

Summary
Connective tissue growth factor (CTGF) plays an important role in the pathogenesis of chronic fibrotic diseases. However, the mechanism by which paracrine effects of CTGF control the cell fate of neighboring epithelial cells is not known. In this study, we investigated the paracrine effects of CTGF overexpressed in fibroblasts of Col1a2-CTGF transgenic mice on epithelial cells of skin and lung. The skin and lungs of Col1a2-CTGF transgenic mice were examined for phenotypic markers of epithelial activation and differentiation and stimulation of signal transduction pathways. In addition to an expansion of the dermal compartment in Col1a2-CTGF transgenic mice, the epidermis was characterized by focal hyperplasia, and basal cells stained positive for αSMA, Snail, S100A4 and Sox9, indicating that these cells had undergone a change in their genetic program. Activation of phosphorylated p38 and phosphorylated Erk1/2 was observed in the granular and cornified layers of the skin. Lung fibrosis was associated with a marked increase in cells co-expressing epithelial and mesenchymal markers in the lesional and unaffected lung tissue of Col1a2-CTGF mice. In epithelial cells treated with TGFβ, CTGF-specific siRNA-mediated knockdown suppressed Snail, Sox9, S100A4 protein levels and restored E-cadherin levels. Both adenoviral expression of CTGF in epithelial cells and treatment with recombinant CTGF induced EMT-like morphological changes and expression of α-SMA. Our in vivo and in vitro data supports the notion that CTGF expression in mesenchymal cells in the skin and lungs can cause changes in the differentiation program of adjacent epithelial cells. We speculate that these changes might contribute to fibrogenesis.
Key words: CTGF, Epithelial–mesenchymal transition, Dermal fibrosis, Pulmonary fibrosis
Introduction
Organ fibrosis is the outcome of a complex set of cellular interactions initially caused by a pathological injury followed by a process of dysregulated healing (Wynn, 2008). Tissue or organ fibrosis results in increased morbidity and mortality in humans. Hallmarks of fibrosis are the accumulation of fibroblasts and their contractile partners, the myofibroblasts. In addition to resident fibroblasts, perivascular fibroblasts or pericytes, mesenchymal stem cells, and circulating fibrocytes derived from the bone marrow compartment also contribute to the enhanced numbers of fibroblasts and to increased collagen production (Abraham et al., 2007; Krieg et al., 2007). A certain proportion of fibroblast-like cells in fibrotic lesions originate from epithelial cells that have acquired migratory and proliferative characteristics (Krieg et al., 2007). These cells are stimulated by a variety of cytokines, with TGFβ being a key player in fibrosis (Pohlers et al., 2009).
CTGF is a secreted matricellular protein that has been implicated as a regulator of cellular proliferation, angiogenesis and remodeling of the extracellular matrix (Friedrichsen et al., 2003; Ivkovic et al., 2003; Leask and Abraham, 2006). CTGF can act on the same cells that produce it in an autocrine manner elevating extracellular matrix (ECM) synthesis in normal fibroblasts to a level seen in cells of fibrotic lesions (Shi-wen et al., 2000). Our recent mouse model, in which CTGF is overexpressed in mesenchymal cells in vivo, clearly demonstrates that fibrosis of multiple organs can be mediated by CTGF alone independently of TGFβ activation (Sonnylal et al., 2010).
During normal development and during wound healing, epithelial mesenchymal transition (EMT) plays a very dynamic role (Kalluri and Neilson, 2003). During EMT, tightly adjoining epithelial cells resting on a basal lamina with apical–basolateral polarization convert to a non-polarized mesenchymal cell phenotype, detach from the epithelial layer, develop migratory properties and express proteins that are typical of mesenchymal cells. In certain pathological situations, such as in kidney and lung fibrosis, epithelial cells acquire mesenchymal characteristics and contribute to enhanced synthesis of collagen. During EMT, epithelial cells acquire the ability to interact with specific interstitial matrices subsequent to traversing the basement membrane. Generally such cell fate switches of cellular identity and function are important for disease progression.
We have reported that transgenic mice expressing high levels of CTGF specifically in mesenchymal cells (Col1a2-CTGF) manifest hallmarks of sustained multi-organ fibrosis recapitulating key aspects of the fibrotic disease systemic sclerosis (SSc, scleroderma) (Sonnylal et al., 2010). In addition to overt changes in the connective tissue compartments, we observed focal hyperplasia in the epidermal layer of the fibrotic skin and in uninvolved areas of fibrotic lung. We hypothesize that the persistent overexpression of CTGF in mesenchymal cells leads to epithelial cell proliferation and differentiation by a paracrine mechanism.
In the present study, we explored the paracrine effects resulting from the production and secretion of CTGF by mesenchymal cells on neighboring epithelial cells in fibrotic skin and lung of Col1a2-CTGF transgenic mice. We found that some basal keratinocytes in the skin and alveolar epithelial cells in fibrotic foci and normal areas of the lung are morphologically and functionally different from epithelial cells in these tissues in wild-type (wt) mice. Cells that were committed to an epithelial lineage are undergoing a change in their genetic program that is exemplified by expression of EMT marker genes. In the skin these changes are associated with activated MAPK and ERK signaling. In vitro studies in lung epithelial cells showed that the onset of EMT marker gene expression due to exogenous TGFβ can be blocked by CTGF knockdown suggesting that CTGF mediates TGFβ-induced EMT. Furthermore, expression of CTGF in lung epithelial cells or treatment with exogenous CTGF also induced EMT-like changes in vitro. Overall, our results provide new evidence that CTGF causes an EMT-like phenotype in skin and lung in vivo, and that these changes are likely to occur through a paracrine mechanism.
Results
Alteration in keratin and integrin β gene expression in the skin of Col1a2-CTGF transgenic mice
Mice overexpressing CTGF in mesenchymal cells developed dramatic and progressive thickening of the dermis. In addition, we observed increased hyperplasia and hypertrophy of keratinocytes in focal areas of the dermal–epidermal junction. Evidence for hyper-proliferation of epidermal cells was shown by the increase in the multilayer structure of the epidermis of Col1a2-CTGF mice. Previous experiments had also shown a threefold increase in BrdU incorporation in the areas of epidermal hyperplasia (data not shown). These results were confirmed by increased expression of the K16 proliferation marker in the interfollicular epidermis of transgenic mice compared to wt littermate controls (Fig. 1A,B, green), with an increase in the number of positive cells to nearly 10% of the total cell frequency.
Fig. 1.

Abnormal expression of hyperproliferative epidermal markers in Col1a2-CTGF transgenic mice. The expression of keratin 16 (a hyperproliferative epidermal marker), integrin β6, and integrin β4 in the skin of wt and Col1a2-CTGF transgenic mice was examined by immunofluorescence. Increased expression of keratin 16 (green) was observed suprabasally in the epidermis in Col1a2-CTGF transgenic mice (B) compared to wt controls (A). Increased expression of integrin β4 (red) was observed in the hyperproliferative epidermis of Col1a2-CTGF transgenic mice (B) whereas in wt skin integrin β4 expression was restricted to the basal side of the basal layer and seemed reduced (A). Epidermal cells also showed a marked increase in the level of integrin β6 in Col1a2-CTGF transgenic mice (D), whereas no staining was observed in control skin (C). Scale bars: A, B = 100 µm (A,B), 50 µm (C,D). Quantification of the expression of these proteins is shown below. Data are means ± s.d.; n = 6 skin sections. *P<0.0001, **P<0.001, ***P<0.0005, Student's t-test.
Since the epidermis of Col1a2-CTGF mice showed increased hyperplasia, we wanted to determine if the apical–basal polarity was maintained by the basal cells in the epithelium. To address this, we analyzed the expression of integrins. Integrins are transmembrane receptors that mediate cell–cell adhesion and terminal differentiation of keratinocytes. In wt skin the distribution of integrin β4 was, as expected, mainly confined to the basal side of the basal epithelial cells. In contrast, the expression of integrin β4 in the epidermis of Col1a2-CTGF transgenic mice was irregularly distributed all around the hyper-proliferative epithelial cells (Fig. 1A,B, red). Another epithelial specific integrin, αvβ6, that is barely detectable in quiescent epithelial cells is rapidly induced upon epithelial injury and imparts increased migratory properties to these cells (Huang et al., 1996). Interestingly, we observed a marked enhancement in the expression of integrin β6 in the epidermis of Col1a2-CTGF transgenic mice compared to littermate controls (Fig. 1C,D) suggesting that these cells might have increased migratory properties. The redistribution of integrin β4 suggests that the basal cells in the epidermis had lost their apical-basal polarity. Based on the marked increase in integrin β6 expression, we hypothesize that these cells had acquired increased migratory properties. Both changes are likely the result of changes in the genetic program of these cells.
Col1a2-CTGF mice exhibit focal hyperplasia of the epidermis associated with cell fate changes in epithelial cells
The increase in collagen in the dermis of Col1a2-CTGF transgenic mice was associated with increased numbers of myofibroblasts (Sonnylal et al., 2010). Immunostaining of skin for αSMA revealed large numbers of cells positive for αSMA in both the papillary and the reticular dermis of Col1a2-CTGF transgenic mice (Sonnylal et al., 2010). Intriguingly, we have observed that αSMA-positive cells are intermingled with basal cells in the hypertrophic epidermis of Col1a2-CTGF transgenic mice (Fig. 2A,C and corresponding DIC images B,D). We explored this finding further and examined the origin of these cells. Immunostaining for Snai1, a marker for EMT (Kalluri and Neilson, 2003), revealed significant Snai1 levels in areas in which there was focal hyperplasia of the epidermis in Col1a2-CTGF transgenic mice. Intense cytoplasmic staining for Snai1 was seen in the basal cells adjacent to the dermis and clear Snai1 staining primarily confined to the nucleus was seen in the upper layers of the epidermis in Col1a2-CTGF transgenic mice. Snai1 staining was absent in wt controls (Fig. 2E,G and corresponding DIC images F,H). To determine if the epidermal cells of Col1a2-CTGF transgenic mice displayed properties of newly formed mesenchymal cells, immunofluorescence was performed with antibodies against S100A4 or FSP-1, a Ca2+ binding protein closely associated with EMT. We observed that cells in both the basal and suprabasal layer of the Col1a2-CTGF epidermis showed robust staining for S100A4, unlike the skin epidermis of wt mice where staining for S100A4 was absent (Fig. 2I,K and corresponding DIC images J,L). Marked staining for αSMA, Snai1 and S100A4 in the epidermal cells within focal areas of hyperplasia in the epidermis of Col1a2-CTGF transgenic mice strongly suggest that these cells were undergoing an EMT-like process.
Fig. 2.

Changes in the genetic program of the basal cells in the epidermis. Immunofluorescence was performed on skin sections of Col1a2-CTGF transgenic mice and wt littermate controls with antibodies to αSMA, Snai1 and S100A4. De novo expression of αSMA was increased in the epidermis of Col1a2-CTGF fibrotic skin (C; corresponding DIC overlay in D), indicated by white arrows, compared with wt littermate controls (A; corresponding DIC overlay in B). Cells in the basal layer also stained positive for Snai1 (G; corresponding DIC overlay in H) indicated by white arrows, whereas no Snai1 staining was observed in controls (E; corresponding DIC overlay in F). Similarly, S100A4 staining was observed in basal cells of the epidermis of Col1a2-CTGF transgenic mice (K; corresponding DIC overlay in L), and absent in control sections (I; corresponding DIC overlay in J). Arrowheads indicate the cells expressing Snai1 and S100A4 in the dermis. The horizontal dashed line marks the dermal–epidermal junction. Scale bar: 50 µm. Quantification of the expression of these proteins is shown on the right. Data are means ± s.d., n = 6 skin sections. *P<0.005, **P<0.001, ***P<0.001, Student's t-test.
We also observed staining for S100A4, a marker of newly formed fibroblasts, in cells of the papillary dermis suggesting that these cells either arose from resident fibroblasts or originated from epithelial cells by EMT (Fig. 2I,K). Taken together these results strongly suggest that overexpression of CTGF in the dermal compartment is able to induce EMT-like changes in the adjacent epithelial cells of the epidermis most likely by paracrine mechanisms.
Aberrant Sox9 expression in the epidermis of adult Col1a2-CTGF transgenic mice
Sox9, which belongs to the HMG box super-family of DNA binding proteins, is a key transcription factor for chondrocytes and several other lineages (Pritchett et al., 2011; Bi et al., 1999). In the skin, and particularly in the hair follicle, Sox9 has essential roles in the development of the outer root sheath (ORS) and of the stem cell compartment (the bulge) (Vidal et al., 2005). Recent evidence in the literature has implicated Sox9 expression in diseases that affect the extracellular matrix such as in skin keloids (Naitoh et al., 2005), glomerular sclerosis of the kidney (Bennett et al., 2007), and activated stellate cells in the liver (Hanley et al., 2008). We wanted therefore to investigate whether the distribution of Sox9 expression in the skin of Col1a2-CTGF transgenic mice was altered. Using immunofluorescence, we showed abundant expression of nuclear Sox9 in the basal cells of the epidermis in Col1a2-CTGF transgenic mice but not in wt mice (Fig. 3A,B and corresponding DIC images C,D). Increased expression of Sox9 (over threefold) was also observed in the ORS and in the bulge of hair follicles (Fig. 3E,F and corresponding DIC images G,H).
Fig. 3.

Abnormal expression of Sox9 in the epidermis of Col1a2-CTGF transgenic mice. Immunofluorescence of skin sections showed abundant Sox9 expression in the basal cells of the epidermis of Col1a2-CTGF mice (B; corresponding DIC overlay in D) compared to wt controls (A; corresponding DIC overlay in C). Increased Sox9 expression was also observed in the ORS and the bulge of hair follicles in Col1a2-CTGF transgenic mice (F; corresponding DIC overlay in H) compared to wt controls (E, corresponding DIC overlay in G). Autofluorescence in the cornified layer of the skin is observed in both transgenic mice and controls. Scale bar: 50 µm. Quantification of the expression is shown below. Data are means ± s.d., n = 6 skin sections. *P<0.0004, **P<0.002; Student's t-test.
Similar to the abnormal expression of αSMA, Snai1 and S100A4, the anomalous Sox9 expression patterns strongly suggest that overexpression of CTGF in mesenchymal cells results in major cell fate changes in the basal layer of the epidermis.
Multiple signaling pathways are activated in the epidermis of Col1a2-CTGF transgenic mice
We have previously provided evidence that increased expression of CTGF in mesenchymal cells causes constitutive activation of multiple signaling molecules including phosphorylated p38 (p-p38), Erk1/2 (pErk1/2), Akt (pAkt), and PI3K (Sonnylal et al., 2010). Because integrin β6 causes the activation of Erk1/2 and p38 signaling molecules in epithelial cells (Sullivan et al., 2011; Ahmed et al., 2002), and because these molecules are known to stimulate the proliferation of these cells, we examined the skin using routine histology (Fig. 4A,B) and the phosphorylation status of Erk1/2 and p38 in the skin of Col1a2-CTGF transgenic mice and wt littermate controls. These experiments would help us gain insights into the mechanism that could contribute to increased proliferation and dedifferentiation of keratinocytes. Immunofluorescence with antibodies against phosphorylated p38 (p-p38) and ERK1/2 (p-ERK1/2) showed three- to sevenfold increases in the levels of both pErk1/2 (Fig. 4C,D,G) and p-p38 (Fig. 4E,F,G) in the granular and cornified layers of the epidermis. We hypothesize that the increased expression of integrin β6 and the increased activation of p38 and Erk1/2 could account for the hyper-proliferation of keratinocytes in Col1a2-CTGF transgenic mice.
Fig. 4.
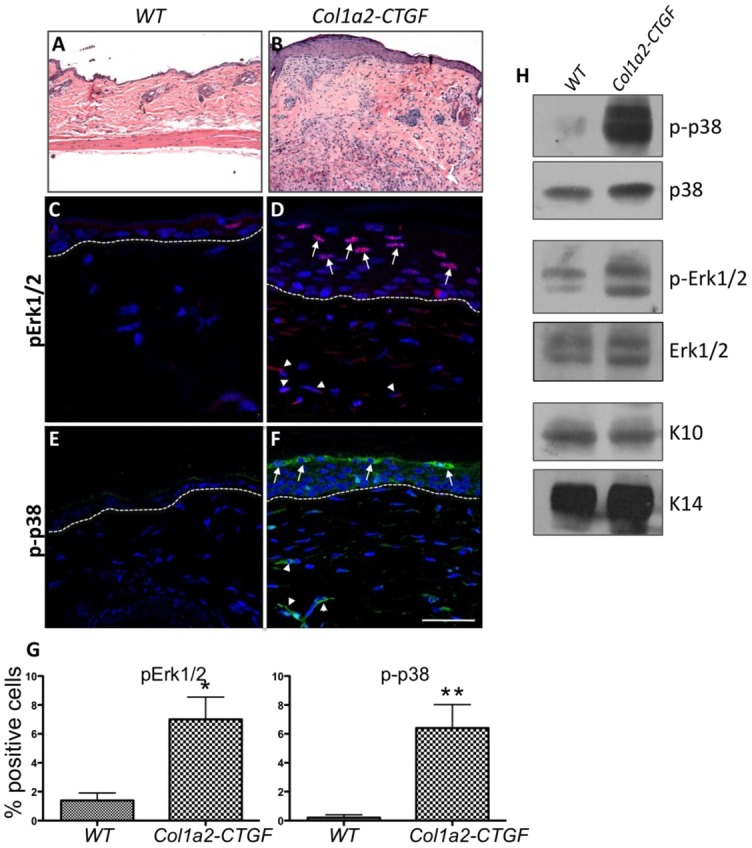
Constitutive activation of MAPK and ERK signaling in the epidermis of Col1a2-CTGF transgenic mice. (A,B) Compared with wt mice, Col1a2-CTGF transgenic mice showed pronounced and progressive dermal fibrosis with focal thickening of the epidermis (Hematoxylin and Eosin staining). (C–F) Skin sections from Col1a2-CTGF transgenic mice and wt littermate controls were stained with antibodies against p-ERK1/2 and p-p38. Increased nuclear localization of p-ERK1/2 and p-p38 was observed in transgenic mice (D,F) compared to wt controls (C,E). Increased p-ERK1/2 and p-p38 was also observed in the dermis (white arrowheads). (H) Western blot analysis of keratinocyte cell extracts from Col1a2-CTGF transgenic mice also revealed increased p-p38 and pERK1/2 compared to control samples. Total p38 and ERK1/2 were unchanged in both samples and keratins 10 and 14 were used as loading controls. (G) Quantification of p-ERK1/2 and p-p38 expression. Data are means ± s.d., n = 6 skin sections. *P<0.01, **P<0.005, Student’s t-test.
Western blot analysis of protein extracts from primary keratinocytes also showed constitutive activation of ERK1/2 and p38 in cells from Col1a2-CTGF transgenic mice (Fig. 4H). Levels of total p38 and ERK1/2 were the same in cells derived from both Col1a2-CTGF and wt mice (Fig. 4H).
Contribution of epithelial cells to fibrotic lung lesions in Col1a2-CTGF transgenic mice
Col1a2-CTGF transgenic mice died early from extensive focal fibrotic lesions within the lung parenchyma. The fibrosis was characteristic with markedly increased Masson’s trichrome staining and increased collagen type I, CTGF expression compared to wt control mice (Sonnylal et al., 2010) (supplementary material Fig. S1A–D). We also observed a three to fourfold increase in BrdU-positive cells in the fibrotic lesions suggesting increased numbers of cells undergoing cell division (data not shown). To determine the proportion of cells that are myofibroblasts and to distinguish them from alveolar epithelial cells we performed co-immunostaining of lung sections from Col1a2-CTGF transgenic mice and wt mice for αSMA and TTF-1, markers for myofibroblasts and alveolar epithelial cells, respectively. The immunofluorescence analysis revealed a marked increase in αSMA-positive cells in the lesional areas of the lungs of Col1a2-CTGF transgenic mice, whereas αSMA-positive cells were lacking in the lungs of wt control mice (Fig. 5A–D,G). A significant proportion of cells in the fibrotic lesions were also positive for TTF-1 (Fig. 5A,C and the corresponding DIC images in B,D) consistent with the BrdU staining suggesting an increased proliferation of epithelial cells within the fibrotic lesions. Most interesting was the observation of cells within the fibrotic foci that were expressing both αSMA and TTF-1 (Fig. 5C,D) suggesting that these cells that were once committed to an epithelial lineage are in an intermediate stage between an epithelial and a mesenchymal cell type. In other areas of the lung in which the overall alveolar architecture was preserved, co-immunostaining for αSMA and TTF-1 revealed abnormal expression of αSMA in epithelial cells lining the alveoli that were also TTF-1 positive (Fig. 5E and corresponding DIC image Fig. 5F,H), similar to the staining pattern in the fibrotic foci. In lung sections from wt control mice cells lining the alveolar wall only expressed TTF-1 (Fig. 5A and corresponding DIC image Fig. 5B). This suggests that at least some of the αSMA expressing myofibroblasts in the lungs of the Col1a2-CTGF transgenic mice are likely to be derived from epithelial cells. Thus, in non-fibrotic areas of the lung αSMA-positive cells are in an intermediate stage, expressing both epithelial and mesenchymal markers. In addition cells in the fibrotic foci also stained for Snai1 suggesting that these cells have undergone EMT (Fig. 5I,J,K). These results strongly suggest that overexpression of CTGF in lung fibroblasts can induce EMT-like changes in alveolar epithelial cells as assessed by co-expression of epithelial and acquisition of mesenchymal cell markers.
Fig. 5.

Alveolar epithelial abnormality in the lungs of Col1a2-CTGF mice. (A–F) Immunofluorescence of lung sections from Col1a2-CTGF mice and wt controls co-immunostained for αSMA and TTF-1 showed increased numbers of αSMA-expressing myofibroblasts in fibrotic lesions of Co1a2-CTGF transgenic lungs (C, arrows; corresponding DIC overlay in D) compared to wt controls (A; corresponding DIC overlay in B). In epithelial cells lining the alveoli in normal areas of lungs from Col1a2-CTGF mice abnormal expression of αSMA was evident (E; corresponding DIC overlay in F) compared to wt control mice (A; corresponding DIC overlay in B). (I,J) Increased expression of Snai1 was observed in fibrotic lesions in lungs from Col1a2-CTGF transgenic mice (J, arrows), compared to wt controls (I). Autofluorescence of red blood cells occurred in wt lung section stained for Snai1 (I). Scale bar: 50 µm. (G,H,K) Quantification of expression was performed on samples from six mice per group. Data are means ± s.d. *P<0.0001, **P<0.0006, ***P<0.0001, Student’s t-test.
EMT in lung fibrosis is associated with increased TGFβ signaling
To further understand the mechanisms of lung fibrosis in Col1a2-CTGF transgenic mice, the expression levels of potential CTGF target genes were measured by qRT-PCR. Since the fibrotic lung had an abundance of macrophages, a known source of TGFβ (Khalil et al., 1996), we also measured the mRNA levels of Tgfb1 and Tgfbr1. In addition to the expected increase in Ctgf mRNA, we observed a marked enhancement of the levels of Tgfb1 and Tgfbr1 mRNAs. Genes that are specific targets of the TGFβ signaling pathway such as Timp1, Serpine1 (PAI-I), Col1a1, Fn1 (fibronectin), Acta1 (alpha smooth muscle actin, αSMA) and Sox9 were also overexpressed (Fig. 6). These alterations in mRNA expression levels suggest that TGFβ may be an important participant in the fibrotic phenotype of the CTGF-induced lung fibrosis and may also play a role in EMT of alveolar epithelial cells described above.
Fig. 6.

Persistent activation of fibrotic and EMT marker genes in the lungs of Col1a2-CTGF transgenic mice. qRT-PCR revealed a marked increase in the expression of CTGF, TGFβ, their target genes, and the EMT markers Sox9 and αSMA in Col1a2-CTGF transgenic mice compared to wt controls. Results are expressed as average fold change compared to wt control mice; n = 4 mice per group.
In vitro TGFβ-mediated EMT in lung epithelial cells is inhibited by specific CTGF knockdown
There is evidence in the literature that TGFβ can induce EMT in lung epithelial cells in vitro (Xu et al., 2009). We wished to determine whether this TGFβ-induced EMT is mediated by CTGF. Lung epithelial cells were treated in vitro with recombinant TGFβ1. We observed changes in cell morphology from a rounded epithelial structure to a more spindle shaped fibroblast-like structure (data not shown). In addition, western blot analysis showed that the epithelial marker e-cadherin was downregulated and other mesenchymal markers, including αSMA, S100A4, SOX9, were upregulated in TGFβ1-treated cells (Fig. 7, lanes 5 and 6), indicating that EMT was occurring. This evidence of EMT-like changes was accompanied by increased expression of CTGF (Fig. 7, lanes 5 and 6). To directly assess the role of increased CTGF in cellular changes in epithelial cells, we used Ctgf siRNA to inhibit CTGF expression after TGFβ1 treatment. Specific CTGF knockdown significantly decreased expression of SNAI1, αSMA and SOX9 expression and increased expression of e-cadherin following TGFβ1 stimulation (Fig. 7, lanes 7 and 8). No effect was observed on SNAI1, αSMA or SOX9 expression when cells were transfected with a non-specific scrambled siRNA (Fig. 7, lane 6). Hence, knockdown of CTGF blocks TGFβ1-induced EMT in epithelial cells in vitro suggesting that CTGF mediates expression of EMT markers in these TGFβ1 treated cells.
Fig. 7.

In vitro TGFβ1-mediated EMT in lung epithelial cells is dependent on CTGF expression. Western blot analysis of lung epithelial cells treated with TGFβ1 (lanes 5–8) shows increased expression of CTGF, αSMA, Sox9, S100A4 and reduced expression E-cadherin (lanes 5 and 6). Specific knockdown using CTGF siRNA significantly inhibited Snai1, αSMA, Sox9, S100A4 overexpression and rescued the downregulation of E-cadherin (lane 7). No effect was observed on Snai1, αSMA, Sox9, S100A4, or E-cadherin expression when cells were either un-transfected or transfected with a non-specific scrambled siRNA (lanes 5, 6). Representative blots are shown. Bar graphs show relative expression of the proteins after the different treatments (normalized to GAPDH loading control; means ± s.d. of three independent experiments).
CTGF treatment induces EMT-like changes in lung epithelial cells in vitro
As the TGFβ1-induced EMT-like changes seen in lung epithelial cells are, at least in part, mediated by CTGF we investigated whether treatment of cells with CTGF alone could induce EMT-like changes. Lung epithelial cells were transduced with an adenovirus expressing CTGF from the CMV promoter (Ad.CTGF). Phase contrast microscopy images of control cells transduced with a GFP-expressing adenovirus (Ad.gfp) showed a typically epithelial cuboidal morphology (Fig. 8A,a), whereas cells transduced with Ad.CTGF showed a change in morphology to a spread, spindly morphology characteristic of mesenchymal cells (Fig. 8A,b–c). In addition, epithelial cells transduced with Ad.CTGF show expression of the mesenchymal marker αSMA (Fig. 8B,f), whereas this marker is absent in control cells transduced with Ad.gfp (Fig. 8B,e). Deconvoluted z-stack images (Fig. 8B,g) demonstrate that the expressed αSMA forms stress fibers. These experiments showed EMT-like changes occur when CTGF is expressed by the epithelial cells themselves. In order to more closely mimic the in vivo situation in the Col1a2-CTGF mice, in which the CTGF is expressed by fibroblast cells neighboring the epithelial cells, we treated epithelial cells in vitro with recombinant human CTGF (rhCTGF). Treatment of cells with rhCTGF in the absence of serum induced expression of αSMA (Fig. 8B,b,c) with no expression seen in untreated cells (Fig. 8B,a), and deconvoluted z-stack images (Fig. 8B,d) demonstrate that the expressed αSMA forms stress fibers. The induced expression seen by immunofluorescence was confirmed by western blot analysis (Fig. 8C) which showed a significant (P = 0.004) induction of α-SMA in treated cells. In order to assess any morphological changes, cells were stained with Rhodamine–phalloidin and co-stained for αSMA (Fig. 8D). Untreated cells (Fig. 8D,a–c) show peripheral actin filaments (Fig. 8D,a) with no αSMA (Fig. 8D,b,c merge). Cells treated with 100 ng/ml rhCTGF (Fig. 8D,d–f) show an increase in phalloidin staining (Fig. 8D,d) and an induction of αSMA (Fig. 8D,e,f merge). Furthermore, deconvoluted z-stack images (Fig. 8D,g–i) show that the induced αSMA is incorporated into the actin filament stress fibers.
Fig. 8.

CTGF induces EMT-like changes in vitro, which are dependent on TGFβ signaling. (A) CTGF expression induces a change in morphology in vitro. Phase-contrast images of epithelial cells transduced with Ad.GFP (a) show a typically epithelial cobblestone morphology, whereas epithelial cells transduced with Ad.CTGF (b,c) show a more spread, spindly morphology typical of mesenchymal cells. Scale bars: 20 µm. (B,C) CTGF induces expression of the mesenchymal marker αSMA in epithelial cells in vitro. Epithelial cells were stained with a Cy3-conjugated anti-αSMA antibody (orange) and nuclei were counter-stained with DAPI (blue). Cells were either exposed to exogenous rhCTGF (b–d) or transduced with adenovirus expressing CTGF (f,g) or gfp control (e). Untreated (a) or Ad.gfp-transduced (e) epithelial cells show no αSMA expression, whereas cells treated with 50 ng/ml (b), 100 ng/ml (c), or 500 ng/ml (d) rhCTGF, or transduced with Ad.CTGF (f,g) express αSMA. Deconvolution of z-stack images (d,g) shows that the expressed αSMA forms stress fibers. Scale bars: 20 µm. In C, western blot analysis of epithelial cells treated with 10 or 50 ng/ml rhCTGF shows a significant (P = 0.004) induction of α-SMA compared to controls. Representative blots for control and 50 ng/ml rhCTGF are shown. Graph shows densitometry of α-SMA (normalized to β-tubulin loading control). Data are means ± s.e.m. of three independent experiments. P-values were determined using Students t-test. (D) Exogenous CTGF induces a change in actin filaments in vitro. Epithelial cells were co-stained with Rhodamine-conjugated phalloidin (red; a,d,g) and an anti-αSMA antibody (green; b,e,h), and images were merged (c,f,i). Nuclei were counter-stained with DAPI (blue). Untreated cells (a–c) show mainly peripheral actin filaments that do not contain αSMA. Cells treated with 100 ng/ml rhCTGF (d–i) show an increase in actin filaments across the cells and these filaments include αSMA. Deconvoluted z-stack images (g–i) show the formation of actin filaments and αSMA into stress fibers. Scale bars: 20 µm. (E) αSMA induction by CTGF is prevented by neutralization of TGFβ or inhibition of TGFβRI. Epithelial cells were stained with an anti-αSMA antibody (green), and nuclei were counter-stained with DAPI (blue). Cells were untreated (a), exposed to rhCTGF (100 ng/ml; b), rhCTGF and a neutralizing anti-TGFB antibody, 1d11 (100 ng/ml and 25 µg/ml, respectively; c); or rhCTGF and the small molecule TGFβRI inhibitor LY-364947 (100 ng/ml and 300 nM, respectively; d). Panel e, shows a graphical representation of the frequency of αSMA-positive cells in treated and untreated cultures (as a percentage of total cell number) as means ± s.e.m. of four fields of view at 20× magnification. Lane 1, control cultures; lanes 2 and 3, cells treated with rhCTGF at 50 and 100 ng/ml respectively; lanes 4 and 5, cells treated with rhCTGF at 100 ng/ml in the presence of 1d11 at 10 or 25 µg/ml, respectively; lanes 6 and 7, cells treated with rhCTGF at 100 ng/ml in the presence of a control IgG at 10 or 25 µg/ml, respectively; lanes 8, 9 and 10, cells treated with rhCTGF at 100 ng/ml in the presence of the TGFβRI inhibitor LY-364947 at 50, 300 or 600 nM, respectively; lane 11, cells treated with TGFβ at 2 ng/ml. *P<0.00005 compared to untreated; #P<0.00005 compared to 100 ng/ml rhCTGF alone; ∧not significant (P>0.3) compared to 100 ng/ml rhCTGF alone; Student’s t-test.
As the fibrotic lungs of the Col1a2-CTGF mice show increased TGFβ signaling (Fig. 6), we investigated whether the EMT-like changes induced by CTGF in vitro may be mediated by increased TGFβ signaling in a positive feedback loop. Epithelial cells stimulated with rhCTGF were exposed to 1d11, a neutralizing antibody to TGFβ, or IgG control. Cells treated with rhCTGF alone induced expression of αSMA (Fig. 8E,b) compared to untreated cells (Fig. 8E,a). Cell counts showed a significantly (P<0.00005) higher percentage of αSMA-positive cells following treatment with 50 or 100 ng/ml rhCTGF (Fig. 8E; Panel e, lanes 2,3). Antibody blockade of TGFβ with 1d11 prevented the CTGF-induced expression of αSMA (Fig. 8E,c). Cell counts showed a significant (P<0.00005) reduction in the percentage of αSMA-positive cells with 10 or 25 µg/ml 1d11 (Panel e, lanes 4,5) compared to rhCTGF alone (Panel e, lanes 2,3), and with 25 µg/ml 1d11(Panel e, lane 5) the percentage of αSMA-positive cells was not significantly (P>0.9) different from unstimulated cells (Fig. 8E, Panel e, lane 1). Treatment of rhCTGF stimulated cells with 10 or 25 µg/ml IgG control did not result in a reduction (P>0.3) in the percentage of αSMA-positive cells (Fig. 8E, Panel e, lanes 6, 7). In addition LY-364947, a TGFβRI inhibitor, also prevented the CTGF-induced expression of αSMA (Fig. 8E,d). Cell counts showed a significant (P<0.00005) reduction in the percentage of αSMA-positive cells with 50, 300 or 600 nM LY-364947 (Panel e, lanes 8,9,10) compared to rhCTGF alone (Panel e, lanes 2,3), and with 300 or 600 nM LY-364947 the percentage of αSMA-positive cells was not significantly (P>0.2) different from unstimulated cells (Fig. 8E, Panel e, lanes 9,10 compared to lane 1). Interestingly, stimulation of epithelial cells with 2 ng/ml TGFβ induced significantly (P<0.003) more αSMA-positive cells than stimulation with 100 ng/ml rhCTGF (Panel e, lane 11 compared to lane 3). These results demonstrate that CTGF, either endogenously expressed by epithelial cells or given exogenously as a recombinant protein, is able to induce EMT-like changes in vitro. These results also indicate that CTGF-induced expression of αSMA is mediated by activation of TGFβ signaling. Taken together our results show that CTGF is able to induce EMT-like changes in epithelial cells and is able to induce αSMA expression in stress fibers.
Discussion
Studies of different fibrotic diseases, as well as the characterization of Col1a2-CTGF transgenic mice, point to CTGF as a key player in the fibrotic disease process. Indeed, the relative levels of CTGF found in fibroblasts from these mice are similar to levels observed in fibroblasts from systemic sclerosis patients (Shi-wen et al., 2000). However, the paracrine effects of CTGF on cellular targets outside of fibroblasts is largely unknown as is the cell and molecular mechanism underlying CTGF action on keratinocytes in skin, alveolar epithelial cells in lung or tubulointerstitial cells in kidney. The current study focuses primarily on the dermal and pulmonary compartment, the most profoundly affected tissues in the Col1a2-CTGF transgenic mice (Sonnylal et al., 2010). In most organs Col1a2 expression is located in cells of mesenchymal origin (Bou-Gharios et al., 1996), however in kidney, expression has also been observed in tubular epithelial cells upon injury (Fragiadaki et al., 2011). Interestingly, in a study using administration of aristolochic acid (AA) to induce renal fibrosis, the Col1a2-CTGF transgenic mice did not exhibit more fibrosis compared with wt AA-treated mice (Fragiadaki et al., 2011). This is however perhaps not surprising as AA-administration at the similar dose range (5–10 mg/kg) has been shown to induce as severe nephropathy resulting in florid interstitial inflammation with immune cell infiltrates and severe tissue damage and necrosis (Pozdzik et al., 2008; Zhou et al., 2010). This pathology was shown to be associated with a profound induction of TGFβ expression and Smad signaling which was shown to be responsible for the collagen matrix accumulation (Wang, Y. et al., 2008; Li et al., 2009). In this study we show that increased expression of CTGF in fibroblasts of Col1a2-CTGF transgenic mice results in impaired or loss of epithelial characteristics in adjacent epithelial structures and acquisition by these cells of mesenchymal properties in vivo in the skin and lung presumably by paracrine mechanisms.
In addition to the fibrotic phenotype, Col1a2-CTGF transgenic mice develop hair loss early on associated with hair follicle abnormalities and focal hyperplasia of the epidermis. This hyper-proliferation was associated with changes in cell morphology and marked changes in gene expression including increased expression of keratin K16. In the normal skin epidermis keratins K5 and K14 are expressed in the basal cells and keratins K1 and K10 are expressed in the suprabasal layers (Koch and Roop, 2004). In some diseased states such as in psoriasis where the epithelial cells undergo hyperproliferation and/or aberrant differentiation, expression of keratins K6 and its partner K16 are induced in the inter-follicular epidermis (Bernot et al., 2002). Recent, proteomic analysis of whole skin lesions from SSc patients shows abnormal expression of K6 and K16 suggesting hyperproliferation of the epidermal component in SSc (Aden et al., 2008).
In the skin of Col1a2-CTGF transgenic mice expression of the epithelial-specific integrin β6 is also markedly increased in focal areas of the epidermis. We speculate that the increase in β6 expression might eventually favor enhanced signaling by CTGF since integrins have previously been shown to be involved in such signaling. Mice in which integrin β6 overexpression is targeted to the basal keratinocytes, showed loss of hair in the lower back associated with fibrosis and chronic ulceration characterized by hyperplasia of epithelial cells and thick connective tissue (Häkkinen et al., 2004), a phenotype that is analogous to that of Col1a2-CTGF transgenic mice.
Furthermore the focal changes in the morphology of epidermal cells and the abnormal expression of αSMA, Snai1 and S100A4 by these cells in Col1a2-CTGF transgenic mice strongly suggests that the increased levels of CTGF that are secreted by the dermal fibroblasts resulting in an EMT-like process in neighboring epithelial cells presumably through a paracrine mechanism. EMT is a process by which epithelial cells undergo a transition into a mesenchymal cell-type giving rise to fibroblasts and myofibroblasts. EMT is characterized by loss of epithelial markers, loss of epithelial cell polarity, acquisition of mesenchymal markers and an invasive phenotype (Kalluri and Neilson, 2003). This process is now being recognized to play an important role in tissue repair and fibrosis. One recent report provides suggestive evidence consistent with EMT in post-menopausal frontal fibrosing alopecia (Nakamura and Tokura, 2011). This disease is characterized by a recession of the hairline, a reduction or loss of terminal hair follicles and fibrosis around the affected hair follicles. Cells expressing Snai1 were present in the fibrotic dermis raising the possibility that these cells might have been derived from epithelial cells.
Emerging evidence from studies of liver fibrosis showed that TGFβ-induced expression of Sox9 in hepatic stellate cells (HSC) in culture correlated with increased type I collagen production (Hanley et al., 2008). This increase can be inhibited by knockdown of Sox9 by a specific siRNA suggesting a role of Sox9 in ECM synthesis (Hanley et al., 2008).
In the Col1a2-CTGF transgenic mice the production of excess CTGF in dermal fibroblasts results in the abundant expression of Sox9 in the basal cells of the epidermis and in the outer root sheath and bulge of hair follicles. We speculate that this occurs through a paracrine mechanism. Interestingly embryonic ablation of Sox9 in K14 expressing epithelial cells of the skin results in defects in the stem cell compartment and subsequent structures that arise from the stem cells. These mice also displayed impaired wound repair (Nowak et al., 2008). Our in vitro experiments in lung A549 epithelial cells also show that knockdown of CTGF in these cells is associated with markedly decreased levels of Sox9. Thus both in vivo and in vitro experiments establish a clear correlation between levels of CTGF and those of Sox9. Increased expression of Sox9 is also observed in different cancers (Wang et al., 2007; Schaeffer et al., 2008). Inhibition of endogenous Sox9 in CWR22Rv1, a human prostate cancer cell line, resulted in decreased cellular proliferation (Wang et al., 2007). Transplantation of these cells in immuno-compromised mice resulted in decreased tumor growth correlating with its role in cellular proliferation (Wang, H. et al., 2008). In in vitro experiments, Sox9 activates the expression of the EMT marker Snail2 (Sakai et al., 2006) and co-expression of Sox9 and Snail2 can induce EMT in neural epithelial cells (Cheung et al., 2005). These lines of evidence would suggest the hypothesis that in our model system increase in expression of Sox9 in the skin might promote proliferation of progenitor cells and changes in the genetic program of existing epithelial cells or of newly formed epithelial cells undergoing EMT. In the epidermis of Col1a2-CTGF transgenic mice skin there was a marked increase in phosphorylated p38 and ERK. We speculate that ERK and p38 activation could mediate the paracrine effects of CTGF and together with the increased levels of β6 integrin have a role in cell proliferation and migration. Previous in vivo studies of BLM-induced lung fibrosis have shown by fate mapping experiments that one third of cells expressing the fibroblast specific marker S100A4 are derived from alveolar epithelial cells indicative of EMT (Tanjore et al., 2009). Similar EMT-like event is also observed in endoplasmic reticulum stress-induced lung fibrosis (Tanjore et al., 2011). Other studies have suggested a link between CTGF and lung fibrosis. Intratracheal bleomycin treatment induces lung fibrosis with an increase in CTGF expression in a bleomycin-sensitive mouse strain (C57BL/6) but not in a bleomycin-resistant mouse strain (BALB/c) (Lasky et al., 1998). Adenoviral-mediated CTGF expression in rat lungs induces a transient fibrosis (Bonniaud et al., 2003). Additionally, adenoviral expression of CTGF in the lungs of bleomycin-resistant BALB/c mice leads to significant lung fibrosis after treatment with bleomycin (Bonniaud et al., 2004). The lung fibrosis seen in Col1a2-CTGF transgenic mice was associated with increased proliferation of epithelial cells in the fibrotic lesions exemplified by the increased number of cells that expresses the epithelial specific transcription factor TTF-1. These fibrotic lesions also contained a large number of myofibroblasts expressing αSMA and a large percentage of these cells were co-expressing both αSMA and TTF-1 suggesting that the myofibroblast population arose from alveolar epithelial cells. This hypothesis was also supported by evidence that in the uninvolved areas of the transgenic mouse lung, which seemingly displayed a normal architecture, epithelial cells lining the alveoli co-expressed αSMA and TTF-1. Fibrotic lesions also displayed cells positive for Snai1 suggesting that EMT is associated with lung fibrosis. We propose that in the lung of Col1a2-CTGF transgenic mice EMT is a consequence of the paracrine effects of the abnormal high levels of secreted CTGF.
The results of the real-time PCR experiments on whole lung RNA of Col1a2-CTGF transgenic mice showed markedly increased RNA levels of TGFβ and its downstream signature targets. This is likely to be a consequence of the abundance of macrophages in the fibrotic lung. In contrast, in the skin of Col1a2-CTGF transgenic mice no macrophages were observed. Microarrays of total skin RNA did not reveal any significant increase in TGFβ and TβR1 RNA levels (data not shown). We propose that the combined increase in TGFβ and CTGF in the fibrotic lung not only causes increased myofibroblast transformation and excess ECM deposition but could also contribute to EMT in the lung epithelial cells. To understand the role of CTGF in mediating EMT in the lung, we have used lung epithelial cells to show that the activation of EMT marker genes by TGFβ can be inhibited by CTGF siRNA. We have also demonstrated that CTGF expression by epithelial cells in vitro causes EMT-like changes. Treatment of lung epithelial cells with recombinant CTGF also induces EMT-like changes. Additionally, we have demonstrated that the EMT-like changes induced by recombinant CTGF in vitro can be prevented by immunoinhibition of TGFβ or by inhibition of TGFβRI suggesting that TGFβ signaling plays a central role in the EMT process in lung epithelial cells. These in vitro findings strongly suggest that paracrine CTGF is likely to have a direct effect on inducing EMT in vivo. We speculate that these CTGF-induced EMT-like changes may be a contributor to the development of lung fibrosis. The results also suggest that the induction of TGFβ and TGFβRI by CTGF may establish a positive feedback loop driving increased expression of endogenous CTGF and further contributing to the EMT-like and fibrotic changes seen in the lungs of Col1a2-CTGF mice.
In summary our in vivo observations strongly suggest that paracrine effects of secreted CTGF produced in fibroblastic cells of Col1a2-CTGF mice stimulate epithelial cells to modify their genetic program in order to undergo changes that mimic features of EMT. The results of our in vitro experiments support these conclusions. Thus CTGF plays a crucial role not only in promoting a profibrotic program in fibroblastic cells but also in the EMT-like transition of epithelial cells into mesenchymal cells. We speculate that the increase in Sox9 expression in skin and lung epithelial cells may be an important factor in the EMT-like phenotype.
Materials and Methods
Transgenic mice
The Col1a2-CTGF transgenic mice used in this study have been described previously (Sonnylal et al., 2010). Briefly, the murine homolog of CTGF (Fisp-12) was cloned into a vector containing the 6-kb enhancer and minimal promoter of the murine Col1a2 gene, an IRES-lacZ reporter, and the murine protamine polyA signal. Transgenic mice were produced by the standard method of pronuclear injection of linear DNA into a fertilized mouse egg. All experiments performed with the mice were in compliance with the standards of care approved by the M. D. Anderson Cancer Center Institutional Animal Care and Use Committee. Genotyping was performed by PCR for the lacZ reporter on DNA extracted from ear biopsies.
Immunofluorescence of tissue sections and quantification
Skin and lung tissues from Col1a2-CTGF transgenic mice and wild-type (wt) littermate controls were fixed overnight in neutral buffered 4% paraformaldehyde at 4°C, processed through graded alcohols, and paraffin embedded. Immunofluorescence was performed on 5 µm sections with the following primary antibodies: Keratin 16 (Novus Biologicals, Littleton, CO, USA), integrin β4 (Southern Biotech, Birmingham, AL, USA), integrin β6 (R&D Systems, Minneapolis, MN, USA), αSMA (Sigma-Aldrich, St. Louis, MO, USA), Snai1 and CTGF (Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA), S100A4 (Abcam, Cambridge, MA, USA), Sox9 (Millipore, Billerica, MA, USA), TTF-1, p-p38, pERK (Cell Signaling, Lake Placid, NY, USA), and type I collagen (Novotec, Lyon, France). The secondary antibodies used were Alexa Fluor 488 goat anti-mouse IgG or Alexa Fluor 555 goat anti-rabbit IgG (Life Technologies, Carlsbad, CA, USA); nuclei were counterstained with Topro (Life Technologies, Carlsbad, CA, USA) and sections were mounted in ProLong Gold (Life Technologies, Carlsbad, CA, USA). Image stacks were collected using a Zeiss LSM 510 confocal microscope (Carl Zeiss, Oberkochen, USA). For type I collagen, sections were stained using the Vectastain ABC kit as previously described (Sonnylal et al., 2010). Quantitation for K16, integrin β4, integrin β6, α-SMA, Snai1, S100A4, Sox9, pErk1/2 and p-p38 staining of skin sections was performed using MetaMorph software by counting the number of positive cells within a 100 µm2 box in at least six different tissue sections. The expression of α-SMA and Snai1 in lung sections was graded on a scale of 0 to 4 with 0 signifying no staining, 2 signifying moderate staining and 3 or 4 signifying extensive staining. The number of α-SMA-positive cells in uninvolved areas of lung was quantified as described above.
Cell culture
The lung epithelial cell lines A549 (ATCC: CCL-185) (Giard et al., 1973) and SV40-T2 (Clement et al., 1991) were maintained in DMEM medium (Life Technologies, Carlsbad, CA, USA) containing 10% FBS (BioSera, Ringmer, UK). For all treatments cells were serum starved overnight (cells were washed twice in PBS to remove serum then grown in DMEM without serum) and treatments were performed in DMEM without serum.
Keratinocytes were isolated from Col1a2-CTGF transgenic mice and wt littermate controls as described previously (Flores et al., 2000). Briefly, the skin biopsies were incubated in dispase II (Roche, Bassel, Switzerland) overnight at 4°C to separate the epidermis from the dermis. The epidermis was carefully peeled off the following day, chopped into small pieces, and incubated in 0.25% trypsin–EDTA (Life Technologies, Carlsbad, CA, USA) for 20 minutes at 37°C. Cells were plated on J2-3T3 feeder cells and cultured in F medium (Sigma-Aldrich, St. Louis, MO, USA) supplemented with 0.4 µg/ml hydrocortisone, 24 ng/ml adenine, 8.4 ng/ml cholera toxin, 5 µg/ml insulin, 13 ng/ml 3,3,5-triiodo-L-thyronine and 10 ng/ml EGF.
Transfection with siRNA
Epithelial cells were transfected with ON-TARGETplus CTGF siRNA or non-targeting control siRNA (Dharmacon) using Fugene6 transfection reagent (Roche) in serum-free medium according to the manufacturers’ instructions. After 48 hours transfected cells were treated with 4 ng/ml recombinant TGFβ for a further 24 hours before proteins were extracted for western blot analysis.
Adenoviral transduction
Epithelial cells were grown in T25 flasks (Corning Inc., Corning, NY, USA) to 70% confluence then transduced at a multiplicity of infection of 100 with either Ad.CTGF (Ad5 serotype adenovirus expressing human CTGF from the CMV promoter) or Ad.gfp (Ad5 serotype adenovirus expressing GFP from the CMV promoter, negative control). Briefly, the cell monolayer was washed in PBS to remove all serum, incubated for 2 hours with adenoviral particles in a minimal volume of DMEM without serum, washed twice in PBS to remove any remaining virus particles, then normal growth media was replaced. After viral transduction the cells were allowed to recover for 24 hours then seeded into chamberslides (BD Bioscience, Franklin Lakes, NJ, USA) at 70% confluence and allowed to attach for 24 hours before fixing and staining.
Treatment with recombinant proteins, neutralizing antibodies and inhibitors
Recombinant human CTGF (rhCTGF) produced in HEK293 cells was bought from EMP Genetech (Ingolstadt, Germany). Recombinant TGFβ was from R&D Systems (Minneapolis, MN, USA). 1d11 (a mouse IgG1 neutralizing antibody against TGFβ) was from Abcam (Cambridge, MA, USA), purified mouse IgG (negative control) was from Vector Labs (Burlingame, CA, USA). LY-364947 (a selective TGFβRI inhibitor) was from Calbiochem (Merck KGaA, Darmstadt, Germany). For immunofluorescence, epithelial cells were seeded at 60% confluence in chamberslides, allowed to attach for 24 hours, serum-starved overnight, then treated with rhCTGF, with and without 1d11, IgG or LY-364947, in DMEM without serum for 48 hours before fixing and staining. For western blot analysis epithelial cells were seeded in six-well plates, allowed to attach for 24 hours, serum-starved overnight, then treated with rhCTGF, with and without 1d11, IgG or LY-364947, in DMEM without serum for 24 hours before protein extraction.
Immunofluorescence of epithelial cell culture
Cells in chamber slides were fixed in ice-cold methanol (for staining with anit-αSMA antibody) or 2% PFA (for anti-αSMA and phalloidin co-staining). Cy3-conjugated anti- αSMA (clone 1A4, Sigma-Aldrich, St. Louis, MO, USA), unconjugated anti- αSMA (clone 1A4, Dako, Glostrup, Denmark), and Rhodamine-conjugated phalloidin (Sigma-Aldrich, St. Louis, MO, USA) were used at 1∶100 dilution. Purified mouse IgG (Vector Labs, Burlingame, CA, USA) was used as negative control. Unconjugated anti-aSMA was detected with Alexa Fluor 488 chicken anti-mouse IgG (Life Technologies, Carlsbad, CA, USA) at 1∶1000 dilution. Nuclei were counterstained with DAPI and mounted in Vectashield (Vector Labs, Burlingame, CA, USA). Stained cells were imaged on an Axioskop Mot2 plus microscope with a monochrome camera and Axiovision microscope control software (Carl Zeiss, Oberkochen, Germany). The IgG control was used to set the exposure time. Z-stack images were taken using automated stage movement and inter-slice distances were optimised to fulfill the Nyquist criterion. Z-stacks were deconvoluted using an iterative point spread function in Axiovision. Cell counts were performed using the cell counter plugin for ImageJ (US National Institutes of Health, Bethesda, MD, USA).
Western blot analysis
Cell extracts for western blot analysis has been described earlier (Sonnylal et al., 2007). Briefly, cells were cultured as monolayers, washed with ice-cold PBS, and proteins were extracted with RIPA buffer with protease inhibitors (50 mM Tris-HCl, pH 8.0, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, 150 mM NaCl, 2 mM phenylmethylsulfonyl fluoride, 0.2 U/ml aprotinin). Cell extracts were resolved on 10% SDS-PAGE and immunoblotted as described earlier (Sonnylal et al., 2007). In addition to the antibodies described above, the following antibodies were also used in western blotting: CTGF (Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA), p38, ERK (Cell Signaling, Lake Placid, NY, USA), e-cadherin (BD Biosciences, Franklin Lakes, NJ, USA), GAPDH, β-tubulin (Abcam, Cambridge, MA, USA). Blots were quantified with respect to the expression of GAPDH by densitometric analysis using the UVP Biospectrum AC image system with VisionWorksLS software. The images present are representative of three independent experiments.
Real-time PCR
Standard RT-PCR was performed with the following primers: TGFβ1 forward, 5′-GAAGGGCCGGTTCATGTCATG-3′; TGFβ1 reverse, 5′-TGTGACAGCAAAGATAACAAACTCCAC-3′; TβR1 forward, 5′-ATTGCTGGTCCAGTCTGCTT-3′; TβR1-reverse, 5′-CCTGATCCAGACCCTGATGT-3′; Col1a1 forward, 5′-CCCGCCGAT GTCGCTAT-3′; Col1a1 reverse, 5′-GCTACGCTG TTCTTGCAGTGAT-3′; Fibronectin forward, 5′-GGTCTGCAGAGGTTGACAGTG-3′, Fibronectin reverse, 5′-GGAGAAGTTTGTGCATGGTGTCC-3′; CTGF forward, 5′-TTGTAATGGCAGGCACAGGTC-3′; CTGF reverse, 5′-CGCACAAGAACCACCACTCTG-3′ PAI 1 forward, 5′-ATGCGGGCTGAGATGACAAAG-3′; PAI 1 reverse, 5′-ACTGCAAAAGGTCAGGATCGA-3′; αSMA forward, 5′-GCCAAGTCCAGACGCATGATG-3′; αSMA reverse, 5′-TATGCCTCTGGACGTACAACT-3′; TIMP 1 forward, 5′-CACAAGCCTGGATTCCGTGG-3′; TIMP 1 reverse, 5′-TCCCTTGCAAACTGGAGAGTGAC-3′; Sox9 forward, 5′-GACGCTGGGCAAGCTCT-3′; Sox9 reverse, 5′-GTAATCCGGGTGGTCCTTCT-3′.
Supplementary Material
Acknowledgments
D.A. and B.de.C. are joint senior authors.
Footnotes
Author contributions
S.S., D.A., S.X., J.N. and B.d.C. developed the hypothesis, and conceived and designed the experiments. S.S., S.X., H.J., A.T., V.R.S. P.A. and M.P. performed the experiments and contributed to writing the paper. S.S., H.J., D.A., J.N., M.P. and B.d.C. contributed to the supervision of the study, and helped in the analysis and interpretation of data from experimental results. S.S., D.A. and B.d.C. wrote the paper. All authors provided critical comments on drafts of the paper, and all read and approved the final manuscript.
Funding
This work was supported by National Institutes of Health/National Institute of Arthritis and Musculoskeletal and Skin Diseases Center of Research Translation in Scleroderma [grant number P50AR054144 to B.d.C.]; Arthritis Research UK [grant numbers 19427 and 18627 to D.A.]; the Medical Research Council [grant number G0801052 to D.A.]; and The Raynaud’s and Scleroderma Association [grant number RF27 to D.A.]. Deposited in PMC for release after 12 months.
Supplementary material available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.111302/-/DC1
References
- Abraham D. J., Eckes B., Rajkumar V., Krieg T. (2007). New developments in fibroblast and myofibroblast biology: implications for fibrosis and scleroderma. Curr. Rheumatol. Rep. 9, 136–143 10.1007/s11926-007-0008-z [DOI] [PubMed] [Google Scholar]
- Aden N., Shiwen X., Aden D., Black C., Nuttall A., Denton C. P., Leask A., Abraham D., Stratton R. (2008). Proteomic analysis of scleroderma lesional skin reveals activated wound healing phenotype of epidermal cell layer. Rheumatology (Oxford) 47, 1754–1760 10.1093/rheumatology/ken370 [DOI] [PubMed] [Google Scholar]
- Ahmed N., Niu J., Dorahy D. J., Gu X., Andrews S., Meldrum C. J., Scott R. J., Baker M. S., Macreadie I. G., Agrez M. V. (2002). Direct integrin alphavbeta6-ERK binding: implications for tumour growth. Oncogene 21, 1370–1380 10.1038/sj.onc.1205286 [DOI] [PubMed] [Google Scholar]
- Bennett M. R., Czech K. A., Arend L. J., Witte D. P., Devarajan P., Potter S. S. (2007). Laser capture microdissection-microarray analysis of focal segmental glomerulosclerosis glomeruli. Nephron Exp. Nephrol. 107, e30–e40 10.1159/000106775 [DOI] [PubMed] [Google Scholar]
- Bernot K. M., Coulombe P. A., McGowan K. M. (2002). Keratin 16 expression defines a subset of epithelial cells during skin morphogenesis and the hair cycle. J. Invest. Dermatol. 119, 1137–1149 10.1046/j.1523-1747.2002.19518.x [DOI] [PubMed] [Google Scholar]
- Bi W., Deng J. M., Zhang Z., Behringer R. R., de Crombrugghe B. (1999). Sox9 is required for cartilage formation. Nat. Genet. 22, 85–89 10.1038/8792 [DOI] [PubMed] [Google Scholar]
- Bonniaud P., Margetts P. J., Kolb M., Haberberger T., Kelly M., Robertson J., Gauldie J. (2003). Adenoviral gene transfer of connective tissue growth factor in the lung induces transient fibrosis. Am. J. Respir. Crit. Care Med. 168, 770–778 10.1164/rccm.200210-1254OC [DOI] [PubMed] [Google Scholar]
- Bonniaud P., Martin G., Margetts P. J., Ask K., Robertson J., Gauldie J., Kolb M. (2004). Connective tissue growth factor is crucial to inducing a profibrotic environment in “fibrosis-resistant” BALB/c mouse lungs. Am. J. Respir. Cell Mol. Biol. 31, 510–516 10.1165/rcmb.2004-0158OC [DOI] [PubMed] [Google Scholar]
- Bou-Gharios G., Garret L. A., Rossert J., Niederreither K., Eberspaecher H., Smith C., et al. (1996). A potent far-upstream enhancer in the mouse proα2(I) collagen gene regulates expression of reporter genes in transgenic mice. J. Cell Biol. 132, 1333–1344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung M., Chaboissier M. C., Mynett A., Hirst E., Schedl A., Briscoe J. (2005). The transcriptional control of trunk neural crest induction, survival, and delamination. Dev. Cell 8, 179–192 10.1016/j.devcel.2004.12.010 [DOI] [PubMed] [Google Scholar]
- Clement A., Steele M. P., Brody J. S., Riedel N. (1991). SV40T-immortalized lung alveolar epithelial cells display post-transcriptional regulation of proliferation-related genes. Exp. Cell Res. 196, 198–205 10.1016/0014-4827(91)90251-O [DOI] [PubMed] [Google Scholar]
- Flores E. R., Allen-Hoffmann B. L., Lee D., Lambert P. F. (2000). The human papillomavirus type 16 E7 oncogene is required for the productive stage of the viral life cycle. J. Virol. 74, 6622–6631 10.1128/JVI.74.14.6622-6631.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fragiadaki M., Witherden A. S., Kaneko T., Sonnylal S., Pusey C. D., Bou-Gharios G., Mason R. M. (2011). Interstitial fibrosis is associated with increased COL1A2 transcription in AA-injured renal tubular epithelial cells in vivo. Matrix Biol. 30, 396–403 10.1016/j.matbio.2011.07.004 [DOI] [PubMed] [Google Scholar]
- Friedrichsen S., Heuer H., Christ S., Winckler M., Brauer D., Bauer K., Raivich G. (2003). CTGF expression during mouse embryonic development. Cell Tissue Res. 312, 175–188 [DOI] [PubMed] [Google Scholar]
- Giard D. J., Aaronson S. A., Todaro G. J., Arnstein P., Kersey J. H., Dosik H., Parks W. P. (1973). In vitro cultivation of human tumors: establishment of cell lines derived from a series of solid tumors. J. Natl. Cancer Inst. 51, 1417–1423 [DOI] [PubMed] [Google Scholar]
- Häkkinen L., Koivisto L., Gardner H., Saarialho-Kere U., Carroll J. M., Lakso M., Rauvala H., Laato M., Heino J., Larjava H. (2004). Increased expression of beta6-integrin in skin leads to spontaneous development of chronic wounds. Am. J. Pathol. 164, 229–242 10.1016/S0002-9440(10)63113-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanley K. P., Oakley F., Sugden S., Wilson D. I., Mann D. A., Hanley N. A. (2008). Ectopic SOX9 mediates extracellular matrix deposition characteristic of organ fibrosis. J. Biol. Chem. 283, 14063–14071 10.1074/jbc.M707390200 [DOI] [PubMed] [Google Scholar]
- Huang X. Z., Wu J. F., Cass D., Erle D. J., Corry D., Young S. G., Farese R. V., Jr, Sheppard D. (1996). Inactivation of the integrin beta 6 subunit gene reveals a role of epithelial integrins in regulating inflammation in the lung and skin. J. Cell Biol. 133, 921–928 10.1083/jcb.133.4.921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivkovic S., Yoon B. S., Popoff S. N., Safadi F. F., Libuda D. E., Stephenson R. C., Daluiski A., Lyons K. M. (2003). Connective tissue growth factor coordinates chondrogenesis and angiogenesis during skeletal development. Development 130, 2779–2791 10.1242/dev.00505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalluri R., Neilson E. G. (2003). Epithelial-mesenchymal transition and its implications for fibrosis. J. Clin. Invest. 112, 1776–1784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalil N., Corne S., Whitman C., Yacyshyn H. (1996). Plasmin regulates the activation of cell-associated latent TGF-beta 1 secreted by rat alveolar macrophages after in vivo bleomycin injury. Am. J. Respir. Cell Mol. Biol. 15, 252–259 [DOI] [PubMed] [Google Scholar]
- Koch P. J., Roop D. R. (2004). The role of keratins in epidermal development and homeostasis—going beyond the obvious. J. Invest. Dermatol. 123, x–xi 10.1111/j.0022-202X.2004.23495.x [DOI] [PubMed] [Google Scholar]
- Krieg T., Abraham D., Lafyatis R. (2007). Fibrosis in connective tissue disease: the role of the myofibroblast and fibroblast-epithelial cell interactions. Arthritis Res. Ther. 9 Suppl 2, S4 10.1186/ar2188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasky J. A., Ortiz L. A., Tonthat B., Hoyle G. W., Corti M., Athas G., Lungarella G., Brody A., Friedman M. (1998). Connective tissue growth factor mRNA expression is upregulated in bleomycin-induced lung fibrosis. Am. J. Physiol. 275, L365–L371 [DOI] [PubMed] [Google Scholar]
- Leask A., Abraham D. J. (2006). All in the CCN family: essential matricellular signaling modulators emerge from the bunker. J. Cell Sci. 119, 4803–4810 10.1242/jcs.03270 [DOI] [PubMed] [Google Scholar]
- Li J., Zhang Z., Wang D., Wang Y., Li Y., Wu G. (2009). TGF-beta 1/Smads signaling stimulates renal interstitial fibrosis in experimental AAN. J. Recept. Signal Transduct. Res. 29, 280–285 10.1080/10799890903078465 [DOI] [PubMed] [Google Scholar]
- Naitoh M., Kubota H., Ikeda M., Tanaka T., Shirane H., Suzuki S., Nagata K. (2005). Gene expression in human keloids is altered from dermal to chondrocytic and osteogenic lineage. Genes Cells 10, 1081–1091 10.1111/j.1365-2443.2005.00902.x [DOI] [PubMed] [Google Scholar]
- Nakamura M., Tokura Y. (2011). Epithelial-mesenchymal transition in the skin. J. Dermatol. Sci. 61, 7–13 10.1016/j.jdermsci.2010.11.015 [DOI] [PubMed] [Google Scholar]
- Nowak J. A., Polak L., Pasolli H. A., Fuchs E. (2008). Hair follicle stem cells are specified and function in early skin morphogenesis. Cell Stem Cell 3, 33–43 10.1016/j.stem.2008.05.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pohlers D., Brenmoehl J., Löffler I., Müller C. K., Leipner C., Schultze-Mosgau S., Stallmach A., Kinne R. W., Wolf G. (2009). TGF-beta and fibrosis in different organs – molecular pathway imprints. Biochim. Biophys. Acta 1792, 746–756 10.1016/j.bbadis.2009.06.004 [DOI] [PubMed] [Google Scholar]
- Pozdzik A. A., Salmon I. J., Husson C. P., Decaestecker C., Rogier E., Bourgeade M. F., Deschodt-Lanckman M. M., Vanherweghem J. L., Nortier J. L. (2008). Patterns of interstitial inflammation during the evolution of renal injury in experimental aristolochic acid nephropathy. Nephrol. Dial. Transplant. 23, 2480–2491 10.1093/ndt/gfn140 [DOI] [PubMed] [Google Scholar]
- Pritchett J., Athwal V., Roberts N., Hanley N. A., Hanley K. P. (2011). Understanding the role of SOX9 in acquired diseases: lessons from development. Trends Mol. Med. 17, 166–174 10.1016/j.molmed.2010.12.001 [DOI] [PubMed] [Google Scholar]
- Sakai D., Suzuki T., Osumi N., Wakamatsu Y. (2006). Cooperative action of Sox9, Snail2 and PKA signaling in early neural crest development. Development 133, 1323–1333 10.1242/dev.02297 [DOI] [PubMed] [Google Scholar]
- Schaeffer E. M., Marchionni L., Huang Z., Simons B., Blackman A., Yu W., Parmigiani G., Berman D. M. (2008). Androgen-induced programs for prostate epithelial growth and invasion arise in embryogenesis and are reactivated in cancer. Oncogene 27, 7180–7191 10.1038/onc.2008.327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi-wen X., Pennington D., Holmes A., Leask A., Bradham D., Beauchamp J. R., Fonseca C., du Bois R. M., Martin G. R., Black C. M. et al. (2000). Autocrine overexpression of CTGF maintains fibrosis: RDA analysis of fibrosis genes in systemic sclerosis. Exp. Cell Res. 259, 213–224 10.1006/excr.2000.4972 [DOI] [PubMed] [Google Scholar]
- Sonnylal S., Denton C. P., Zheng B., Keene D. R., He R., Adams H. P., Vanpelt C. S., Geng Y. J., Deng J. M., Behringer R. R. et al. (2007). Postnatal induction of transforming growth factor beta signaling in fibroblasts of mice recapitulates clinical, histologic, and biochemical features of scleroderma. Arthritis Rheum. 56, 334–344 10.1002/art.22328 [DOI] [PubMed] [Google Scholar]
- Sonnylal S., Shi-Wen X., Leoni P., Naff K., Van Pelt C. S., Nakamura H., Leask A., Abraham D., Bou-Gharios G., de Crombrugghe B. (2010). Selective expression of connective tissue growth factor in fibroblasts in vivo promotes systemic tissue fibrosis. Arthritis Rheum. 62, 1523–1532 10.1002/art.27382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan B. P., Kassel K. M., Manley S., Baker A. K., Luyendyk J. P. (2011). Regulation of transforming growth factor-β1-dependent integrin β6 expression by p38 mitogen-activated protein kinase in bile duct epithelial cells. J. Pharmacol. Exp. Ther. 337, 471–478 10.1124/jpet.110.177337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanjore H., Xu X. C., Polosukhin V. V., Degryse A. L., Li B., Han W., Sherrill T. P., Plieth D., Neilson E. G., Blackwell T. S. et al. (2009). Contribution of epithelial-derived fibroblasts to bleomycin-induced lung fibrosis. Am. J. Respir. Crit. Care Med. 180, 657–665 10.1164/rccm.200903-0322OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanjore H., Cheng D. S., Degryse A. L., Zoz D. F., Abdolrasulnia R., Lawson W. E., Blackwell T. S. (2011). Alveolar epithelial cells undergo epithelial-to-mesenchymal transition in response to endoplasmic reticulum stress. J. Biol. Chem. 286, 30972–30980 10.1074/jbc.M110.181164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal V. P., Chaboissier M. C., Lützkendorf S., Cotsarelis G., Mill P., Hui C. C., Ortonne N., Ortonne J. P., Schedl A. (2005). Sox9 is essential for outer root sheath differentiation and the formation of the hair stem cell compartment. Curr. Biol. 15, 1340–1351 10.1016/j.cub.2005.06.064 [DOI] [PubMed] [Google Scholar]
- Wang H., McKnight N. C., Zhang T., Lu M. L., Balk S. P., Yuan X. (2007). SOX9 is expressed in normal prostate basal cells and regulates androgen receptor expression in prostate cancer cells. Cancer Res. 67, 528–536 10.1158/0008-5472.CAN-06-1672 [DOI] [PubMed] [Google Scholar]
- Wang H., Leav I., Ibaragi S., Wegner M., Hu G. F., Lu M. L., Balk S. P., Yuan X. (2008). SOX9 is expressed in human fetal prostate epithelium and enhances prostate cancer invasion. Cancer Res. 68, 1625–1630 10.1158/0008-5472.CAN-07-5915 [DOI] [PubMed] [Google Scholar]
- Wang Y., Zhang Z., Shen H., Lu Y., Li H., Ren X., Wu G. (2008). TGF-beta1/Smad7 signaling stimulates renal tubulointerstitial fibrosis induced by AAI. J. Recept. Signal Transduct. Res. 28, 413–428 10.1080/10799890802176741 [DOI] [PubMed] [Google Scholar]
- Wynn T. A. (2008). Cellular and molecular mechanisms of fibrosis. J. Pathol. 214, 199–210 10.1002/path.2277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J., Lamouille S., Derynck R. (2009). TGF-β-induced epithelial to mesenchymal transition. Cell Res. 19, 156–172 10.1038/cr.2009.5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L., Fu P., Huang X. R., Liu F., Chung A. C., Lai K. N., Lan H. Y. (2010). Mechanism of chronic aristolochic acid nephropathy: role of Smad3. Am. J. Physiol. Renal Physiol. 298, F1006–F1017 10.1152/ajprenal.00675.2009 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.