

Summary
Trafficking of the chitin synthase Chs2p from the endoplasmic reticulum (ER) to the bud-neck in late mitosis is tightly regulated by the cell cycle via phosphorylation of serine residues in the N-terminus of the protein. Here, we describe the effects of Chs2p phosphorylation on the interaction with coat protein complex II (COPII). Identification of a cdc5ts mutant, which fails to transport Chs2p–3xGFP to the bud-neck and instead accumulates the protein in intracellular puncta, led us to discover that Chs2p–3xGFP accumulates at ER exit sites in metaphase-arrested wild-type cells. Using an in vitro ER vesicle formation assay we showed that phosphorylation of Chs2p by the cyclin-dependent kinase CDK1 prevents packaging into COPII vesicles, whereas dephosphorylation of Chs2p by the phosphatase Cdc14p stimulates selection into the vesicles. We found that the cytoplasmic N-terminal domain of Chs2p, which contains the CDK1 phosphorylation sites, interacts with the COPII component Sec24p in a yeast two-hybrid assay and that phosphomimetic substitutions of serines at the CDK1 consensus sites reduces the interaction. Our data suggest that dephosphorylation functions as a molecular switch for regulated ER exit of Chs2p.
Key words: COPII, Secretory pathway, ER, Protein phosphorylation, Cell cycle, Chs2p
Introduction
One of the yeast chitin synthases, Chs2p, synthesizes the primary septum which separates mother and daughter cells at the bud-neck late in mitosis. Chs2p is transported to the bud-neck via the secretory pathway (Chuang and Schekman, 1996; Zhang et al., 2006) but in contrast to other cargoes such as invertase and alpha factor, which are constitutively secreted, localization of Chs2p to the bud-neck is regulated temporally by the cell cycle. Chs2p accumulates in the endoplasmic reticulum (ER) during metaphase when mitotic kinase activity is high, and is transported to the bud neck only upon activation of the mitotic exit network (MEN) (Zhang et al., 2006; Meitinger et al., 2010).
Retention of Chs2p in the ER is dependent on N-terminal phosphorylation sites (Teh et al., 2009). Mutation of serine residues to alanines at four perfect CDK1 sites results in constitutive export of Chs2p out of the ER whereas phosphomimetic serine to glutamate mutations prevent Chs2p from exiting the ER when mitotic kinase activity is absent (Teh et al., 2009). Chs2p is a substrate of CDK1 in vivo (Ubersax et al., 2003) and tandem MS analysis confirms that Chs2p is phosphorylated at all six Ser-Pro sites in the N-terminus (Chin et al., 2012). Recently, it was shown that Chs2p can be dephosphorylated by the phosphatase Cdc14p (Chin et al., 2012), an essential effector of MEN. Chs2p is retained in the ER in a strain carrying the temperature-sensitive allele cdc14-3 and a truncated version of SIC1, sic1-NTdelta, allowing bypass of the mitotic exit defects of the cdc14 allele. This implies that Cdc14p is required for ER exit of Chs2p.
In this paper, we further explore the role of Cdc14p in regulation of Chs2p ER exit. We show that mutation of polo-like kinase Cdc5p, the kinase controlling Cdc14p release from the nucleolus to cytoplasm through MEN, prevents Chs2p trafficking to the bud-neck. We provide evidence that Cdc14p acts by promoting capture of Chs2p into COPII transport vesicles and that Chs2p dephosphorylation stimulates interaction with the COPII component Sec24p. Our data demonstrates that Chs2p phosphorylation/dephosphorylation regulates interaction with the COPII machinery.
Results and Discussion
Chs2–3xGFP trafficking to the bud-neck is compromised in a novel cdc5 mutant
Two yeast chitin synthases, CHS2 and CHS3 are partially overlapping in function such that deletion of the gene for one of them alone is not lethal, whereas deletion of both is synthetically lethal. To identify genes required for Chs2p trafficking through the secretory pathway, we performed a chs3Δ synthetic lethal screen by screening UV-mutagenized chs3Δ CHS3-URA3-CEN CHS2–3xGFP cells for 5-fluoro-orotic acid (5-FOA) sensitivity. 5-FOA-sensitive cells were then screened by microscopy to identify mutants that failed to transport Chs2–3xGFP to the bud-neck. As a result of the screen, we identified mutant strain 5-32. The out-crossed mutant was unable to grow on 5-FOA (Fig. 1A). To complement the mutant, the strain was transformed with a yeast genomic library (ATCC 77162), and transformants were screened for complementation of 5-FOA sensitivity. A single library plasmid was isolated that complemented 5-FOA-sensitive growth and further subcloning analyses showed that CDC5 retained complementing activity (Fig. 1A). Sequencing of the mutant allele revealed a singe base pair substitution, resulting in a glycine to valine substitution at amino acid 92 in the predicted ATP-binding site of the protein (Fig. 1B,C). Because CDC5 is an essential gene, we tested if the cdc5-32 allele identified is temperature sensitive. The mutant strain is able to grow at 24°C and 30°C, but not at 37°C (Fig. 1D). Fluorescence microscopy revealed that the cdc5-32ts mutant failed to transport Chs2–3xGFP to the bud-neck and the enzyme instead accumulated intracellularly (Fig. 2A,B).
Fig. 1.
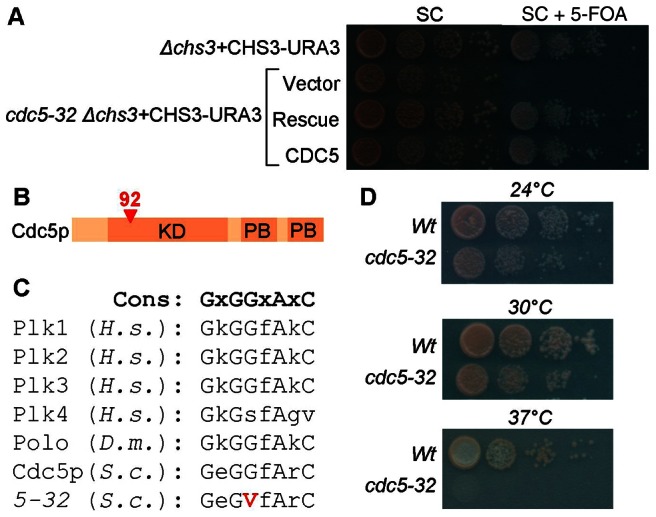
The cdc5-32 mutation is synthetic lethal with chs3Δ. (A) 5-FOA sensitivity of the chs3Δ CHS3-URA3 background strain used in the chs3Δ synthetic lethal screen and of mutant cdc5-32 chs3Δ CHS3-URA3 transformed with the empty library vector (Vector), the rescue LEU2 plasmid from the genomic library containing CDC5 and four other open reading frames (Rescue), or a LEU2 plasmid with the wild-type CDC5 (CDC5). Since 5-FOA selects against the URA3-marked plasmid, the lack of growth of cdc5-32 in the presence of the drug indicates that cell viability is dependent on the wild-type copy of CHS3 present on the URA3 plasmid. Growth of the mutant on 5-FOA is restored by CDC5 on a LEU2 plasmid. (B) Cartoon of Cdc5p showing the kinase domain (KD) and polo-box domains (PB) and position of the mutation (red triangle). (C) ATP-binding sites in selected eukaryotes, with the position of the G to V mutation, encoded by cdc5-32. (D) The cdc5-32 allele is temperature sensitive.
Fig. 2.
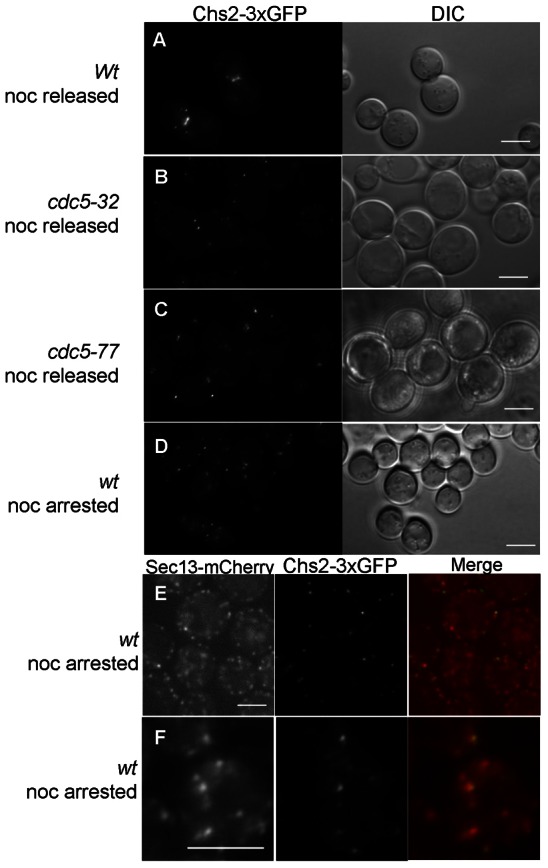
Chs2p accumulates at ER exit sites in metaphase-arrested cells. (A-C) Localization of Chs2–3xGFP 60 minutes after release from nocodazole arrest in (A) wild type, (B) cdc5-32 and (C) a previously characterized mutant allele cdc5-77 with defective Cdc14p release from the nucleolus. (D) Punctate localization of Chs2–3xGFP in nocodazole-arrested wild-type cells. (E) Colocalization of Sec13–mCherry and Chs2–3xGFP in wild-type cells visualized using spinning disk confocal microscopy. (F) Colocalization of Sec13–mCherry and Chs2–3xGFP in wild-type cells following glucose deprivation (Levi et al., 2010). Scale bars: 5 µm.
One of the key functions of Cdc5p in cell division is to promote release of Cdc14p from the nucleolus through the mitotic exit network (Visintin et al., 2003). In light of the role of Cdc14p in Chs2p export from the ER (Chin et al., 2012), we hypothesized that the Chs2p trafficking defects observed by mutation of CDC5 are caused by defects in release of Cdc14p from the nucleolus. To explore this, we took advantage of a cdc5 mutant strain, cdc5-77ts, which carries a single amino acid mutation D263Q in the kinase domain resulting in a diminished MEN-induced nucleolar Cdc14p release (Ratsima et al., 2011). Trafficking of Chs2–3xGFP to the bud-neck is also compromised in cdc5-77ts (Fig. 2C). Thus, it seems likely that the Chs2–3xGFP trafficking defects observed in the cdc5 mutants result from defective Cdc14p release from the nucleolus. This finding supports previous reports on MEN and Cdc14p control of Chs2p localization (Zhang et al., 2006; Meitinger et al., 2010; Chin et al., 2012).
Chs2p accumulates at ER exit sites in metaphase-arrested cells
Chs2p has previously been reported to accumulate in the ER in metaphase and when expressed from a galactose-inducible promoter, Chs2–YFP exhibits a mostly uniform ER localization in nocodazole-arrested cells (Zhang et al., 2006; Teh et al., 2009). In S. cerevisiae, ER exit sites appear as numerous small puncta continuous with the ER when visualized with a fluorescently tagged Sec13p (Rossanese et al., 1999; Castillon et al., 2009; Levi et al., 2010; Shindiapina and Barlowe; 2010). Because of the punctate appearance of Chs2–3xGFP in arrested cells (Fig. 2D), we asked whether these puncta represent Chs2–3xGFP accumulation at ER exit sites. To address this question, we examined the coincident localization of Chs2–3xGFP and Sec13–mCherry using a spinning disk confocal microscope. Following 3 h of nocodazole arrest, 58% of Chs2–3xGFP puncta colocalized with Sec13p–mCherry (n = 60; Fig. 2E). After a brief incubation of the culture in glucose-free medium prior to microscopy, which results in coalescence of ER exit sites (Levi et al., 2010), cells with especially evident Chs2–3xGFP colocalization with Sec13–mCherry were observed (Fig. 2F). Thus, Chs2–3xGFP accumulates at ER exit sites prior to being exported out of the ER, which to our knowledge has not previously been reported for a cargo protein and may be unique to cargo subjected to regulated secretion from the ER.
Packaging of Chs2p in COPII vesicles is stimulated by Chs2p dephosphorylation in vitro
Export of secretory proteins from the ER is mediated by coat protein complex II (COPII) components, which assemble on the ER membrane leading to formation of COPII coated vesicles (Lee et al., 2004). Because ER export of Chs2p is dependent on Cdc14p we examined whether packaging of Chs2p into COPII vesicles at the ER exit sites is regulated by dephosphorylation using an in vitro assay that recapitulates COPII-dependent vesicle budding (Barlowe et al., 1994). Briefly, COPII vesicle budding from the ER was induced by addition of purified COPII proteins (Sar1p, Sec23p/Sec24p and Sec13p/Sec31p), GTP and an ATP regeneration system to ER microsomal membranes purified from a strain expressing 6xMYC-Chs2p from the endogenous promoter. After incubation to allow vesicle formation, membranes were removed by sedimentation at low speed, and budded vesicles were collected by sedimentation at high speed. The packaging of 6xMYC-Chs2p was compared with a positive control protein, Sec22p, a V-snare that cycles constitutively between the ER and Golgi, and a negative control, Sec63p, a resident ER protein.
First, we examined the budding efficiency of phosphorylated Chs2p. Purified ER membranes were treated with purified CDK1 kinase complex (Clb2p-Cdc28p) and then a phosphatase-dead mutant of Cdc14p, GST–Cdc14C283S, was included in the budding reaction. Under these conditions, budding of 6xMYC-Chs2p was not detectable, whereas the positive control protein Sec22p exhibited robust packaging into COPII vesicles (Fig. 3, lane 1). The ER resident protein Sec61p was absent from the vesicles generated, confirming that the incorporation of Sec22p in the budded vesicle fraction was not due to fragmentation of the ER microsomes during the procedure. To examine the effect of Chs2p dephosphorylation on budding efficiency, we treated ER membranes with CDK1, and then included purified GST–Cdc14p in the budding reaction. Following phosphatase treatment, the budding efficiency of 6xMYC-Chs2p was ∼1% (Fig. 3, lane 2). Sec22p budding efficiency was unaffected by addition of Cdc14p (Fig. 3). These results show that COPII-mediated export of Chs2p from ER exit sites is stimulated by Cdc14p-dependent dephosphorylation of Chs2p.
Fig. 3.
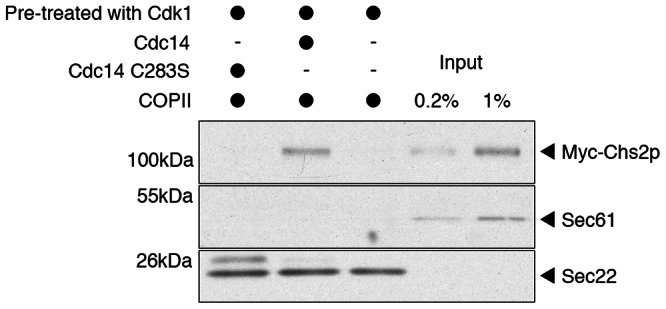
Packaging of Chs2p into COPII vesicles is stimulated by Cdc14p. Microsomal membranes isolated from cells expressing 6xMYC–Chs2p were pretreated with CDK1 and incubated with nucleotides, COPII recombinant proteins and GST–Cdc14C283S (lane 1) or GST–Cdc14p (lane 2) or no Cdc14p (lane 3). After incubation, vesicles were collected by differential centrifugation and analyzed by SDS-PAGE and immunoblotting. For comparison, 0.2% (lane 4) and 1% (lane 5) of total microsome input was loaded.
Dephosphorylation of Chs2p stimulates interaction with Sec24p in vivo
Cargo capture of secretory membrane proteins into COPII vesicles is mediated by the COPII protein Sec24p (Miller et al., 2002). Because dephosphorylation of Chs2p stimulates packaging of the protein in COPII vesicles in our in vitro budding assay, we asked if a dephosphorylation-dependent interaction between Chs2p and Sec24p could be observed in vivo using the yeast two-hybrid assay. The N-terminal cytosolic domain (1–640) of Chs2p containing the CDK1 phosphorylation sites was fused to the Gal4 activation domain and probed for interaction with full-length Sec24p fused to the Gal4 DNA-binding domain. As evident from Fig. 4A, the Chs2p N-terminal domain interacts strongly with Sec24p.
Fig. 4.

Chs2p interaction with Sec24p is modulated by phosphorylation. (A) Yeast two-hybrid analysis. Wild-type Chs2p N-terminus, a mutant without phosphorylation sites (6S-6A) or with phosphomimetic mutations (6S-6E) of the CDK1 phosphorylation sites were fused to the GAL4 activation domain (AD) and probed for interaction with Sec24p fused to the GAL4 binding domain. (B) Pulldown assay with MBP fusion proteins, and quantification of immunoprecipitated Sec24p normalized to MBP–Chs2p signals. (C) Yeast two-hybrid analysis. Wild-type Chs2p N-terminus were fused to GAL4-AD and probed for interaction with Sec24p cargo binding mutants L616W, R342A or W897A fused to the GAL4-BD. (D) Localization of Chs2–3xGFP 60 min after release from nocodazole arrest in cells expressing Sec24-R342A or Sec24-W897A in a sec24Δ background. (E)Yeast two-hybrid analysis using truncated versions of the Chs2p N-terminal fragment and wild-type Sec24p.
Chs2p has six putative CDK1 phosphorylation sites (S14, S60, S69, S86, S100, S133) in the N-terminal region. All six sites are rapidly dephosphorylated by Cdc14p in vitro (Chin et al., 2012). To study the effect of dephosphorylation on interaction of Chs2p with Sec24p, we constructed a phosphomimetic Ser to Glu mutant where the serine residues of the six CDK1 sites in the N-terminal of Chs2p were mutated to glutamates (6S-6E), and likewise constructed a Ser to Ala mutant to remove the phosphorylation sites (6S-6A). Both of these mutant Chs2p peptides interacted with Sec24p in the yeast two-hybrid assay (Fig. 4A). However, compared to the wild-type Chs2p peptide, the strength of the interaction was reduced by introduction of the phosphomimetic Ser to Glu mutations, and conversely, the mutant without phosphorylation sites displayed a stronger interaction with Sec24p than the wild-type Chs2p peptide. Wild-type, 6S-6A and 6S-6E mutant forms of Chs2(1–640) were expressed as maltose binding protein (MBP) fusion proteins and the amounts of Sec24p bound by MBP–Chs2 were quantified. The results of the biochemical assay are in agreement with the results of the yeast two-hybrid experiment. The MBP–Chs2(1–640)_6S-6A fusion protein co-precipitated more Sec24p than the wild type, and Sec24p binding to MBP–Chs2(1–640)_6S-6E was reduced compared to the wild type (Fig. 4B). These results suggest that Chs2p dephosphorylation stimulates the interaction between Chs2p and Sec24p. This represents the first direct evidence that protein phosphorylation negatively regulates the interaction of a membrane cargo protein with the sorting subunit of the COPII machinery.
Three independent sites on Sec24p have been implicated in cargo selection by recognition of distinct ER export motifs (Miller et al., 2003; Mossessova et al., 2003; Mancias and Goldberg, 2007). The A-site binds the sequence YxxxNPF on the Golgi protein Sed5p. The B-site recognizes three different motifs: DxE, LxxLe and LxxME found on Sys1p, Bet1p and Sed5p, respectively. The C-site on Sec24p interacts with the SNARE Sec22p via a conformational epitope so that only the correctly folded Sec22p is captured into COPII vesicles (Mancias and Goldberg, 2007). If one of the known cargo binding sites on Sec24p were to impact Chs2p interaction, this would expand our knowledge on the tolerance of the known sites for the different cargoes. To test this, we carried out the yeast two-hybrid assay using three known Sec24p cargo-binding mutants, L616W, R342A and W897A. No differences in interaction between the Chs2p N-terminal 640 amino acids and Sec24p carrying one of the three mutations could be detected using the yeast two-hybrid assay (Fig. 4C). Consistent with this observation, Chs2–3xGFP trafficking to the bud-neck was unaffected in strains expressing Sec24-R342A or Sec24-W897A in a sec24Δ background (Fig. 4D). These data may suggest that Chs2p is recognized by a hitherto unidentified cargo binding domain on Sec24p.
To define the region of the Chs2p N-terminus required for interaction with Sec24p we made a serial truncation along the Chs2p N-terminal peptide and tested the strength of Sec24p interaction of the truncations in the yeast two-hybrid assay. Truncation of the N-terminus up to amino acid 143 did not impair Sec24p interaction (Fig. 4E). Truncation to amino acid 109 slightly reduced growth of the cells suggesting that the region between amino acids 109 and 143 may be important for Sec24p interaction. Yeast cells carrying constructs with the N-terminal 109, 95 or 76 amino acids of Chs2p displayed similar growth rates, but further truncation to amino acid 47 completely abolished growth under the most stringent conditions (Fig. 4E) suggesting that the main Sec24p interaction site is found in the region between amino acids 47 and 76. This stretch of amino acids overlaps with two of the CDK1 phosphorylation sites (S60 and S69). It would be of interest, in a future structural study, to determine if binding is based on a conformational epitope regulated by phosphorylation.
Our study suggests that Chs2p accumulates at ER exit sites during metaphase, until MEN-induced Cdc14p release to the cytoplasm results in Chs2p dephosphorylation and ER export by COPII. The in vitro budding assay demonstrates that dephosphorylation of Chs2p is required for the protein to be packaged into COPII vesicles. Because Chs2p is phosphorylated in metaphase-arrested cells (Chin et al., 2012) our results imply that although packaging of Chs2p into COPII vesicles is clearly dependent on COPII, concentration of Chs2p at the ERES is independent of Sec24p binding. Using spinning disk confocal microscopy we showed that 58% of Chs2–3xGFP puncta colocalized with Sec13–mCherry in arrested cells, but the remaining 42% of Chs2p puncta did not. We suggest that cargo concentration at the ERES may precede capture into a COPII vesicle. Castillon et al. reported that the GPI-anchored protein Cwp2p becomes concentrated in puncta in sec12-4 and sec16-2 mutants, showing that although Cwp2p is dependent on COPII to exit the ER it does not require the early components of the COPII machinery to be concentrated (Castillon et al., 2009). In mammalian cells, procollagen secretion requires an ER sorting receptor, TANGO1, that is organized in ERES and yet is not captured in COPII vesicles (Saito et al., 2009). The mechanism leading to Chs2p concentration is currently unknown, however identification of the components required for Chs2p concentration may provide important insights into ERES biogenesis.
Materials and Methods
Yeast strains
A chs3Δ CHS2-3xGFP::HIS3::TRP1 CHS3::URA3 strain for the chs3Δ synthetic lethal screen was constructed in yPH499 by integration of a chs3::LYS disruption cassette (Brachmann et al., 1998) followed by transformation with CHS3-URA3-CEN plasmid RSB1608. To make a 3xGFP construct for COOH-terminal tagging of Chs2p, the last 549 bp of the CHS2 coding region except the stop codon were amplified from a plasmid containing the CHS2 ORF by PCR using a forward oligo with an AscI site: 5′-AGGCGCGCCACACTTCCCGCCTTTACGTGTACT-3′ and a reverse oligo with a BamHI site: 5′-TGGATCCGCCCTTTTTGTGGAAAACATTTGG-3′. After PCR amplification, the product was digested with AscI and BamHI and ligated into the 3xGFP plasmid pDD2205 to create pMJ79. This plasmid was digested with Bsu361 which cuts at a natural site in the CHS2 sequence and integrated into the genome of yPH499 by transformation. Transformants were selected on –HIS. Cassettes for chromosomal tagging of SEC13 and SEC63 with mCherry and tdTOMATO, respectively, were constructed by PCR using plasmids pKS390 and pKS392 (Snaith et al., 2005) as templates and the method of Longtine et al. (Longtine et al., 1998). All insertions and deletions were confirmed by PCR. Construction of the 6xMYC-Chs2p strain is described elsewhere (Chuang and Schekman, 1996).
Cell synchronization
To visualize Chs2p–3xGFP, we grew cells in synthetic defined (SD) medium minus uracil plus 4 µg/ml adenine to OD600 ∼0.4 at 24°C. The culture was synchronized with 7.5 µg/ml nocodazole (Sigma-Aldrich, St. Louis, MO) for 1.5 h followed by further addition of 7.5 µg/ml nocodazole for another 1.5 h. To visualize bud-neck staining, we washed cells twice in growth medium and Chs2–3xGFP bud-neck localization was imaged 60 min after release from nocodazole arrest.
Fluorescence microscopy
Cells were imaged on a fluorescence microscope (IX71; Olympus, Melville, NY) equipped with a 100×1.4 numerical aperture objective and a charge-coupled device camera (Orca II; Hamamatsu, Bridgewater, NJ). Spinning disk confocal images were captured using a 100×1.4 NA oil immersion objective on a Leica SD6000 microscope attached to a Yokogawa CSU-X1 spinning disc head with a 561 nm laser for mCherry and 488 nm laser for GFP. All images were acquired with MetaMorph software (Molecular Devices, Sunnyvale, CA) and processed in Photoshop (Adobe Systems, Mountain View, CA).
Cell-free COPII vesicle budding reaction
Budding reactions were carried out as described (Barlowe et al., 1994) using ER membranes purified from a strain expressing 6xMYC-Chs2p (Chuang and Schekman, 1996). GAL-Clb2p-TAP was purified as described (Puig et al., 2001). For phosphorylation, an aliquot (100 µl) of the microsomal suspension was incubated with 0.6 µg/ml CDK1, 1 mM MnCl2 and 1 mM ATP and an ATP regeneration system at 22°C for 15 min prior to the budding reaction. Where indicated, 25 ng of GST–Cdc14p or GST–Cdc14C283 was included in the budding assay.
Yeast two-hybrid assay
The two-hybrid strain pJ69-4A (James et al., 1996) was co-transformed with DNA-binding domain (pGBT9 empty vector or pGBT9-SEC24) and activation domain (pACT2-CHS2) fusion protein expression vectors. Transformants were grown to saturation in SD medium lacking leucine and tryptophan and serial dilutions were spotted onto plates as indicated.
MBP fusion protein pulldown
MBP and MBP–Chs2(1–640) proteins expressed in E. coli were attached to amylose resins (New England Biolab) by incubation at 4°C for 2 h. Next, yeast cell lysate was added to the protein-attached resins and incubated at 25°C for 30 min. Resins were washed and bound proteins were analyzed by SDS-PAGE. Immunoprecipitated Sec24p signals were quantified and normalized to the MBP–Chs2(1–640) signals.
Acknowledgments
We thank Anthony Cormier and David Drubin (Department of Molecular and Cell Biology, UC Berkeley) for the generous gift of purified GST–Cdc14p and GST–Cdc14C283S, and Damien D'Amours for providing strain cdc5-77ts. S.K.L. was a fellow of the Human Frontier Science Program and he and Z.C. are Associates of the Howard Hughes Medical Institute. R.S. is an Investigator of the Howard Hughes Medical Institute and a Senior Fellow of the UC Berkeley Miller Institute.
Footnotes
Author contributions
M.K.J., Z.C., J.K.L. and E.R.J. performed experiments, M.K.J., Z.C., R.M.B. and R.S. planned the experiments and discussed the results, M.K.J. wrote the manuscript.
Funding
This work was supported by the Danish Research Council [grant number 09-062083 to M.K.J.]; the Carlsberg Foundation [grant number 2011_01_0700 to M.K.J.]; the Howard Hughes Medical Institute (to R.S); and the Miller Institute, UC Berkeley, where R.S. is a Senior Fellow. Deposited in PMC for release after 6 months.
References
- Barlowe C., Orci L., Yeung T., Hosobuchi M., Hamamoto S., Salama N., Rexach M. F., Ravazzola M., Amherdt M., Schekman R. (1994). COPII: a membrane coat formed by Sec proteins that drive vesicle budding from the endoplasmic reticulum. Cell 77, 895–907 10.1016/0092-8674(94)90138-4 [DOI] [PubMed] [Google Scholar]
- Brachmann C. B., Davies A., Cost G. J., Caputo E., Li J., Hieter P., Boeke J. D. (1998). Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast 14, 115–132 [DOI] [PubMed] [Google Scholar]
- Castillon G. A., Watanabe R., Taylor M., Schwabe T. M., Riezman H. (2009). Concentration of GPI-anchored proteins upon ER exit in yeast. Traffic 10, 186–200 10.1111/j.1600-0854.2008.00857.x [DOI] [PubMed] [Google Scholar]
- Chin C. F., Bennett A. M., Ma W. K., Hall M. C., Yeong F. M. (2012). Dependence of Chs2 ER export on dephosphorylation by cytoplasmic Cdc14 ensures that septum formation follows mitosis. Mol. Biol. Cell 23, 45–58 10.1091/mbc.E11-05-0434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang J. S., Schekman R. W. (1996). Differential trafficking and timed localization of two chitin synthase proteins, Chs2p and Chs3p. J. Cell Biol. 135, 597–610 10.1083/jcb.135.3.597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- James P., Halladay J., Craig E. A. (1996). Genomic libraries and a host strain designed for highly efficient two-hybrid selection in yeast. Genetics 144, 1425–1436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M. C., Miller E. A., Goldberg J., Orci L., Schekman R. (2004). Bi-directional protein transport between the ER and Golgi. Annu. Rev. Cell Dev. Biol. 20, 87–123 10.1146/annurev.cellbio.20.010403.105307 [DOI] [PubMed] [Google Scholar]
- Levi S. K., Bhattacharyya D., Strack R. L., Austin J. R., 2nd, Glick B. S. (2010). The yeast GRASP Grh1 colocalizes with COPII and is dispensable for organizing the secretory pathway. Traffic 11, 1168–1179 10.1111/j.1600-0854.2010.01089.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longtine M. S., McKenzie A., 3rd, Demarini D. J., Shah N. G., Wach A., Brachat A., Philippsen P., Pringle J. R. (1998). Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14, 953–961 [DOI] [PubMed] [Google Scholar]
- Mancias J. D., Goldberg J. (2007). The transport signal on Sec22 for packaging into COPII-coated vesicles is a conformational epitope. Mol. Cell 26, 403–414 10.1016/j.molcel.2007.03.017 [DOI] [PubMed] [Google Scholar]
- Meitinger F., Petrova B., Lombardi I. M., Bertazzi D. T., Hub B., Zentgraf H., Pereira G. (2010). Targeted localization of Inn1, Cyk3 and Chs2 by the mitotic-exit network regulates cytokinesis in budding yeast. J. Cell Sci. 123, 1851–1861 10.1242/jcs.063891 [DOI] [PubMed] [Google Scholar]
- Miller E., Antonny B., Hamamoto S., Schekman R. (2002). Cargo selection into COPII vesicles is driven by the Sec24p subunit. EMBO J. 21, 6105–6113 10.1093/emboj/cdf605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller E., Beilharz T. H., Malkus P. N., Lee M. C. S., Hamamoto S., Orci L., Schekman R. (2003). Multiple cargo binding sites on the COPII subunit Sec24p ensure capture of diverse membrane proteins into transport vesicles. Cell 114, 497–509 10.1016/S0092-8674(03)00609-3 [DOI] [PubMed] [Google Scholar]
- Mossessova E., Bickford L. C., Goldberg J. (2003). SNARE selectivity of the COPII coat. Cell 114, 483–495 10.1016/S0092-8674(03)00608-1 [DOI] [PubMed] [Google Scholar]
- Puig O., Caspary F., Rigaut G., Rutz B., Bouveret E., Bragado-Nilsson E., Wilm M., Séraphin B. (2001). The tandem affinity purification (TAP) method: a general procedure of protein complex purification. Methods 24, 218–229 10.1006/meth.2001.1183 [DOI] [PubMed] [Google Scholar]
- Ratsima H., Ladouceur A-M., Pascariu M., Sauvé V., Salloum Z., Maddox P. S., D'Amours D. (2011). Independent modulation of the kinase and polo-box activities of Cdc5 protein unravels unique roles in the maintenance of genome stability. Proc. Natl. Acad. Sci. USA 108, E914–E923 10.1073/pnas.1106448108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossanese O. W., Soderholm J., Bevis B. J., Sears I. B., O'Connor J., Williamson E. K., Glick B. S. (1999). Golgi structure correlates with transitional endoplasmic reticulum organization in Pichia pastoris and Saccharomyces cerevisiae. J. Cell Biol. 145, 69–81 10.1083/jcb.153.1.47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito K., Chen M., Bard F., Chen S., Zhou H., Woodley D., Polischuk R., Schekman R., Malhotra V. (2009). TANGO1 facilitates cargo loading at endoplasmic reticulum exit sites. Cell 136, 891–902 10.1016/j.cell.2008.12.025 [DOI] [PubMed] [Google Scholar]
- Shindiapina P., Barlowe C. (2010). Requirements for transitional endoplasmic reticulum site structure and function in Saccharomyces cerevisiae. Mol. Biol. Cell 21, 1530–1545 10.1091/mbc.E09-07-0605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snaith H. A., Samejima I., Sawin K. E. (2005). Multistep and multimode cortical anchoring of tea1p at cell tips in fission yeast. EMBO J. 24, 3690–3699 10.1038/sj.emboj.7600838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teh E. M., Chai C. C., Yeong F. M. (2009). Retention of Chs2p in the ER requires N-terminal CDK1-phosphorylation sites. Cell Cycle 8, 2964–2974 10.4161/cc.8.18.9542 [DOI] [PubMed] [Google Scholar]
- Ubersax J. A., Woodbury E. L., Quang P. N., Paraz M., Blethrow J. D., Shah K., Shokat K. M., Morgan D. O. (2003). Targets of the cyclin-dependent kinase Cdk1. Nature 425, 859–864 10.1038/nature02062 [DOI] [PubMed] [Google Scholar]
- Visintin R., Stegmeier F., Amon A. (2003). The role of the polo kinase Cdc5 in controlling Cdc14 localization. Mol. Biol. Cell 14, 4486–4498 10.1091/mbc.E03-02-0095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G., Kashimshetty R., Ng K. E., Tan H. B., Yeong F. M. (2006). Exit from mitosis triggers Chs2p transport from the endoplasmic reticulum to mother-daughter neck via the secretory pathway in budding yeast. J. Cell Biol. 174, 207–220 10.1083/jcb.200604094 [DOI] [PMC free article] [PubMed] [Google Scholar]