

Abstract
The micro-exon genes (MEG) of Schistosoma mansoni, a parasite responsible for the second most widely spread tropical disease, code for small secreted proteins with sequences unique to the Schistosoma genera. Bioinformatics analyses suggest the soluble domain of the MEG-14 protein will be largely disordered, and using synchrotron radiation circular dichroism spectroscopy, its secondary structure was shown to be essentially completely unfolded in aqueous solution. It does, however, show a strong propensity to fold into more ordered structures under a wide range of conditions. Partial folding was produced by increasing temperature (in a reversible process), contrary to the behavior of most soluble proteins. Furthermore, significant folding was observed in the presence of negatively charged lipids and detergents, but not in zwitterionic or neutral lipids or detergents. Absorption onto a surface followed by dehydration stimulated it to fold into a helical structure, as it did when the aqueous solution was replaced by nonaqueous solvents. Hydration of the dehydrated folded protein was accompanied by complete unfolding. These results support the identification of MEG-14 as a classic intrinsically disordered protein, and open the possibility of its interaction/folding with different partners and factors being related to multifunctional roles and states within the host.
Introduction
Schistosomiasis is the second most widely spread tropical disease worldwide. It affects >200 million people (1) in over 70 countries, especially those in South and Central America and most of the African continent, where 93% of the infected people live (2). One of the species responsible for causing this disease is Schistosoma mansoni (3), a member of the trematode flatworm family, which can live parasitically in a human host for decades without being eliminated by its immune system. It causes severe abdominal pain and chronic infections in liver and intestines that can lead to death. The sequencing of the S. mansoni genome (4) has enabled the identification of new genes that are involved in the host-parasite relationship. An important finding was the novel class of genes, called micro-exon genes (MEG) (5), which are composed of several very small symmetrical exons. A total of 25 different MEG families have been identified (5,6). The MEG proteins are small molecules (6–20 kDa) that exhibit high variability in their primary structures, and have no homology with any other known families of proteins in any organism yet sequenced outside the Schistosoma genera (5). Due to the variability of the proteins, it was hypothesized that MEGs might be involved in an escape mechanism from the human host defenses.
In this study, MEG-14, a member of this group of proteins, was studied to gain insight into its native conformation and molecular interactions. Its sequence includes a high proportion of charged and hydrophilic residues, as well as a significant number of prolines (Fig. 1 a), suggesting it might have a natively unfolded structure (7,8) as observed in the intrinsically disordered proteins (IDPs). The IDPs tend to lack hydrophobic cores, and hence do not have stable ordered secondary and tertiary structures in solution, but can undergo disorder-to-order transitions upon binding to other molecules (9). As a result, they are often involved in molecular recognition functions (10).
Figure 1.
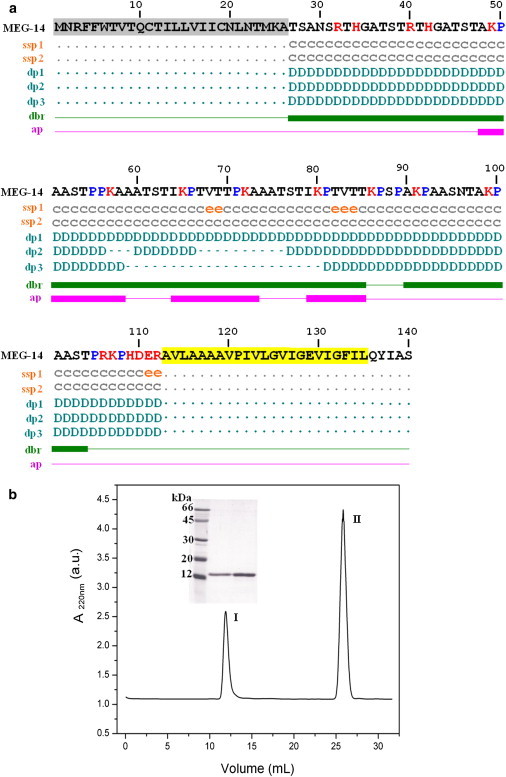
(a) The sequence of MEG-14 (line 1). Residues from the signal peptide and transmembrane tether are highlighted in gray and yellow, respectively. For the soluble domain, the secondary structure predictions with GOR4 (ssp1, line 2) and with PSIPRED (ssp2, line3) have the code c, coil; e, extended strand; the disorder prediction with RONN and Spritz (dp1, line 4), FoldIndex (dp2, line 5) and GlobPlot2 (dp3, line 6) has the code = D, disordered; -; ordered; the disorder binding region prediction (dbr, line 7) is in green; and prediction of antigenic peptides (ap, line 8) is in pink. Proline residues are colored in blue and charged residues in red. (b) Size exclusion chromatography of MEG-14 soluble domain on Superdex-75 column. Peak I represents MEG-14 and peak II the elution of imidazole. Inset: SDS-PAGE of MEG-14.
Synchrotron radiation circular dichroism (SRCD) spectroscopy was employed to investigate the structure of the MEG-14 protein and conformational changes produced by different physical-chemical environments. MEG-14 appears to be an IDP in aqueous solution, but with specific partners and under different conditions, it can fold into forms with well-ordered secondary structures. The variety of different structures that are produced under diverse conditions suggest that this protein, as has been suggested for other IDPS, uses partnering interactions to produce different structures with both different roles within the host, and ones that can effectively evade the immune system surveillance.
Materials and Methods
Materials
The soluble domain of MEG-14 was expressed in Escherichia coli and purified as described below. Chemicals for cloning/expression were purchased from Promega (Madison, WI) unless otherwise stated. Because of the absence of aromatic amino acids in MEG-14 (hence prohibiting the use of the A280 for quantitation), protein concentrations were measured using the BCA method (11).
All reagents and solvents (acetonitrile (ACN), methanol, and trifluoroethanol (TFE)) were analytical grade. Surfactants and phospholipids were purchased from Sigma-Aldrich (United Kingdom) and Avanti Polar Lipids (Alabaster, AL), respectively.
Cloning, expression, and purification of the MEG-14 soluble domain
Primers were designed to amplify the gene transcript coding for the MEG-14 soluble domain (residues 27 to 112) of the predicted sequence from the genome XP_002571808.1 (Fig. 1 a). The signal peptide at the N-terminus predicted by SignalP (12) and the C-terminal tether predicted by the TMHMM program (13) were not included in the construct. The amplicon was cloned into the pTZ57RT plasmid (Fermentas) and sequenced. The sequence of the cloned transcript was deposited into GenBank under accession no. KC311556, which corresponds to a previously deposited EST sequence (accession no. EX708308.1). The cloned isoform, which represents the most abundant isoform of this transcript in adult worms (Orcia, D. and DeMarco, R., unpublished data), differs from the sequence predicted from the deposited gene in its C-terminal 23 amino acids (residues 89 to 112). The cDNA was subcloned into pET-28a (Novagen) expression vector to produce a fusion protein with a hexahistidine tag at the N-terminus. This was used to transform E. coli BL21 (DE3) cells for expression. Bacteria were grown at 37°C in lysogeny broth (LB) medium to an O.D.600nm of 0.6 and then induced at 37°C with 0.5 mM isopropylthio-β-galactoside (IPTG). After 4 h cells were harvested and lysed in a 50 mM Tris-HCl (pH 8.0) buffer, containing 100 mM NaCl. The protein was purified by affinity chromatography using a nickel-NTA column. The hexahistidine tag was removed by an overnight digestion with bovine thrombin (1U/250 μg MEG-14). Thrombin was removed by passing the solution through a benzamidine column. The flow-through was applied to a nickel-NTA column to remove the hexahistidine tag from the solution. The soluble domain of MEG-14 was further purified by size exclusion chromatography on a Superdex 75 10/300 in 20 mM sodium phosphate, pH 7.0. The results of the purification are shown in Fig. 1 b. Direct infusion mass spectrometry analyses of the purified fraction of MEG-14 (60 μM) were performed on a micrOTOF-Q II ESI-TOF (Bruker Daltonics) in 50 mM ammonium acetate (pH 7.0).
Predictions of secondary structure, disorder, disorder binding regions, and antigenic peptides in MEG-14
The sequence-based predictions of the secondary structure were performed using the Expasy server (14) with GOR4 (15) and PSIPRED (16) algorithms. The prediction of disordered segments used the GlobPlot2 (17), RONN (18), Spritz (19), and FoldIndex (20) methods. The potential binding regions of MEG-14 were predicted with ANCHOR (21) and the search for antigenic peptides was performed using the method of Kolaskar and Tongaonkar (22).
SRCD spectroscopy and analyses
The SRCD spectra of MEG-14 (0.1 mM) were obtained at the CD1 beamline located at the ISA synchrotron (University of Aarhus, Denmark). Similar results were also obtained at the DISCO (Soleil Synchrotron, France) and the CD12 (ANKA Synchtrotron, Germany) beamlines. Three scans were collected over the wavelength range from 280 to 170 nm with 1 nm step size, 2 s dwell time, at 25°C in a 0.009 cm pathlength demountable Suprasil quartz cell (Hellma Ltd, UK); the baseline for each sample consisted of the buffer and all components present in the sample other than the protein. The spectra were processed using CDTools software (23); this included averaging, baseline-subtracting, smoothing with a Savitsky-Golay filter, and calibration against a spectrum of a camphorsulfonic acid standard taken at the beginning of the data collection. Final processed SRCD spectra were expressed in delta epsilon units, using a mean residue weight of 99. The CONTINLL algorithm (24) with database 6 (25) in the DichroWeb analysis server (26) was used to calculate the secondary structure content. The normalized root mean-square deviation (NRMSD) goodness-of-fit parameter (26) was calculated for each analysis. As all NRMSD values were <0.1, this suggests the calculated spectra correspond well with the experimental data.
Thermal stability measurements
To assess the effect of temperature on the conformation of MEG-14 (0.1 mM) in aqueous solution, SRCD spectra were taken over the temperature range from 5 to 85°C, at 10°C intervals, with an equilibration time of 5 min at each step. Three repeat spectra were obtained at each temperature, and the first and last spectra were compared to show that the protein had reached its equilibrium folded state before the measurement at that temperature. Data were processed as described previously. After heating to 85°C, the sample was cooled back to 25°C and an additional spectrum was measured to assess whether the process was reversible. The magnitudes of the 184, 198, and 222 nm peaks were plotted and a linear fit was produced in Origin 8.5.
Effect of solvents, pH, and metal ions
MEG-14 (0.1 mM) was incubated in solutions (from 0% to 100% vol/vol) of aqueous/TFE. Similarly, samples in aqueous methanol (MeOH) and ACN were examined over a range of 0–50% solvent.
To investigate the effect of pH, protein (0.01 mM) was incubated over the pH range of 2.0–12, using 10 mM buffers of sodium phosphate acid (pH values of 2.0, 2.5, 3.0, and 3.5), sodium acetate (pHs of 4.0, 4.5, 5.0, and 5.5), sodium phosphate (6.0, 6.5, 7.0, 7.5, 8.0), sodium borate (8.5, 9.0, 9.5, 10, 10.5), and sodium dibasic phosphate (11, 11.5, 12), and the spectrum was obtained for each condition.
The effect of divalent ions was examined by incubating MEG-14 with solutions ranging from 1 to 10 mM CaCl2 or ZnCl2.
Effects of detergents and lipids
Detergent micelles of cetyltrimethylammonium bromide (CTAB), sodium dodecyl sulfate (SDS), and N-hexadecyl-N,N-dimethyl-3-ammonio-1-propanesulfonate (HPS) were prepared in 10 mM sodium phosphate buffer and incubated with MEG-14 at protein/detergent molar ratios of 1:25, 1:50, 1:100, 1:200, and 1:500.
Large unilamellar vesicles (LUVs) composed of 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC); 1,2-dimyiristoyl-sn-glycero-3-phosphocholine (DMPC), 1-palmitoyl-2-oleoyl-sn-glicero-3-phosphocholine (POPC), 1,2-distearoyl-sn-glycero-3- phosphoethanolamine (DSPE), 1-palmitoyl-2-oleoyl-sn-glycerol-3-phosphoethanolamine (POPE), 1,2-dipalmitoyl-sn-glycero-3-[phospho-L-serine] (DPPS), N-[1-(2,3-dioleoyloxy)propyl]-N,N,N-trimethylammonium methyl-sulfate (DOTAP); and 1-palmitoyl-2-oleoyl-glycero-3-phospho-rac-glycerol (POPG) were prepared as described in Lopes et al. (27) in sodium phosphate buffer (pH 7.0). MEG-14 (0.01 mM) was added to each of these samples at a 1:100 protein/lipid ratio. In addition, the POPG liposomes were incubated with MEG-14 at different ratios, over the range from 1:50 to 1:1000.
Oriented-SRCD spectroscopy (o-SRCD)
A dried film of MEG-14 was formed on the surface of a Suprasil quartz circular plate by the deposition of an aqueous solution of the protein (2 mg/mL, 10 μL), followed by evaporation of the solvent. Cells were mounted in a specially designed cell chamber (28), which would later allow rehydration in situ. o-SRCD spectra were measured over the wavelength range from 280 to 160 nm, in 1 nm increments, at 25°C. To determine whether the dried samples were oriented along the direction perpendicular to the beam in such a way that they would display linear dichroism (they did not), measurements were taken at six different rotation angles on the plate (incremented by 60 deg). The final spectrum reported (in mdeg units) is an average of all the scans, after the subtraction of the SRCD signal of the plate. For the secondary structure calculations, because the amount of material in the beam could not be quantified, the goodness-of-fit scaling minimization procedure was used to determine the effective concentration of the dried samples (29). To observe the process of rehydration, 20 μL of water was added to the cell chamber, which was then sealed. The water did not come in contact with the film, but the water vapor slowly rehydrated the protein in the film, which was monitored as a function of time.
To check if the surface property of the material on which the film was deposited had an effect on the protein structure, films were also formed on CaF2 plates (28) and treated in the same way described previously. In addition, films were formed on the Suprasil quartz plate from suspensions of MEG-14 and liposomes composed of either POPG or POPC.
Results and Discussion
Structure predictions
The primary structure of the soluble domain of MEG-14 is composed of a high percentage of polar (58%), charged (21%), and proline (14%) residues (Fig. 1 a). Moreover, it contains no aromatic resides and a low content (4.5%) of Val and Ile, the most hydrophobic residues present. This type of amino acid composition suggests that MEG-14 should have an extended rather than compact globular conformation due to the potential for interactions between the polar residues and water molecules, and the lack of residues suitable for producing a stable hydrophobic core. This feature of MEG-14 is shared with the IDPs (7,8,30–33), which generally have a high relative content of charged and polar residues and a low content of hydrophobic residues. The high proline content in MEG-14 also limits the number of backbone hydrogen bonds necessary for forming regular secondary structures.
Indeed, sequence-based secondary structure predictions do not identify any regular secondary structure for between 92% and 100% of the protein (depending on the method used), and there is no predicted helix content anywhere in the protein (Fig. 1 a). Several types of disorder predictions suggested high probabilities for MEG-14 to be a fully disordered protein (Fig. 1 a). The area with the lowest index of disorder (i.e., least disordered) in MEG-14 is in the region between the Ala-58 and Pro-81, which also corresponds to the segment with the highest probability of forming a disordered binding region.
The soluble domain of MEG-14 includes two potential disordered binding regions; together these almost completely cover the protein sequence (86% of the residues). These regions extend from residues 27 to 85 and from 90 to 104. These regions are predicted to undergo disorder-to-order transitions upon binding to partners. The MEG-14 sequence also contains three segments that are likely to be antigenic in eliciting an antibody response. They are found in segments that include residues 48 to 58, 64 to 73, and 79 to 85. All these segments also correspond to the regions predicted to refold upon binding to a partner molecule; hence, it may be that in situ they are not in an antigenically accessible conformation.
Solution behavior of MEG-14
The molecular mass of the recombinant MEG-14 was determined to be 10,613 ± 7 Da by mass spectrometry, which corresponds to the monomer state of the protein (expected as 10,612 Da), whereas SDS-PAGE analyses gave an apparent molecular mass of 13.7 kDa (Fig. 1 b). As observed for other IDPs (34,35), the discrepancies between the sequence and hydrodynamic methods are a consequence of the extended (not globular) conformation of MEG-14 in solution and its high content of hydrophilic amino acids, which reduce its mobility on polyacrylamide gels because it binds less SDS.
SRCD spectroscopy was employed in this study, as opposed to conventional CD spectroscopy, for several reasons: it enabled monitoring the low wavelength peaks, which provide more accurate information on proteins that are primarily disordered, because the peaks that are characteristic for unordered or coil structures are found primarily in the range from 180 to 200 nm (36), and because some of the conditions used to examine the conformational behavior such as detergent micelles and liposomes can produce significant absorption/scattering effects that can be ameliorated in the SRCD measurements due to the instrument geometry (36).
The SRCD spectrum of MEG-14 in aqueous solution (Fig. 2) exhibits a strong negative peak between 195 and 198 nm and a small more positive peak between 180 and 185 nm that are characteristic of proteins with a low content of ordered secondary structure (Table 1). Indeed, the calculated amount of unordered secondary structure from these data (∼80%) is consistent with the sequence-based predictions. The small amount of measured canonical secondary structure is primarily sheet or turn, again consistent with the predictions. The SRCD measurements confirm experimentally that MEG-14 should be classified as an IDP.
Figure 2.
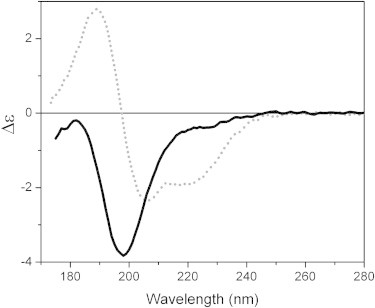
SRCD spectra of MEG-14 in sodium phosphate 50 mM (pH 7.0) (bold, black) and in 100% TFE (dot, gray).
Table 1.
Experimental determination of the secondary structure content of MEG-14
| α-helix | β-strand | Turns | Disordered | NRMSD | |
|---|---|---|---|---|---|
| Aqueous solution | 2 | 10 | 10 | 78 | 0.034 |
| 100% TFE | 26 | 13 | 13 | 48 | 0.054 |
| 50% ACN | 6 | 17 | 15 | 62 | 0.049 |
| 50% MeOH | 7 | 15 | 15 | 63 | 0.042 |
| LUVs of POPG | 6 | 17 | 15 | 62 | 0.044 |
| Micelles of SDS | 8 | 17 | 15 | 60 | 0.034 |
| Dehydrated on plate | 38 | 3 | 0 | 59 | 0.056 |
Values are given as percentages. The protein/detergent and protein/lipid ratios were 1:1000.
Interestingly, even in the presence of TFE (Fig. 2, Table 1), a solvent that tends to induce helical structure in proteins that are not natively helical, the secondary structure of MEG-14 was only slightly helical (26%). TFE has been proposed to (37,38) produce regular secondary structures as a consequence of the stabilizing of intraprotein hydrogen bonds rather than protein-solvent hydrogen bonds. Perhaps a reason for the low helix content of MEG-14 even in TFE is the high number of proline residues, known to be helix breakers. However, the TFE studies suggest that under the appropriate conditions, MEG-14 does have the capacity to fold to at least a partially ordered helical structure, even though the predictions for the native protein are that it will not form any helical structure. To examine if these results were specific for TFE, spectra were obtained by titration into other nonaqueous solvents (methanol and ACN). In both cases, although the extent of ordering was less, decreases in disorder and increases in ordered secondary structures were also observed (Fig. S1 a, Table 1). This may suggest that the ordering is at least in part a consequence of decrease in the water-protein interactions due to dehydration.
The effect of the temperature on the conformation of MEG-14 (Fig. 3 a) was radically different from that generally observed in soluble globular proteins (39,40). Instead of unfolding as the temperature is raised, MEG-14 gained (albeit a small amount of) ordered secondary structure. The changes in the SRCD spectrum (a decrease of the minimum at ∼198 nm, and small increase in the peaks around 184 and 222 nm (more typically associated with helical structures) occurred over the temperature range of 5–85°C (Fig. 3 b). A similar type of behavior has been observed for other IDPs (41,42); it is proposed to result from the increased strength of the hydrophobic interactions at higher temperatures being a major driving force for folding/condensation of the polypeptide chain. For MEG-14 this process was reversible, with the 25°C spectrum obtained at the end of the process matching that of the initial 25°C spectrum.
Figure 3.
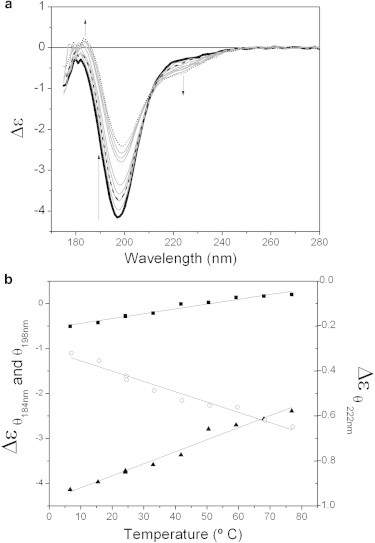
(a) Thermal melt series for MEG-14 in aqueous solution from 5°C (bold) to 85°C (dotted), in 10°C steps (intermediate temperatures in gray, the arrows indicate the direction of the spectral changes). The spectrum taken at 25°C after heating is the dashed line. (b) Plots of the magnitudes of the peaks at 184 nm (square), 198 nm (triangle), and 222 nm (circle) during the heating process.
Although solvation and temperature appear to have effects on the conformation of MEG-14, only very small spectral changes were observed in aqueous solutions of different pHs in the range from 2 to 12 (Fig. S1 b in the Supporting Material), and no changes were detected in the presence of the divalent ions of CaCl2 and ZnCl2. (Fig. S1 c).
Specific interactions with detergents and lipids
To examine whether specific binding of MEG-14 to potential partner molecules could influence its structure, protein was incubated in the presence of detergent micelles and phospholipid liposomes with different headgroup compositions. No changes in the SRCD spectra of MEG-14 were observed either in the presence of the zwitterionic micelles composed of HPS or in the positively charged CTAB (Fig. 4, a, b). However, incubation of MEG-14 with the negatively charged detergent SDS promoted conformational changes, yielding protein with a partially folded conformation (Fig. 4, c).
Figure 4.
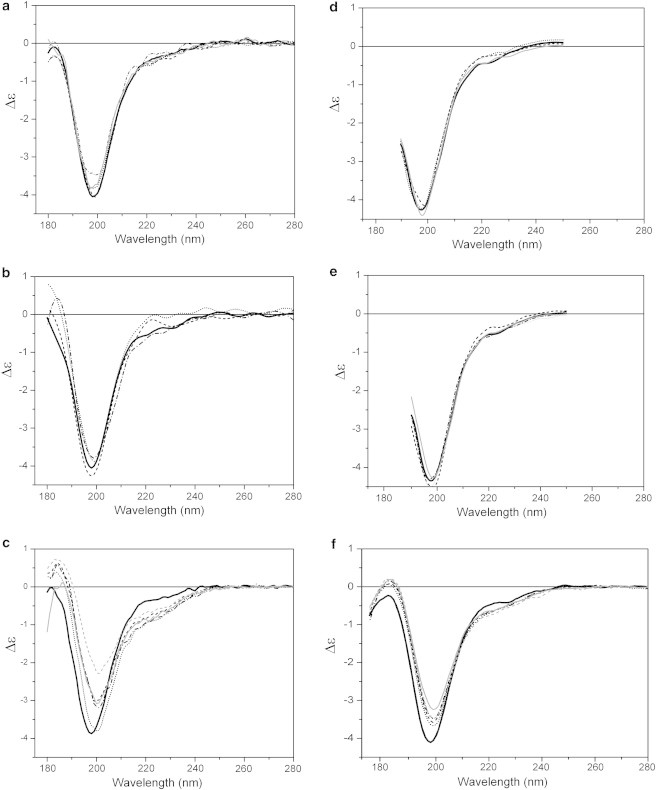
SRCD spectra of MEG-14 in aqueous solution (bold) and in the presence of different detergents and lipids. (a–c) MEG-14 in the presence of micelles: (a) zwitterionic HPS at mole ratios of 1:25 (dot), 1:100 (dash), 1:250 (dot dash), 1:500 (gray); (b) cationic CTAB at 1:25 (dot), 1:50 (dash), 1:250 (dot dash); (c) anionic SDS: 1:25 (dot), 1:100 (dash), 1:250 (dot dash), 1:500 (gray), 1:750 (dashed, gray) protein/detergent molar ratios. (d–f) MEG-14 in the presence of the liposomes: (d) from the PC series: DPPC (dot), DMPC (dash), and with POPC (gray); e) zwitterionic: DSPE (dot), POPE (dash), DPPS (dot dash), and positively charged DOTAP (gray); (f) negatively charged POPG at 1:50 (dot), 1:100 (dash), 1:200 (gray), 1:400 (dot dash), 1:1000 (dash, gray) protein/lipid molar ratios.
Similar behavior was noted following incubation with liposomes composed of phospholipids with different headgroups. None of the zwitterionic or positively charged liposomes changed the conformation of MEG-14 (Fig. 4, d and e). However, incubation with negatively charged liposomes formed from POPG promoted an increase of the intensity of the peak around 222 nm and concomitant decrease in the minimum at 198 nm as the protein/lipid ratio decreased, reflecting the acquisition of partial ordered conformation) (Fig. 4 f). In addition, changing the length or nature of the lipid fatty acid chains had no effect on the conformation (Fig. 4 d).
The ordering of MEG-14 in the negatively charged detergents and lipids was most likely influenced by electrostatic attraction of the 13 positively charged Lys and Arg residues present.
Dehydration/rehydration effects on folding
MEG-14 was deposited as dehydrated films on both quartz silica and CaF2 plates, enabling spectroscopic measurements in the dehydrated state. The two types of plates have very different surface properties (the former hydrophilic and the latter hydrophobic due to the crystal plane exposed on the surface (28)). In both cases following dehydration and film formation (Fig. 5 a), MEG-14 was shown to have refolded to structure with considerable helical content (similar, but even more helical than that found in TFE solution). This suggests that for this IDP, the process of complete desolvation/dehydration favors the formation of intramolecular hydrogen bonds, much in the manner of the dehydration in the presence of nonaqueous solvents. This was very different from the behavior seen for films formed by dehydration of soluble proteins, where the dehydration has either no effect on the conformation (43) or tends to unfold the protein due to surface interactions. These results suggest that interactions with other macromolecules in vivo, which produce nonhydrated interfaces or the displacement of water molecules from the protein in the process of anhydrobiosis, may similarly act as a factor in refolding of the MEG-14 protein.
Figure 5.
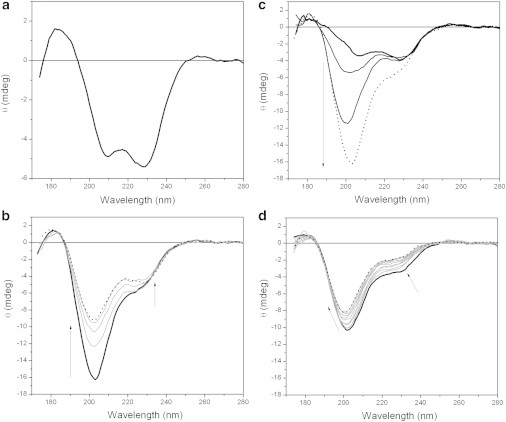
(a) o-SRCD spectrum of dehydrated MEG-14 deposited on a quartz silica plate. (b–d) The process of rehydration was monitored as a function of time. Initially (bold, black), a huge increase of the band at 200 nm region is noted (bold to dash); (c) At a later time the observed reductions of the intensities of the spectral bands are most likely due to the protein dilution in the hydrated cell (as evidenced by the decrease in the HT signal (not shown)); (d) Finally, there is a decrease of the 222 nm minimum and a shift of the 202 peak to 198 nm, indicating the acquisition of a more disordered conformation in the protein.
When the films were slowly rehydrated, huge and gradual conformational changes were observed, with the spectrum changing from that of a helical structure to the type of disordered spectrum originally observed for the protein in solution (Fig. 5, b–d). Hence, the folding/unfolding process is reversible and is a further indication of the role water molecules play in the disordered state of this IDP.
To further investigate the nature of the substrate-protein on the dehydration effect, MEG-14 was dehydrated in the presence of zwitterionic lipids (POPC) (Fig. 6). This resulted in the same type of spectral changes as seen for MEG-14 dehydration/rehydration alone, indicating that this lipid surface did not change the ordering effect due to the removal of water molecules. However, when the dehydration of MEG-14 was undertaken in the presence of negatively charged lipids (POPG), the spectrum remained that of a disordered polypeptide, suggesting that the disorder-to-order transition in MEG-14 can be influenced by the nature of the molecular interactions available during dehydration and is further evidence that either physical or chemical factors of the environment may influence the type of conformation MEG-14 adopts. This conformational plasticity in vitro has been observed for other IDPs (44) and has been suggested to be the basis for induced fit interactions with partners in vivo (10,45). It is exactly the type of behavior that would be expected for a molecule that can adopt many different conformations depending on the availability of binding partners, and could be one way in which it eludes antibody surveillance of a specific conformation.
Figure 6.
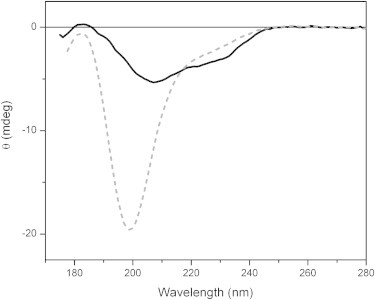
o-SRCD spectra of dehydrated films deposited in the presence of the phospholipids vesicles of POPC (black) and POPG (gray).
Conclusions
Both the predictions and experimental studies on its secondary structure support the classification of the micro-exon gene protein MEG-14 from Schistosoma mansoni as an intrinsically disordered protein. Furthermore, its propensity to adopt different conformations in the presence of different partner molecules or other environmental factors is a characteristic that enables IDPs to be promiscuous in the types of in vivo complexes they can form. Binding of MEG-14 specifically to negatively charged partner molecules observed in this study may be yet another suggestion of the potential for this molecule to undergo complex formation.
The disordered-to-ordered transitions seen here in MEG-14 were a consequence of the modifications of external factors, such as temperature, solvent, charge, and state of hydration. Although, the conditions used here to produce these changes may be different from those in cells, the dynamics of the conformations of MEG-14 seen indicate this type of behavior may be involved in the recruitment of binding partners within a host cell.
Acknowledgments
This work was supported by grants from Conselho Nacional de Densenvolvimento Científico e Tecnológico (CNPq) (INCT-INBEQMeDI), Fundacão de Amparo a Pesquisa do Estado de São Paulo (FAPESP) (to R.D.M., D.O., and J.L.S.L.), the Biotechnology and Biological Sciences Research Council (BBSRC) (to B.A.W.), UK-Brazil Partnering grants from the BBSRC and CNPq (to B.A.W. and A.P.U.A.), and beamtime grants from the ANKA, ISA, and Soleil synchrotrons (to B.A.W. and J.L.S.L.).
Footnotes
This is an Open Access article distributed under the terms of the Creative Commons-Attribution Noncommercial License (http://creativecommons.org/licenses/by-nc/2.0/), which permits unrestricted noncommercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Supporting Material
References
- 1.Emery A.M., Allan F.E., Rollinson D. Schistosomiasis collection at NHM (SCAN) Parasit. Vectors. 2012;5:185. doi: 10.1186/1756-3305-5-185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schistosomiasis. Situation and trends. 2012. http://www.who.int/gho/neglected_diseases/schistosomiasis/en/index.html.
- 3.Gryseels B. Schistosomiasis. Infect. Dis. Clin. North Am. 2012;26:383–397. doi: 10.1016/j.idc.2012.03.004. [DOI] [PubMed] [Google Scholar]
- 4.Berriman M., Haas B.J., El-Sayed N.M. The genome of the blood fluke Schistosoma mansoni. Nature. 2009;460:352–358. doi: 10.1038/nature08160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.DeMarco R., Mathieson W., Wilson R.A. Protein variation in blood-dwelling schistosome worms generated by differential splicing of micro-exon gene transcripts. Genome Res. 2010;20:1112–1121. doi: 10.1101/gr.100099.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Almeida G.T., Amaral M.S., Verjovski-Almeida S. Exploring the Schistosoma mansoni adult male transcriptome using RNA-seq. Exp. Parasitol. 2012;132:22–31. doi: 10.1016/j.exppara.2011.06.010. [DOI] [PubMed] [Google Scholar]
- 7.Uversky V.N., Gillespie J.R., Fink A.L. Why are “natively unfolded” proteins unstructured under physiologic conditions? Proteins. 2000;41:415–427. doi: 10.1002/1097-0134(20001115)41:3<415::aid-prot130>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 8.Campen A., Williams R.M., Dunker A.K. TOP-IDP-scale: a new amino acid scale measuring propensity for intrinsic disorder. Protein Pept. Lett. 2008;15:956–963. doi: 10.2174/092986608785849164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Uversky V.N. Intrinsically disordered proteins from A to Z. Int. J. Biochem. Cell Biol. 2011;43:1090–1103. doi: 10.1016/j.biocel.2011.04.001. [DOI] [PubMed] [Google Scholar]
- 10.Hsu W.L., Oldfield C.J., Dunker A.K. Exploring the binding diversity of intrinsically disordered proteins involved in one-to-many binding. Protein Sci. 2013;22:258–273. doi: 10.1002/pro.2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Smith P.K., Krohn R.I., Klenk D.C. Measurement of protein using bicinchoninic acid. Anal. Biochem. 1985;150:76–85. doi: 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- 12.Petersen T.N., Brunak S., Nielsen H. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat. Methods. 2011;8:785–786. doi: 10.1038/nmeth.1701. [DOI] [PubMed] [Google Scholar]
- 13.Krogh A., Larsson B., Sonnhammer E.L. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J. Mol. Biol. 2001;305:567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- 14.Gasteiger E., Hoogland C., Bairoch A. Protein identification and analysis tools on the ExPASy server. In: Walker J.M., editor. The Proteomics Protocols Handbook. Humana Press; Totowa, NJ: 2005. pp. 571–607. [Google Scholar]
- 15.Garnier J., Gibrat J.F., Robson B. GOR method for predicting protein secondary structure from amino acid sequence. Methods Enzymol. 1996;266:540–553. doi: 10.1016/s0076-6879(96)66034-0. [DOI] [PubMed] [Google Scholar]
- 16.Buchan D.W., Ward S.M., Jones D.T. Protein annotation and modelling servers at University College London. Nucleic Acids Res. 2010;38(Web Server issue):W563–W568. doi: 10.1093/nar/gkq427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Linding R., Russell R.B., Gibson T.J. GlobPlot: exploring protein sequences for globularity and disorder. Nucleic Acids Res. 2003;31:3701–3708. doi: 10.1093/nar/gkg519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang Z.R., Thomson R., Esnouf R.M. RONN: the bio-basis function neural network technique applied to the detection of natively disordered regions in proteins. Bioinformatics. 2005;21:3369–3376. doi: 10.1093/bioinformatics/bti534. [DOI] [PubMed] [Google Scholar]
- 19.Vullo A., Bortolami O., Tosatto S.C. Spritz: a server for the prediction of intrinsically disordered regions in protein sequences using kernel machines. Nucleic Acids Res. 2006;34(Web Server issue):W164–W168. doi: 10.1093/nar/gkl166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Prilusky J., Felder C.E., Sussman J.L. FoldIndex: a simple tool to predict whether a given protein sequence is intrinsically unfolded. Bioinformatics. 2005;21:3435–3438. doi: 10.1093/bioinformatics/bti537. [DOI] [PubMed] [Google Scholar]
- 21.Dosztányi Z., Mészáros B., Simon I. ANCHOR: web server for predicting protein binding regions in disordered proteins. Bioinformatics. 2009;25:2745–2746. doi: 10.1093/bioinformatics/btp518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kolaskar A.S., Tongaonkar P.C. A semi-empirical method for prediction of antigenic determinants on protein antigens. FEBS Lett. 1990;276:172–174. doi: 10.1016/0014-5793(90)80535-q. [DOI] [PubMed] [Google Scholar]
- 23.Lees J.G., Smith B.R., Wallace B.A. CDtool-an integrated software package for circular dichroism spectroscopic data processing, analysis, and archiving. Anal. Biochem. 2004;332:285–289. doi: 10.1016/j.ab.2004.06.002. [DOI] [PubMed] [Google Scholar]
- 24.Sreerama N., Woody R.W. Estimation of protein secondary structure from circular dichroism spectra: comparison of CONTIN, SELCON, and CDSSTR methods with an expanded reference set. Anal. Biochem. 2000;287:252–260. doi: 10.1006/abio.2000.4880. [DOI] [PubMed] [Google Scholar]
- 25.van Stokkum I.H.M., Spoelder H.J.W., Groen F.C. Estimation of protein secondary structure and error analysis from circular dichroism spectra. Anal. Biochem. 1990;191:110–118. doi: 10.1016/0003-2697(90)90396-q. [DOI] [PubMed] [Google Scholar]
- 26.Whitmore L., Wallace B.A. Protein secondary structure analyses from circular dichroism spectroscopy: methods and reference databases. Biopolymers. 2008;89:392–400. doi: 10.1002/bip.20853. [DOI] [PubMed] [Google Scholar]
- 27.Lopes J.L., Nobre T.M., Beltramini L.M. Disruption of Saccharomyces cerevisiae by Plantaricin 149 and investigation of its mechanism of action with biomembrane model systems. Biochim. Biophys. Acta. 2009;1788:2252–2258. doi: 10.1016/j.bbamem.2009.06.026. [DOI] [PubMed] [Google Scholar]
- 28.Wien F., Wallace B.A. Calcium fluoride micro cells for synchrotron radiation circular dichroism spectroscopy. Appl. Spectrosc. 2005;59:1109–1113. doi: 10.1366/0003702055012546. [DOI] [PubMed] [Google Scholar]
- 29.Miles A.J., Whitmore L., Wallace B.A. Spectral magnitude effects on the analyses of secondary structure from circular dichroism spectroscopic data. Protein Sci. 2005;14:368–374. doi: 10.1110/ps.041019905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tompa P. Intrinsically disordered proteins: a 10-year recap. Trends Biochem. Sci. 2012;37:509–516. doi: 10.1016/j.tibs.2012.08.004. [DOI] [PubMed] [Google Scholar]
- 31.Uversky V.N. Intrinsically disordered proteins and novel strategies for drug discovery. Expert Opin. Drug. Discov. 2012;7:475–488. doi: 10.1517/17460441.2012.686489. [DOI] [PubMed] [Google Scholar]
- 32.Dunker A.K., Lawson J.D., Obradovic Z. Intrinsically disordered protein. J. Mol. Graph. Model. 2001;19:26–59. doi: 10.1016/s1093-3263(00)00138-8. [DOI] [PubMed] [Google Scholar]
- 33.Radivojac P., Obradovic Z., Dunker A.K. Protein flexibility and intrinsic disorder. Protein Sci. 2004;13:71–80. doi: 10.1110/ps.03128904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weinreb P.H., Zhen W., Lansbury P.T., Jr. NACP, a protein implicated in Alzheimer’s disease and learning, is natively unfolded. Biochemistry. 1996;35:13709–13715. doi: 10.1021/bi961799n. [DOI] [PubMed] [Google Scholar]
- 35.Chakrabortee S., Meersman F., Tunnacliffe A. Catalytic and chaperone-like functions in an intrinsically disordered protein associated with desiccation tolerance. Proc. Natl. Acad. Sci. USA. 2010;107:16084–16089. doi: 10.1073/pnas.1006276107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wallace B.A. Protein characterization by synchrotron radiation circular dichroism spectroscopy. Q. Rev. Biophys. 2009;42:317–370. doi: 10.1017/S003358351000003X. [DOI] [PubMed] [Google Scholar]
- 37.Luo P., Baldwin R.L. Mechanism of helix induction by trifluoroethanol: a framework for extrapolating the helix-forming properties of peptides from trifluoroethanol/water mixtures back to water. Biochemistry. 1997;36:8413–8421. doi: 10.1021/bi9707133. [DOI] [PubMed] [Google Scholar]
- 38.Cammers-Goodwin A., Allen T.J., Kemp D.S. Mechanism of stabilization of helical conformations of polypeptides by water containing trifluoroethanol. J. Am. Chem. Soc. 1996;118:3082–3090. [Google Scholar]
- 39.Catanzano F., Graziano G., Barone G. Circular dichroism study of ribonuclease A mutants containing the minimal structural requirements for dimerization and swapping. Int. J. Biol. Macromol. 1998;23:277–285. doi: 10.1016/s0141-8130(98)00060-9. [DOI] [PubMed] [Google Scholar]
- 40.Meersman F., Atilgan C., Koch M.H. Consistent picture of the reversible thermal unfolding of hen egg-white lysozyme from experiment and molecular dynamics. Biophys. J. 2010;99:2255–2263. doi: 10.1016/j.bpj.2010.07.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Uversky V.N., Dunker A.K. Understanding protein non-folding. Biochim. Biophys. Acta. 2010;1804:1231–1264. doi: 10.1016/j.bbapap.2010.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Uversky V.N. Intrinsically disordered proteins and their environment: effects of strong denaturants, temperature, pH, counter ions, membranes, binding partners, osmolytes, and macromolecular crowding. Protein J. 2009;28:305–325. doi: 10.1007/s10930-009-9201-4. [DOI] [PubMed] [Google Scholar]
- 43.Ikeda T., Kuroda A. Why does the silica-binding protein “Si-tag” bind strongly to silica surfaces? Implications of conformational adaptation of the intrinsically disordered polypeptide to solid surfaces. Colloids Surf. B Biointerfaces. 2011;86:359–363. doi: 10.1016/j.colsurfb.2011.04.020. [DOI] [PubMed] [Google Scholar]
- 44.Uversky V.N. A protein-chameleon: conformational plasticity of alpha-synuclein, a disordered protein involved in neurodegenerative disorders. J. Biomol. Struct. Dyn. 2003;21:211–234. doi: 10.1080/07391102.2003.10506918. [DOI] [PubMed] [Google Scholar]
- 45.Oldfield C.J., Meng J., Dunker A.K. Flexible nets: disorder and induced fit in the associations of p53 and 14-3-3 with their partners. BMC Genomics. 2008;9(Supp. 1):S1. doi: 10.1186/1471-2164-9-S1-S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.