −1, whereas only a few interactions exist with site −3 (Fig. 4). Indeed, five hydrogen bonds link the NAG moiety at the C site with Asp-312, Thr-315, Ala-351, and Gln-347 (Fig. 4). At site −1, NAG moiety forms both hydrogen bonds (to Glu-292 and Gln-310), hydrophobic interactions (with the side chain of Pro-353), and stacking interactions with the side chain of Tyr-305. On the other hand, the NAG moiety at the −3 site forms only hydrogen bonds to the Gln-347 side chain (Fig. 4).
Figure 4.
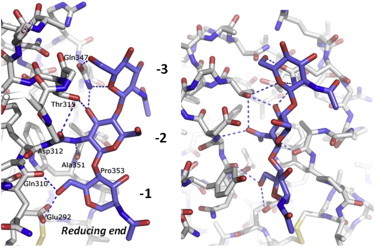
Interactions of NAG3 with RpfBcat. Stick representation of the NAG3-binding site in two perpendicular views (left and right). Interacting residues with NAG at the A, B, and C sites are labeled.
Dynamics of the RpfBcat – NAG6 complex
The crystal structure of RpfBcat in complex with NAG3 showed that the central NAG moiety is the key anchoring point of the molecule, as judged both by electron densities and the number of hydrogen-bonding interactions with RpfBcat. NAG3 conformation was used as a template to model a NAG6 sugar chain, which saturated all six potential binding subsites (−4 to +2) of the enzyme. We performed MD simulations of this model with a twofold goal: i), to achieve a dynamic view of RpfBcat – NAG6 interactions and ii), to evaluate the impact on molecular recognition of all potential sites, including those (−4, +1, and +2) not characterized in the crystallographic study. The analysis of the trajectory structures shows that the RMSD profile reached a plateau value of ∼1.0 Å in ∼10 ns. Therefore, all analysis was performed on the 10–100 ns interval of the simulation. A comparison of the RMSF values of free and NAG6-bound RpfBcat shows that fluctuations are significantly stifled upon NAG6 binding, in particular for the loop Gly-348-Pro-353 close to the catalytic site (Fig. S2).
Analysis of the MD trajectory evidences a set of hydrogen-bonding interactions between NAG6 sugar chain and RpfBcat binding cleft that remain stable throughout the simulation (Fig. 5). These interactions mainly occur at the −2 site and essentially involve the main-chain atoms of Asp-312 and Ala-351 and the side chain of Thr-315. Hydrogen-bonding interactions at the −3 site are more transient and mainly involve the side chains of Gln-347 and Asn-319. This latter residue also forms transient interactions with the NAG moiety at the −4 site (Fig. 5). Notably, hydrogen-bonding interactions with the −1 and +1 sites mainly involve the catalytic Glu-292, which anchors both sugars (Fig. 5). On the other hand, the NAG moiety in the +2 site only occasionally forms hydrogen bonds with the side chain of Asn-303. It is worth noting that the identified set of interactions, which include most of those observed in the crystal structure with NAG3 (Fig. 4 and Fig. S2 B), keep NAG6 tightly bound, as no sliding of the sugar chains is observed in the MD trajectory. These interactions, which underline a strong involvement of NAG moiety at the −2 site and a marginal role of external moieties in protein binding (29), are in line with the RMSF values calculated for NAG6 and with the distribution of φ-ψ angles of glycosidic bonds between subsequent NAG moieties throughout the simulation. Indeed, the lowest RMSF are observed for NAG−2 and highest for NAG+2 (Fig. S2 C). In addition, wider distributions of φ-ψ dihedral angles corresponds to glycosidic bonds involving external sugars, compared to internal ones (Fig. S2 D). In particular, a broader φ distribution characterizes the torsion angle between NAG−4 and NAG−3, whereas the ψ distribution is broader for the torsion angles between NAG+1 and NAG+2 (Fig. S2 D). In addition, analysis of RMSDs computed on ring atoms of each NAG moiety shows that no major distortion of the pyranose rings occurs during the MD simulation (Fig. S3). Similar to the crystal structure of the RpfB-NAG3 complex, all acetamide moieties in the typical trans conformation, except for the NAG group at position −4. For this external sugar, both cis and trans conformations are observed in the MD trajectories, a feature associated with the wider distribution of φ angles characterizing the glycosidic bond between NAG−4 and NAG−3 (Fig. S2 D).
Figure 5.
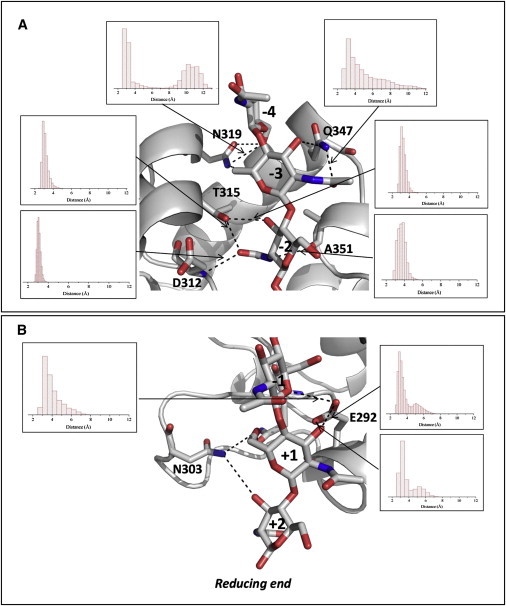
Hydrogen-bonding (including stable and transient) interactions between NAG6 and RpfBcat in a representative structure of the equilibrated region of MD simulation trajectory. Panels A and B describe A–C and D–F NAG moieties, respectively. Insets show histograms of the distribution of hydrogen-bonding distances throughout the simulation.
Structure of Rpf homologs: insights from molecular modeling
RpfB is one of the five resuscitation-promoting factors encoded in Mtb genome. These proteins share a rather similar catalytic domain, which is inserted into a variety of diverse structural contexts in the different members of the Rpf family (Fig. S4 A). The availability of a high-resolution model for RpfBcat provided the opportunity to generate 3D-models for the catalytic domains of the other members of the family.
Pairwise sequence alignments indicate that the catalytic domains share identities in the range of 54–67%, with RpfE and RpfC, RpfA being the closest to RpfB (65% identity) (Table 2). It is worth noting that sequence similarities of the catalytic domains do not correlate with the complexity of the overall proteins. The multiple alignments of these sequences indicate the occurrence of a single significant insertion/deletion region (Fig. S4 B). Indeed, residues 325–327 of RpfB, which are present in RpfA, are missing in the three other members of the family.
Table 2.
Sequence identities (%) of catalytic domains of the five Mtb Rpfs (black)
| Rpf | A | B | C | D | E |
|---|---|---|---|---|---|
| A | – | 65 | 64 | 54 | 66 |
| B | 71 | – | 59 | 56 | 64 |
| C | 73 | 73 | – | 66 | 67 |
| D | 72 | 73 | 73 | – | 61 |
| E | 74 | 74 | 70 | 70 | – |
The number of residues used in each pairwise alignment is reported in gray.
Reliable models of the catalytic domains of the other Rpfs were generated by using the program Modeler (26) and the crystallographic structure of RpfBcat free form as a starting model (see Materials and Methods section for further details). The analysis of the models indicate that the four α-helices (residues 283–294, 312–319, 332–348, and 354–359) that constitute the major secondary structure elements of RpfBcat are well conserved in its homologs. In addition to these elements, RpfBcat also contains two 310 helices at the C-terminus of helix 2 and at the N-terminus of helix 3. The deletion of the region 325–327 in RpfC, RpfD, and RpfE affects the local structure of the region connecting helix 2 and helix 3 (Fig. 6). This variation, however, does not produce any significant perturbation of the overall structure of the domain (Fig. 6).
Figure 6.
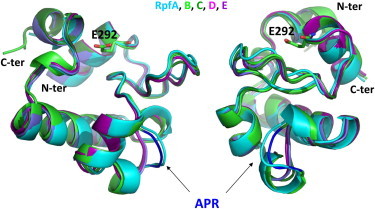
Modeling of RpfBcat homologs in two perpendicular views. Homology models of RpfA, RpfC, RpfD, and RpfE are superposed to the crystal structure of RpfB. The location of the region connecting helix 2 and helix 3 (APR in RpfB), which is deleted in RpfC, RpfD, and RpfE, is indicated by arrows.
An intriguing feature of the RpfBcat domain is the occurrence of a large number of Gly residues in the region 294–321 (nine Gly out of 28 residues). It is important to note that these residues are essential for maintaining the structure of the domains as six of them present positive φ angles, which are rarely assumed by nonglycine residues. The importance of these residues for the Rpf domain structural integrity suggests that they are a fingerprint of these domains.
The analysis of the active site of RpfBcat homologs indicate that Glu-292 (RpfB sequence) is strictly conserved. This residue adopts in all Rpf models the same conformation as in RpfBcat; this conformation also leaves room for docking a water molecule equivalent to W1 in each Rpf active site. Interestingly, the hydrophobic pocket of RpfBcat, made by Ile-288, Trp-297, Val-309, Phe-311, Ile-337, and Trp-352, is well conserved in the other Rpfs. The preservation of this pocket in all Rpfs (A–E) suggests that a hydrophobic environment for Glu-292 is important for the catalytic mechanism of these enzymes. Similar to lysozymes and lytic transglycosylases, this hydrophobic environment likely increases the pKa of Glu-292, thus allowing this residue to engage a hydrogen-bonding interaction with glycosidic oxygen of the scissile bond (30).
Discussion
Mtb resuscitation from dormancy is a complex mechanism, which requires the action of cell wall lytic enzymes as well as of STPK kinases (31). RpfB plays a fundamental role in this process, because it is the sole of the five Rpf of M. tuberculosis to be not dispensable for resuscitation in vivo (12). The study of interaction modes of this key drug target with oligosaccharides represents an important approach for the development of inhibitors of potential therapeutic interest (32,33).
The catalytic site of RpfB structurally resembles lysozyme (13–15). However, different than lysozyme types, no experimental evidence is so far available for the mechanism of action of RpfB. Here, we here report structural investigations, conducted by combining experimental and theoretical approaches, aimed at a), elucidating the determinants of ligand recognition by RpfBcat, and b), gaining insights into the catalytic mechanism of this enzyme. Crystallographic studies and MD simulations carried out on the ligand-free form of the protein provided information on protein dynamics and hydration. These investigations highlighted the presence in the catalytic site of a conserved and highly resident water molecule that mediates the interactions of the side chain of the catalytic Glu-292 with the rest of the protein. Intriguingly, this interaction maintains Glu-292 side chain in a rigid conformation that is similar to that adopted by this residue in the complex with its substrate. Therefore, in the ligand-free enzyme, this water molecule is able to predispose Glu-292 side chain in its active conformation.
Crystallographic and MD simulations on the complexes between RpfBcat and NAG3/NAG6 provided the first representation at atomic level of the key enzyme-carbohydrate interactions. The crystal structure of RpfBcat in complex with NAG3 evidenced that the sugar moieties occupy the subsites from −3 to −1, which establish the strongest interactions with the substrate. MD simulation carried out on the complex between RpfBcat and NAG6 extended the analysis to the other subsites and provided a dynamical view of the recognition process. Overall, these data indicate that most stable interactions involved the side chain of Thr-315 and the backbone atoms of Asp-312 and Ala-351 in the −2 site. Consistently, Thr-315 is strictly conserved in all Mtb homologs of RpfB (Fig. S4) as well as in several RpfB homologs from other bacteria (Fig. S5). Similarly, Ala-351 is strictly conserved, likely because a larger residue would hinder NAG6 binding. Sequence conservation grades mapped onto RpfBcat molecular surface clearly show that whereas residues belonging to subsites from −3 to +1 sites are conserved, a poor conservation characterizes binding sites −4 and +2. In the −4 site, the highest sequence conservation characterizes Gln-347 (Fig. S4). This is consistent with its key contribution to binding, observed both in the crystal structure of the NAG3 complex and in the MD simulations with NAG6 (Fig. 5). Notably, the equivalent conserved glutamine in the lysozyme (Gln-104) is a key residue in oligosaccharide binding (34). Trajectory structures also provided a clear suggestion of the site of substrate cleavage by RpfBcat, so far experimentally unknown. Indeed, the glycosidic bond closest to Glu-292 is that between the sugar moieties in the −1 and the +1 site, both involved in hydrogen-bonding interactions with Glu-292 (Fig. 5). This finding suggests that RpfB cleaves PGN at the glycosidic bond between sites −1 and +1, on analogy with c-type lysozyme (35).
The role of the glutamic acid (Glu-292 in RpfB) in catalysis is shared by all mechanisms of action proposed for lysozymes. Different than HEWL, RpfB has no equivalent of c-type lysozyme Asp-52, which acts as a nucleophile to generate a covalent intermediate (17,18). In RpfB, only one further acidic amino acid, Asp-312, exists in the substrate binding cleft. However, this residue exists only in RpfB and is not conserved in its Mtb homologs (Fig. S4 B). Furthermore, both in the crystal state and in MD models, Asp-312 side chain is far from the C-l carbon of the scissible bond between sugar moieties in the −1 and +1 sites (∼9 Å), too far for covalent stabilization. Altogether, these findings suggest that Glu-292 is the sole residue to be not dispensable for catalysis and that catalysis does not proceed through the formation of a covalent intermediate, but likely through the formation of an oxocarbenium ion.
We also analyzed a possible contribution of solvent molecules to RpfB catalytic mechanism. Water molecules play an important role in biological structure and function, because they can mediate enzymatic catalysis either directly by taking part in the reaction or indirectly through providing a solvation medium for reactants, transition state, and products (36–39). Common to all catalytic mechanisms proposed for members of the lysozyme family (18,40), the second step of lysozyme reaction involves water addition to the accessible face of the reaction intermediate (18,35). As mentioned previously, our hydration analysis evidenced the existence of a water site, W1, which is a constitutive part of the catalytic site (Fig. 1) and is conserved in both the crystal structures of free and NAG3-bound RpfBcat and in MD simulated models. Of importance, W1 becomes totally buried upon substrate ligation, as suggested by MD simulations of the complex between RpfBcat and NAG6 molecule (Fig. S6). In all states of the enzyme, it plays a major role in anchoring Glu-292 in a proper conformation for catalytic function. Indeed, different than in HEWL lysozyme, in which the catalytic Glu-35 is involved in hydrogen-bonding interactions Ala-110 backbone, Glu-292 is not directly anchored to the protein and its conformation is locked solely via interactions mediated by the water site W1 (Fig. 1). Furthermore, W1 is a potential candidate water site for hydrolysis of the enzyme reaction intermediate, because it is in the proper position to be deprotonated by Glu-292 and become the attacking hydroxyl group.
Using the structural information derived from the crystallographic and MD studies, we modeled the large muropeptide (NAG3NAM3Ala6Glu3DAP3) in RpfB binding cleft. In RpfBcat-NAG3 structure and in the RpfBcat-NAG6 model, both the acetyl moiety and the OH group on the adjacent carbon (C3) of NAG moiety at the −2 site form stable hydrogen bonds with Thr-315 (Fig. 5). Therefore, a NAM chain, which would carry a peptide stem attached to the OH group, would not fit (Fig. 5). This observation implies that NAG moieties of PGN occupy pockets −4, −2, +1, whereas NAM moieties occupy sites −3, −1 and +2. Consequent modeling of peptide stems on the NAM moieties at the B, D, and F positions shows that only minimal interactions exist between RpfBcat and the peptide stems of PGN (Fig. S7). Indeed, peptide stems protrude outward to the protein surface, in directions compatible with the currently accepted honeycomb pattern of peptidoglycan polymer (41).
Finally, we used the structure of RpfBcat as a scaffold to model the catalytic domains of its four homologs in Mtb. Whether the existence of five Rpfs (A–E) is the result of full redundancy or not is a debated issue. Indeed, the five Rpfs show differential expression patterns, which vary depending upon the growth phase of the cultures, suggesting that they may serve distinct functions at specific phases of infection (42). Despite their different structural complexities and domain organizations, the five Rpf homologs present the same fold for their catalytic domains (Fig. S4 and Fig. 6). However, significant differences exist among their electrostatic potential surfaces (Fig. S8). The highest discrepancy is observed for the catalytic domain of RpfE, which presents a highly basic pI and a positively charged electrostatic potential with a sole negative patch at the catalytic site. It is worth noting that a negatively charged catalytic site is functional to the mechanism of action of the enzyme, as it likely stabilizes the positively charged reaction intermediate. A more negatively charged electrostatic potential at the catalytic site of RpfB (due to Asp-312) may be associated with a higher activity of this enzyme, but further experiments are required to analyze this issue (Fig. S8). Indeed, RpfB has been shown to rank first among the five homologs, although this result has been ascribed both to its structural complexity and to its ability to interact with the cell division protein RipA (8,43). The functional impact of the distinctive structural features of Rpf (A–E) has not been investigated, but it is tempting to speculate that they may be associated with modulating Rpf function, substrate specificity/selectivity and/or interaction with other proteins.
In conclusion, by adopting a combined experimental-computational approach, we provided the first, to our knowledge, structural information on interactions of RpfB with its substrate analogs. Our results confirm the role of solvation for protein function and structure (24,25,44,45) and provide a set of information that could be exploited in future studies for the development of RpfB inhibitors of therapeutic interest.
Acknowledgments
The authors thank the staff at the European Synchrotron Radiation Facility (ESRF) beamline BM14. We also acknowledge the European Cooperation in Science and Technology (COST) Action BM1003 (COST-grants-BM1003-00772) and Nicole Balasco for help with sequence alignment.
This work has been funded by the Ministero Italiano dell’Istruzione, dell’Università e della Ricerca (PRIN 2009 - prot. 200993WWF9) and by Mizutani Foundation of glycoscience (ref. n. 120012).
Contributor Information
Alfonso De Simone, Email: a.de-simon@imperial.ac.uk.
Rita Berisio, Email: rita.berisio@cnr.it.
Supporting Material
References
- 1.Velayati A.A., Masjedi M.R., Hoffner S.E. Emergence of new forms of totally drug-resistant tuberculosis bacilli: super extensively drug-resistant tuberculosis or totally drug-resistant strains in Iran. Chest. 2009;136:420–425. doi: 10.1378/chest.08-2427. [DOI] [PubMed] [Google Scholar]
- 2.Rowland, K. 2012. Totally drug-resistant TB emerges in India. NATNEWS. http://www.nature.com/news/totally-drug-resistant-tb-emerges-in-india-1.9797.
- 3.Migliori G.B., De Iaco G., Cirillo D.M. First tuberculosis cases in Italy resistant to all tested drugs. Euro. Surveill. 2007;12 doi: 10.2807/esw.12.20.03194-en. E070517.1. [DOI] [PubMed] [Google Scholar]
- 4.Koul A., Arnoult E., Andries K. The challenge of new drug discovery for tuberculosis. Nature. 2011;469:483–490. doi: 10.1038/nature09657. [DOI] [PubMed] [Google Scholar]
- 5.Kaufmann S.H., McMichael A.J. Annulling a dangerous liaison: vaccination strategies against AIDS and tuberculosis. Nat. Med. 2005;11(4, Suppl):S33–S44. doi: 10.1038/nm1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kell D.B., Young M. Bacterial dormancy and culturability: the role of autocrine growth factors. Curr. Opin. Microbiol. 2000;3:238–243. doi: 10.1016/s1369-5274(00)00082-5. [DOI] [PubMed] [Google Scholar]
- 7.Squeglia F., Marchetti R., Silipo A. Chemical basis of peptidoglycan discrimination by PrkC, a key kinase involved in bacterial resuscitation from dormancy. J. Am. Chem. Soc. 2011;133:20676–20679. doi: 10.1021/ja208080r. [DOI] [PubMed] [Google Scholar]
- 8.Ruggiero A., Marasco D., Berisio R. Structure and functional regulation of RipA, a mycobacterial enzyme essential for daughter cell separation. Structure. 2010;18:1184–1190. doi: 10.1016/j.str.2010.06.007. [DOI] [PubMed] [Google Scholar]
- 9.Ruggiero A., Squeglia F., Berisio R. X-ray structural studies of the entire extracellular region of the serine/threonine kinase PrkC from Staphylococcus aureus. Biochem. J. 2011;435:33–41. doi: 10.1042/BJ20101643. [DOI] [PubMed] [Google Scholar]
- 10.Mukamolova G.V., Turapov O.A., Young M. A family of autocrine growth factors in Mycobacterium tuberculosis. Mol. Microbiol. 2002;46:623–635. doi: 10.1046/j.1365-2958.2002.03184.x. [DOI] [PubMed] [Google Scholar]
- 11.Cole S.T., Brosch R., Barrell B.G. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998;393:537–544. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- 12.Tufariello J.M., Mi K., Chan J. Deletion of the Mycobacterium tuberculosis resuscitation-promoting factor Rv1009 gene results in delayed reactivation from chronic tuberculosis. Infect. Immun. 2006;74:2985–2995. doi: 10.1128/IAI.74.5.2985-2995.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ruggiero A., Tizzano B., Berisio R. Crystal structure of the resuscitation-promoting factor (DeltaDUF)RpfB from M. tuberculosis. J. Mol. Biol. 2009;385:153–162. doi: 10.1016/j.jmb.2008.10.042. [DOI] [PubMed] [Google Scholar]
- 14.Cohen-Gonsaud M., Barthe P., Keep N.H. The structure of a resuscitation-promoting factor domain from Mycobacterium tuberculosis shows homology to lysozymes. Nat. Struct. Mol. Biol. 2005;12:270–273. doi: 10.1038/nsmb905. [DOI] [PubMed] [Google Scholar]
- 15.Ruggiero A., Marchant J., Berisio R. Molecular determinants of inactivation of the resuscitation promoting factor B from Mycobacterium tuberculosis. J. Biomol. Struct. Dyn. 2013;31:195–205. doi: 10.1080/07391102.2012.698243. [DOI] [PubMed] [Google Scholar]
- 16.Phillips D.C. The hen egg white lysozyme molecule. Proc. Natl. Acad. Sci. USA. 1967;57:483–495. doi: 10.1098/rspb.1967.0034. [DOI] [PubMed] [Google Scholar]
- 17.Koshland D.E. Stereochemistry and mechanism of enzymatic reactions. Biol. Rev. Camb. Philos. Soc. 1953;28:416–436. [Google Scholar]
- 18.Vocadlo D.J., Davies G.J., Withers S.G. Catalysis by hen egg-white lysozyme proceeds via a covalent intermediate. Nature. 2001;412:835–838. doi: 10.1038/35090602. [DOI] [PubMed] [Google Scholar]
- 19.Otwinowski Z., Minor W. Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 20.Murshudov G.N., Vagin A.A., Dodson E.J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 21.Morris R.J., Perrakis A., Lamzin V.S. ARP/wARP and automatic interpretation of protein electron density maps. Methods Enzymol. 2003;374:229–244. doi: 10.1016/S0076-6879(03)74011-7. [DOI] [PubMed] [Google Scholar]
- 22.Kirschner K.N., Yongye A.B., Woods R.J. GLYCAM06: a generalizable biomolecular force field. Carbohydrates. J. Comput. Chem. 2008;29:622–655. doi: 10.1002/jcc.20820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Berisio R., Vitagliano L. Polyproline and triple helix motifs in host-pathogen recognition. Curr. Protein Pept. Sci. 2012;13:855–865. doi: 10.2174/138920312804871157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vitagliano L., Berisio R., De Simone A. Role of hydration in collagen recognition by bacterial adhesins. Biophys. J. 2011;100:2253–2261. doi: 10.1016/j.bpj.2011.03.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.De Simone A., Vitagliano L., Berisio R. Role of hydration in collagen triple helix stabilization. Biochem. Biophys. Res. Commun. 2008;372:121–125. doi: 10.1016/j.bbrc.2008.04.190. [DOI] [PubMed] [Google Scholar]
- 26.Eswar N., Eramian D., Sali A. Protein structure modeling with MODELLER. Methods Mol. Biol. 2008;426:145–159. doi: 10.1007/978-1-60327-058-8_8. [DOI] [PubMed] [Google Scholar]
- 27.Laskowski R.A., MacArthur M.W., Thornton J.M. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Cryst. 1993;26:283–291. [Google Scholar]
- 28.Harpaz Y., Elmasry N., Henrick K. Direct observation of better hydration at the N-terminus of an alpha-helix with glycine rather than alanine as the N-cap residue. Proc. Natl. Acad. Sci. USA. 1994;91:311–315. doi: 10.1073/pnas.91.1.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Perić-Hassler L., Hansen H.S., Hünenberger P.H. Conformational properties of glucose-based disaccharides investigated using molecular dynamics simulations with local elevation umbrella sampling. Carbohydr. Res. 2010;345:1781–1801. doi: 10.1016/j.carres.2010.05.026. [DOI] [PubMed] [Google Scholar]
- 30.Inoue M., Yamada H., Imoto T. Multiple role of hydrophobicity of tryptophan-108 in chicken lysozyme: structural stability, saccharide binding ability, and abnormal pKa of glutamic acid-35. Biochemistry. 1992;31:5545–5553. doi: 10.1021/bi00139a017. [DOI] [PubMed] [Google Scholar]
- 31.Ruggiero A., De Simone P., Berisio R. Bacterial cell division regulation by Ser/Thr kinases: a structural perspective. Curr. Protein Pept. Sci. 2012;13:756–766. doi: 10.2174/138920312804871201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fernandez-Alonso M.D., Diaz D., Jimenez-Barbero J. Protein-carbohydrate interactions studied by NMR: from molecular recognition to drug design. Curr. Protein Pept. Sci. 2012;13:816–830. doi: 10.2174/138920312804871175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Asensio J.L., Arda A., Jimenez-Barbero J. Carbohydrate-aromatic interactions. Acc. Chem. Res. 2013;46:946–954. doi: 10.1021/ar300024d. [DOI] [PubMed] [Google Scholar]
- 34.Muraki M., Harata K., Sato K.I. Protein-carbohydrate interactions in human lysozyme probed by combining site-directed mutagenesis and affinity labeling. Biochemistry. 2000;39:292–299. doi: 10.1021/bi991402q. [DOI] [PubMed] [Google Scholar]
- 35.Maenaka K., Matsushima M., Kumagai I. Dissection of protein-carbohydrate interactions in mutant hen egg-white lysozyme complexes and their hydrolytic activity. J. Mol. Biol. 1995;247:281–293. doi: 10.1006/jmbi.1994.0139. [DOI] [PubMed] [Google Scholar]
- 36.Sterpone F., Stirnemann G., Laage D. Magnitude and molecular origin of water slowdown next to a protein. J. Am. Chem. Soc. 2012;134:4116–4119. doi: 10.1021/ja3007897. [DOI] [PubMed] [Google Scholar]
- 37.Bianco V., Iskrov S., Franzese G. Understanding the role of hydrogen bonds in water dynamics and protein stability. J. Biol. Phys. 2012;38:27–48. doi: 10.1007/s10867-011-9235-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mazza M.G., Stokely K., Franzese G. More than one dynamic crossover in protein hydration water. Proc. Natl. Acad. Sci. USA. 2011;108:19873–19878. doi: 10.1073/pnas.1104299108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sharma S., Debenedetti P.G. Evaporation rate of water in hydrophobic confinement. Proc. Natl. Acad. Sci. USA. 2012;109:4365–4370. doi: 10.1073/pnas.1116167109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hirakawa H., Ochi A., Kuhara S. Catalytic reaction mechanism of goose egg-white lysozyme by molecular modelling of enzyme-substrate complex. J. Biochem. 2008;144:753–761. doi: 10.1093/jb/mvn133. [DOI] [PubMed] [Google Scholar]
- 41.Meroueh S.O., Bencze K.Z., Mobashery S. Three-dimensional structure of the bacterial cell wall peptidoglycan. Proc. Natl. Acad. Sci. USA. 2006;103:4404–4409. doi: 10.1073/pnas.0510182103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tufariello J.M., Jacobs W.R., Jr., Chan J. Individual Mycobacterium tuberculosis resuscitation-promoting factor homologues are dispensable for growth in vitro and in vivo. Infect. Immun. 2004;72:515–526. doi: 10.1128/IAI.72.1.515-526.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kana B.D., Gordhan B.G., Mizrahi V. The resuscitation-promoting factors of Mycobacterium tuberculosis are required for virulence and resuscitation from dormancy but are collectively dispensable for growth in vitro. Mol. Microbiol. 2008;67:672–684. doi: 10.1111/j.1365-2958.2007.06078.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Venditti V., Bernini A., Niccolai N. MD and NMR studies of alpha-bungarotoxin surface accessibility. Biochem. Biophys. Res. Commun. 2007;356:114–117. doi: 10.1016/j.bbrc.2007.02.094. [DOI] [PubMed] [Google Scholar]
- 45.Biedermann F., Uzunova V.D., De Simone A. Release of high-energy water as an essential driving force for the high-affinity binding of cucurbit[n]urils. J. Am. Chem. Soc. 2012;134:15318–15323. doi: 10.1021/ja303309e. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.