

Summary
In their mammalian host trypanosomes generate ‘stumpy’ forms from proliferative ‘slender’ forms as an adaptation for transmission to their tsetse fly vector. This transition is characterised by the repression of many genes while quiescent stumpy forms accumulate during each wave of parasitaemia. However, a subset of genes are upregulated either as an adaptation for transmission or to sustain infection chronicity. Among this group are ESAG9 proteins, whose genes were originally identified as a component of some telomeric variant surface glycoprotein gene expression sites, although many members of this diverse family are also transcribed elsewhere in the genome. ESAG9 genes are among the most highly regulated genes in transmissible stumpy forms, encoding a group of secreted proteins of cryptic function. To understand their developmental silencing in slender forms and activation in stumpy forms, the post-transcriptional control signals for a well conserved ESAG9 gene have been mapped. This identified a precise RNA sequence element of 34 nucleotides that contributes to gene expression silencing in slender forms but also acts positively, activating gene expression in stumpy forms. We predict that this bifunctional RNA sequence element is targeted by competing negative and positive regulatory factors in distinct developmental forms of the parasite. Analysis of the 3′UTR regulatory regions flanking the highly diverse ESAG9 family reveals that the linear regulatory sequence is not highly conserved, suggesting that RNA structure is important for interactions with regulatory proteins.
Key words: Differentiation, Gene expression, Trypanosoma brucei
Introduction
African trypanosomes are parasites of sub-Saharan Africa that survive in the bloodstream of their mammalian hosts through their expression of a variable surface glycoprotein (VSG) coat (Rudenko, 2011). In order to evade immunity, this coat can be changed through the expression of different representatives of a large repertoire of vsg genes, that are expressed from telomeric expression sites, only one of which is active at any one time. The activity of vsg expression sites is controlled at the level of transcription, this being apparently linked to their physical association with a sub-nuclear expression site body (Navarro and Gull, 2001; Navarro et al., 2007), which ensures monoallelic expression (Borst, 2002). In the tsetse fly vector, responsible for the transmission of trypanosomes, the VSG coat is replaced by a coat of procyclin molecules, this also being linked to the activation of the procyclin gene promoter and the inactivation of the vsg gene expression site promoter (Landeira and Navarro, 2007). In both the case of vsg and procyclin gene expression, transcription is unusual in being driven by RNA polymerase I (Günzl et al., 2003; Rudenko et al., 1989), this being made possible because all trypanosome mRNAs are capped by trans splicing of an RNA polymerase II transcribed leader sequence (Clayton, 2002; Kooter et al., 1987; Zomerdijk et al., 1991).
<@?twb=.3w?>Although vsg and procyclin mRNAs are the major products of their respective transcription units, other genes are also co-transcribed in each locus. Most notably, the vsg expression site contains a number of expression site associated genes (ESAG) positioned between the upstream vsg promoter and telomeric vsg gene (Cully et al., 1985; Kooter et al., 1987; Pays et al., 1989). The function of these is largely unknown, although ESAG6 and 7 (positioned closest to the expression site promoter) encode a heterodimeric transferrin receptor (Salmon et al., 1994), ESAG4 encodes an adenylate cyclase activity (Paindavoine et al., 1992) and, exclusively in Trypanosoma brucei rhodesiense, SRA encodes a gene that enables resistance to the trypanolytic component of human serum (Pays et al., 2001; Xong et al., 1998). Although expression site associated genes are defined by their co-association with the vsg gene, several ESAG gene families also have representatives positioned outside expression sites. These are transcribed as part of RNA-polymerase-II-driven polycistronic arrays that generate the mRNAs for all non-expression site protein coding genes. These genes are often termed GRESAGs (gene related to ESAGs) (Pays et al., 2001). Although positioned outside vsg expression sites, GRESAGs can also be functional; members of the ESAG4 family have a function in both cytokinesis and early establishment of trypanosome infections in the mammalian host, for example (Salmon et al., 2012a; Salmon et al., 2012b).
One unusual ESAG family is the ESAG9 gene family (Florent et al., 1991). These genes are highly diverse and rarely found as components of conventional vsg expression sites, most copies being located within polycistronic transcription units positioned at chromosomal internal and sub-telomeric regions (Barnwell et al., 2010; Hertz-Fowler et al., 2008). The protein products of ESAG9 genes are of unknown function, although they are secreted by bloodstream form parasites, potentially to assist infection chronicity or transmission (Barnwell et al., 2010). Most interestingly, however, ESAG9 genes show developmental expression in the mammalian bloodstream, being expressed only upon development of the bloodstream parasites toward stumpy forms (Barnwell et al., 2010; Jensen et al., 2009), transmission stages that accumulate at the peak of each wave of parasitaemia (MacGregor et al., 2012). This differentiation event occurs in response to a density sensing mechanism that the parasites use to monitor the parasitaemia and thereby ensure the production of transmission stages (Reuner et al., 1997; Vassella et al., 1997). This serves both to prevent the parasite from overwhelming the host and also to prepare the parasite for transmission. These developmental processes involve a form of quorum sensing in which a parasite-derived signal (stumpy induction factor) is detected, this stimulating the cell cycle arrest of parasites, and their morphological progression from proliferative slender forms, through intermediate forms to stumpy forms (Fig. 1A) (MacGregor et al., 2011; MacGregor et al., 2012). These events occur via a defined developmental pathway in the bloodstream, some aspects of which can be mimicked in vitro using cell permeable cAMP or AMP analogues (Laxman et al., 2006; MacGregor and Matthews, 2012; Vassella et al., 1997). In vivo, the upregulation of ESAG9 transcripts is one of the earliest events during stumpy formation, with these genes being among the most highly expressed mRNAs during the transition from intermediate to stumpy cells. This involves the elevated expression of many different ESAG9 family members and so is not restricted to expression associated copies of the gene (Barnwell et al., 2010). Hence, the ESAG9 genes provide an example of a stringently and coordinately regulated gene family that is developmentally expressed in response to the parasites' quorum-sensing signal.
Fig. 1.
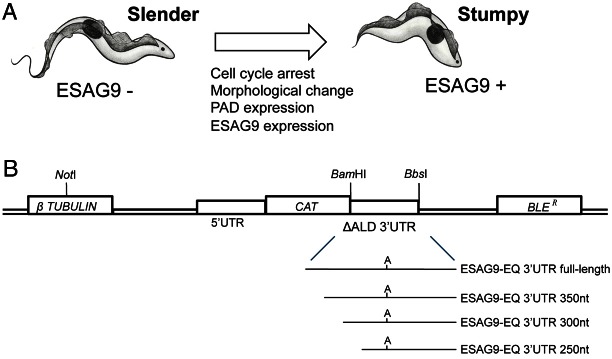
The biological and experimental basis of this study. (A) Representation of the events accompanying the differentiation from slender to stumpy forms during trypanosome development in the mammalian bloodstream. (B) Schematic diagram of the subcloning strategy for 3′UTR analysis with a CAT reporter gene assay, showing the region of the CAT449 vector with the chloramphenicol acetyl transferase (CAT), phleomycin resistance (BLER), and β-tubulin genes. The aldolase (ALD) 5′UTR and truncated 3′UTR (ΔALD) flank the CAT gene, with the ΔALD 3′UTR conferring constitutive expression (Mayho et al., 2006) since it is not developmentally regulated (Biebinger et al., 1997). Replacement of the 3′UTR with an experimental UTR used the BamHI and BbsI restriction enzyme sites. The 3′UTR deletion series (350 nt, 300 nt, 250 nt 3′UTR sequences) were generated with PCRs using forward primers designed to create ∼50 nt sequential deletions along the UTR from the 5′ end. The diagram is not to scale.
In contrast to differential gene regulation between bloodstream and tsetse midgut forms, developmental gene expression in stumpy forms presents some interesting challenges to the parasite. Firstly, stumpy forms are quiescent such that stumpy expressed molecules must escape the translational repression enforced on most genes (Brecht and Parsons, 1998; P. Capewell, S. M., A. Ivens, P. MacGregor, K. Fenn, P. Walrad, F. Bringaud, T. Smith and K. R. M., unpublished data). Secondly, stumpy-enriched gene expression is exclusively the product of a developmental signalling pathway that is parasite intrinsic, contrasting with the wide-ranging environmental signals that govern development upon entry to the tsetse fly (e.g. temperature, glucose levels, pH etc.) (Schwede et al., 2011). Finally, genes that are activated in stumpy forms must also be held silent in slender forms, and this repression must be alleviated upon development in the bloodstream. For these reasons, we have sought to understand developmental gene expression in the mammalian bloodstream, particularly focusing on the small set of genes enriched in stumpy forms. To date, one stumpy enriched molecule has been characterised – PAD1, a carboxylate transporter protein important in the perception of the differentiation signal as stumpy forms enter the tsetse fly (Dean et al., 2009). For this molecule, an analysis of the regulatory signals confirmed the importance of its 3′UTR, but sequences important in developmental regulation were difficult to precisely define, being complex and dispersed (MacGregor and Matthews, 2012).
Here, we have exploited the striking developmental control of the co-regulated ESAG9 gene family to better understand developmental regulation in the mammalian bloodstream. This has identified a highly defined short regulatory RNA motif that ensures ESAG9 gene silencing in slender forms. Moreover, the same element is responsive to cAMP analogues that intersect with the stumpy induction pathway and also to SIF itself, such that it promotes gene expression in stumpy forms. This identifies this bifunctional RNA element as among the most precisely defined post-transcriptional regulatory elements identified in trypanosomes to date and the first well characterised element operating in the disease and transmission relevant bloodstream forms of the parasite.
Results
The ESAG9-EQ 3′UTR contributes to gene expression silencing in slender forms
In order to analyse ESAG9 gene regulation, we used as a model Tb927.5.4620 (henceforth termed ESAG9-EQ), which is most closely related to ESAG9 genes originally identified as an expression site component in Trypanosoma equiperdum [67% and 84% amino acid similarity to ESAG9u and ESAG9c (Florent et al., 1991), respectively], and also highly related to an expression site linked copy in T. brucei s427 identified by TAR cloning (Young et al., 2008) (88% similarity). This gene was also confirmed as being stumpy-enriched in different strains of T. brucei (Barnwell et al., 2010), with Digital SAGE analysis indicating fivefold higher mRNA expression in stumpy forms than in slender forms (data not shown). Initially, a CAT reporter gene assay approach was used to assess the contributions of the ESAG9 3′UTR sequence to the control of gene expression (Fig. 1B). This approach has previously been used successfully to map 3′UTR control elements of COX genes (Mayho et al., 2006) and PAD1 (MacGregor and Matthews, 2012). The nearest gene annotated downstream of ESAG9-EQ in the T. b. brucei TREU 927/4 genome sequence is Tb927.5.4630, generating an intergenic region of 3057 bp. Since this provided too large a region for further manipulation, only 400 nucleotides (nt) downstream of the ESAG9-EQ gene was included for analysis, this extending beyond the mapped polyadenylation site for the ESAG9-EQ transcript (positioned at ∼380 nt relative to the stop codon; K. M. and S. M., unpublished observations). This generated the construct ‘ESAG9-EQ 3′UTR full length’, which was transfected in to monomorphic bloodstream form T. brucei Lister 427 in parallel with the same construct bearing the truncated aldolase 3′UTR as a control (ΔALD; unmodified vector; Fig. 1B). Monomorphic lines are laboratory-adapted forms that have lost the ability to produce stumpy forms but which are much more readily cultured and transfected than pleomorphic lines that can generate stumpy forms. For this and all subsequent analyses, at least two independent cell lines were analysed for each construct, and protein assays were carried out in triplicate.
In comparison to the ΔALD construct, the ESAG9-EQ 3′UTR full length construct showed reduced CAT protein (26±1.7% of ΔALD, which was normalised to 100% in all experiments; Fig. 2A) and mRNA expression (34.5% of ΔALD; Fig. 2A,B). This indicated that the ESAG9-EQ 3′UTR repressed CAT expression in monomorphic slender forms. In order to map sequences that might contribute to this repression a deletion series was constructed that progressively deleted, from the CAT gene stop codon, 50–150 nt of the 400 nt ESAG9 intergenic sequence. Each of these sequences was predicted to retain the ESAG9 polyadenylation site. Interestingly, investigation of the deletion series constructs after transfection into T. brucei revealed that the repression seen with the intact 3′UTR was reduced with the 350 nt and 250 nt constructs, where CAT protein expression increased to 47±2.5% and 77±3.2%, respectively. In contrast, repression was retained with the 300 nt construct, since CAT protein expression was only 32% of that seen for the control vector containing the ΔALD 3′UTR (Fig. 2A), similar to that of the intact ESAG9 3′UTR (26%). Fig. 2B shows that for each deletion construct, the RNA expression level followed the same trend as the pattern of protein expression, such that CAT mRNA expression increased with the 350 nt deletion construct (45% the level CAT mRNA of ΔALD), decreased with the 300 nt construct (34%) and increased again with the 250 nt construct (60%). In each case, the sizes of the transcripts observed were consistent with the 3′UTR deletions incorporated into the reporter construct, indicating use of the endogenous polyadenylation site.
Fig. 2.

Deletion analysis of regulatory regions in the ESAG9-EQ 3′UTR. (A). CAT protein (upper panel) and mRNA (lower panel) expression from the constructs shown, as a percentage of expression from one clone of the ΔALD 3′UTR control. The number of biological replicates (independently derived clones) used was: full-length: 5, 350 nt: 2, 300 nt: 3, 250 nt: 3; each with 1–2 experimental replicates. Values are means ± standard error of the mean (s.e.m.) of biological replicates for the protein expression, and means ± the lower and upper values from two replicates is shown for mRNA expression, these being derived from B). At the left of the graphs are schematic representations of the sequence analysed in each construct, where ‘A’ indicates the polyadenylation site location and the red line indicates the regulatory element location. (B) Northern blot analysis of CAT mRNA, with RNA from the ΔALD vector and two biological replicates of each of the full-length, 350 nt, 300 nt and 250 nt constructs. The lower panel shows the ethidium bromide staining of the rRNA present in each sample, analysed to indicate the loading of each lane. Below the lower blot are values of CAT mRNA abundance following normalisation to rRNA as a percentage of that obtained for the unmodified vector. The upper bands in all lanes represent a bicistronic transcript always detected using this construct (e.g. Mayho et al., 2006).
These results could indicate that regions that negatively regulate gene expression are deleted with the first 50 nt deletion (350 nt) and final 50 nt deletion (250 nt), with an additional region that positively regulates expression being deleted within the second 50 nt deletion (300 nt). Alternatively, the same outcome could be generated if a single negative regulatory secondary structure was disrupted in the 350 nt and 250 nt deletion, but able to form in the 300 nt deletion. Therefore, Sfold analysis (Ding et al., 2004) was carried out to identify potential structures within the ESAG9 3′UTR and its deletion mutants.
The Sfold ensemble centroid RNA structure prediction for the intact 3′UTR is provided in Fig. 3A, whereas the other deletion mutants are shown in supplementary material Fig. S2. Interestingly, the two constructs that confer repression of CAT gene expression, the full length 3′UTR and 300 nt 3′UTR, are predicted to possess a stem-loop structure (boxed in Fig. 3A; supplementary material Fig. S2) that is absent from the other two ensemble centroid structures, either because it is not predicted to form (350 nt 3′UTR construct) or because some of the sequence forming the structure has been deleted (250 nt 3′UTR construct). Although such predictions are speculative they provided a framework for further investigation of regulatory sequences within the ESAG9-EQ 3′UTR. Hereafter, the sequence contained within the boxed region contributing to the potential structure is referred to as the ESAG9-EQ regulatory element (‘e’).
Fig. 3.

RNA secondary structure predictions for ESAG9-EQ 3′UTR sequences. The Sfold (http://sfold.wadsworth.org/cgi-bin/index.pl) software package Srna was used to predict the structure of the ESAG9-EQ 3′UTR. Shown are the Ensemble Centroid structures generated for the full-length ESAG9-EQ 3′UTR (A) and the ESAG9-EQ eΔ 3′UTR (B).
Is ESAG9 3′UTR ‘e’ responsible for the repression of gene expression in monomorphic slender forms?
To assess whether the ESAG9-EQ 3′UTR potential regulatory element contributed to the repression of CAT reporter gene expression, this 34 nt sequence was deleted from the full-length 3′UTR sequence, removing nt 132–164. Initially, the predicted structure of the deleted 3′UTR sequence was analysed by Sfold, the resulting centroid structure demonstrating that the overall folding prediction for the remaining 3′UTR sequence was similar to that of the intact 3′UTR with the exception of the removed regulatory element (Fig. 3B). The resulting deletion construct (ESAG9-EQ 3′UTR ‘eΔ’) was therefore transfected into monomorphic cells and level of CAT reporter protein and RNA analysed in independent clonal lines.
Fig. 4 demonstrates that in comparison to the full-length ESAG9 3′UTR, the eΔ construct alleviated some of the repression of CAT expression, such that the level of CAT protein was 53±5.2% of the ΔALD control, compared with 26±1.7% for the intact ESAG9-EQ 3′UTR. In contrast, when the 34 nt regulatory element was inserted into the 3′UTR of the truncated aldolase 3′UTR (generating construct ‘CAT ΔALD eI’) at a position predicted to conserve its structure, and the overall structure of the remainder of the truncated aldolase 3′UTR, little overall repression of reporter expression was observed (CAT ΔALD eI 90±6% with respect to the ΔALD expression). RNA levels also followed the same trend as protein levels (Fig. 4B).
Fig. 4.
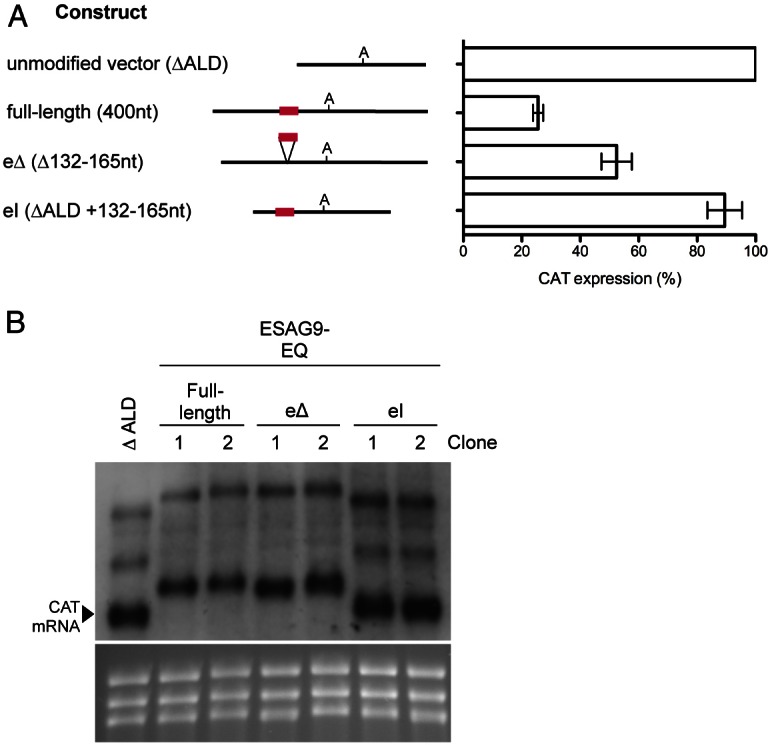
Analysis of the ESAG9-EQ 3′UTR element sequence using a CAT reporter gene assay. (A) CAT protein expression from the constructs shown, as a percentage of expression from one clone of unmodified vector (control). The number of biological replicates (independently derived clones) used was: full-length: 5; element deletion (eΔ): 6; and element insertion (eI): 4; each with 1–2 experimental replicates. Values are means ± s.e.m. of biological replicates. The data for the full-length 3′UTR construct clones from Fig. 2A is repeated here. At the left of the graph are schematic representations of the sequence analysed in each construct, where ‘A’ signifies the polyadenylation site location and the red line indicates the regulatory element location. (B) Northern blot analysis of CAT mRNA, with RNA from the ΔALD vector and two biological replicates of each of the full-length, eΔ, and eI constructs. The lower panel shows the ethidium bromide-staining of the rRNA present in each sample analysed, to indicate the loading of each lane.
These experiments indicated that in monomorphic cells the regulatory element contributed to the repression of reporter gene expression in the ESAG9-EQ 3′UTR. However, insertion of this element into another 3′UTR from a constitutively expressed gene did not repress expression.
Is the ESAG9-EQ 3′UTR responsible for the control of gene expression in ‘stumpy-like’ forms?
The developmental expression of genes in bloodstream stumpy form trypanosomes could result from a combination of both repression in slender forms (developmental negative regulation) and/or activation in stumpy forms (developmental positive regulation). We decided to examine the potential for positive regulation in the monomorphic reporter lines generated earlier by inducing cells to become ‘stumpy-like’ using the cell permeable cAMP analogue 8-pCPT-2′-O-Me-cAMP (Laxman et al., 2006; MacGregor and Matthews, 2012; Vassella et al., 1997). Although this analogue provides a limited proxy for stumpy formation, it promotes cell growth arrest and enhances differentiation to procyclic forms, and also activates the expression of a reporter gene linked to another stumpy enriched transcript, PAD1 (MacGregor and Matthews, 2012). Hence, it provided a useful tool for the analysis of gene expression activation in stumpy-like forms driven by the ESAG9-EQ 3′UTR.
The results for the various 3′UTR constructs exposed, or not, to 10 µM 8-pCPT-2′-O-Me-cAMP for 48 hours are shown in Fig. 5. For each cell line, the response to exposure to the drug is presented as the fold change in CAT protein levels compared to cells exposed to DMSO in place of 8-pCPT-2′-O-Me-cAMP, with the analysis being corrected for cell number under each treatment. For the ΔALD reporter, treatment with 8-pCPT-2′-O-Me-cAMP generated a 1.3-fold elevation in reporter activity, which was reproducible between different cell lines (supplementary material Fig. S1). In contrast, the full-length ESAG9-EQ 3′UTR reporter line exhibited 2.2-fold increase in CAT protein expression after 8-pCPT-2′-O-Me-cAMP treatment, compared to the same cells exposed to DMSO. This demonstrated that the ESAG9-EQ 3′UTR could confer enhanced expression of the linked reporter gene when stimulated to generate stumpy-like forms.
Fig. 5.
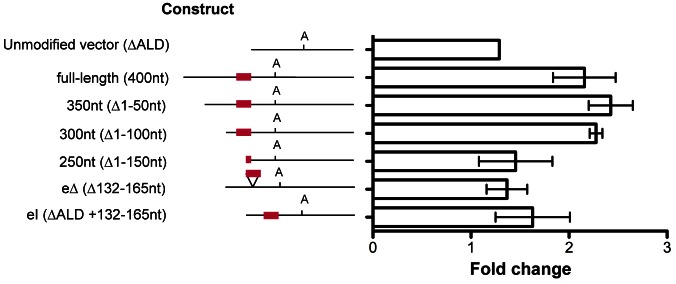
Response of CAT reporter gene expression to 8-pCPT-2′-O-Me-cAMP treatment for ESAG9-EQ 3′UTR-based constructs. Fold change in CAT protein expression levels between cells treated with 10 µM 8-pCPT-2′-O-Me-cAMP and control cells are shown. Values are means ± s.e.m. of biological replicates; unmodified vector, n = 1; full-length and eΔ, n = 3; other constructs, n = 2. For each cell line there were two experimental replicates, meaning that the CAT ELISA assays were performed with samples taken from two separate 8-pCPT-2′-O-Me-cAMP treatment experiments, except for the unmodified vector clone, which had one biological replicate and five experimental replicates, this line being included in each 8-pCPT-2′-O-Me-cAMP treatment experiment as a control. At the left of the graph are schematic representations of the sequence analysed in each construct, with the polyadenylation site location indicated with ‘A’ and the regulatory element location indicated by the red line.
Analysis of the respective deletion mutants of the ESAG9-EQ 3′UTR is also shown in Fig. 5. For the 350 nt construct (removing the first 50 nt of the 3′UTR), there was no difference in CAT protein expression upon 8-pCPT-2′-O-Me-cAMP treatment compared to the full-length construct, with a 2.4-fold increase observed. Similarly, when the next 50 nt of 3′UTR sequence was removed, a similar response was seen (2.3-fold change). However, further deletion of the ESAG9-EQ 3′UTR largely abolished the enhanced expression in response to 8-pCPT-2′-O-Me-cAMP, such that the fold change in CAT decreased to 1.5-fold with the 250 nt construct, similar to the ΔALD 3′UTR response (1.3-fold). This indicated that between 100 and 150 nt into the ESAG9-EQ 3′UTR there existed a positive sequence element responsive to 8-pCPT-2′-O-Me-cAMP. Since this region coincided with the regulatory element observed to repress slender form expression, the response of the ESAG9-EQ 3′UTR eΔ (which deletes nt 132–164 in the ESAG9-EQ 3′UTR) construct was also analysed, as was the insertion of the same region into the ΔALD 3′UTR. These analyses demonstrated that while insertion of the regulatory element into the ΔALD 3′UTR had no effect out of context, removal of the regulatory element from the ESAG9-EQ 3′UTR abolished responsiveness to 8-pCPT-2′-O-Me-cAMP (Fig. 5). This indicated that the regulatory element region in the ESAG9-EQ 3′UTR contributed to both gene repression in slender forms and gene activation when monomorphic slender cells were induced to generate stumpy-like forms. However, the overall magnitude of the effect in both cases was relatively modest, with both repression and enhancement exerting only approximately twofold regulation. Moreover when RNA expression from the different constructs was analysed in different experiments quite variable results were obtained, limiting our ability to usefully interpret the effects of the 8-pCPT-2′-O-Me-cAMP on transcript levels derived from each construct.
The ESAG9 3′UTR regulatory element governs gene regulation in stumpy forms
Although easier to genetically manipulate, monomorphic cells have lost the ability to generate stumpy forms and represent a poor model for gene regulation of trypanosomes in the mammalian bloodstream. Therefore, having identified a region potentially important in gene regulation between slender and stumpy forms, it was important to analyse whether the same regulatory element functioned in pleomorphic cells capable of the biologically relevant developmental transition. Hence, pleomorphic T. brucei AnTat1.1 cells were transfected with constructs where the reporter gene was under the control of the ESAG9-EQ 3′UTR, the mutant of this 3′UTR lacking the 34 nt regulatory element (eΔ), or the ΔALD 3′UTR in which the 34 nt regulatory element had been inserted (ΔALD eI). The resulting cell lines (two independent cell lines per construct) were then inoculated in to mice and RNA and protein isolated either after 3 days of infection (when slender forms predominated) or after 6 days of infection when the cells had uniformly progressed to morphologically stumpy forms. Fig. 6 demonstrates that the truncated aldolase 3′UTR did not show any developmental regulation at either the protein or RNA level, as expected. Also as expected, the intact ESAG9-EQ 3′UTR resulted in a strong developmental regulation, with RNA increasing 4.75-fold (Fig. 6B) and protein increasing 8.92-fold (Fig. 6A) as the cells developed from slender to stumpy forms. This profile closely agrees with the fivefold elevation of ESAG9-EQ mRNA observed by Digital SAGE transcriptome analysis during the slender–stumpy transition (K. M., unpublished observations). However, when the 34 nt regulatory sequence was deleted, the developmental regulation of the ESAG9-EQ 3′UTR was almost completely lost, such that RNA induction upon stumpy formation was only 1.3-fold, whereas the reporter protein expression actually decreased below the level in slender forms. Interestingly, when the regulatory element sequence was inserted into the ΔALD 3′UTR (ΔALD eI), although no developmental enhancement of reporter protein was observed upon stumpy formation, the transcript levels from this construct increased 3.8-fold (Fig. 6B). This revealed an uncoupling between mRNA and protein levels when the element was out of its normal context, indicating that other ESAG9 3′UTR sequences may contribute to translational efficiency when the 34 nt element is intact. These results indicated that the regulatory element acted to repress gene expression in slender forms at the RNA level and strongly activated gene expression at both the RNA and protein levels upon development to stumpy forms. These results contrasted with the relatively subtle effects observed in monomorphic cells and established that the 34 nt element in the ESAG9-EQ 3′UTR acts as a negative regulatory element in slender forms and positive regulatory element in stumpy forms, with translational regulation being particularly important in the latter. Combined these results indicate that the element is entirely responsible for developmental gene regulation of ESAG9-EQ during parasite development in the mammalian bloodstream.
Fig. 6.
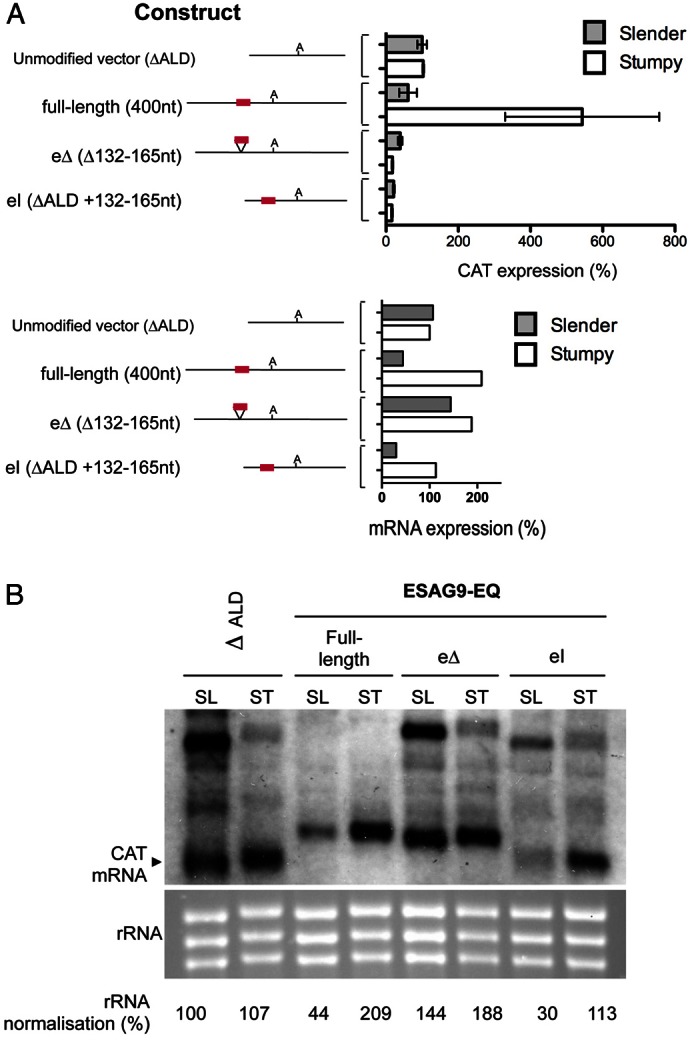
Analysis of the ESAG9-EQ 3′UTR and associated element in true slender and stumpy forms using a CAT reporter gene assay. (A) CAT protein expression (upper panel) and mRNA expression (lower panel) from the constructs in slender form (grey bars) and stumpy form (white bars) cells, shown as a percentage of expression from cell lines of the unmodified vector slender form cells. For protein data, values are the means ± s.e.m. of two biological replicates (cell lines) for each construct. Therefore, because n = 2 for each, the standard error bars represent the data range. To the left of the graph are schematic representations of the sequence analysed in each construct, with the polyadenylation site location indicated with ‘A’ and regulatory element location indicated by the red line. mRNA values are derived by quantification of the signal from the blot shown in B. (B) Northern blot analysis of CAT mRNA, with RNA from the ΔALD vector, full-length ESAG9-EQ 3′UTR, element deletion (eΔ) and element insertion (eI) constructs in slender (SL) and stumpy (ST) forms. Shown in the lower panel is the ethidium bromide staining of the rRNA present, to indicate the loading of each lane.
Folding analysis and conservation of the ESAG9 gene regulatory element
The ESAG9 gene family comprises at least nine intact genes in the T. brucei genome, and all tested genes have demonstrated stumpy-enriched gene expression (Barnwell et al., 2010). Hence, having defined a precise regulatory element controlling the developmental gene expression of ESAG9-EQ we investigated whether the sequence was highly conserved among the 3′UTR of other ESAG9 family members. Firstly a linear alignment of the different 3′UTR sequences (up to 250 nucleotides downstream of the ESAG9 gene in each) was carried out using CLUSTAL Omega. This revealed that the region of the ESAG9-EQ 3′UTR containing the regulatory element showed 47–91% similarity between family members. Interestingly, the 3′UTR sequence of the T. brucei ESAG9-EQ gene was most similar to the 3′UTR from both of the characterised T. equiperdum ESAG9 gene copies (90% and 91% similarity over the regulatory element for ESAG9u and ESA9c, respectively; 86% and 90% over the first 250 nucleotides of the 3′UTR; alignments are shown in Fig. 7A), matching their coding region similarity. This suggests that these gene and regulatory sequences have been conserved since the divergence of these species, and have remained more similar to each other than to other ESAG9 gene sequences within the same genome. Apparently therefore, ESAG9 genes and regulatory sequences are not evolving at a very high frequency despite the diversity of this sequence family.
Fig. 7.

Identification of potential conserved elements in the ESAG9 family 3′UTRs. (A) The regulatory region in different ESAG9 gene 3′UTRs aligned using CLUSTAL Omega. Yellow and purple shaded residues are the nucleotides that form ascending and descending stem structures, determined from analysis of consensus-predicted RNA structures, as detailed in the text. (B) Predicted RNA structures of sequences downstream of ESAG9 genes with similarity to that of the ESAG9-EQ element. These represent the region of the ESAG9-EQ regulatory element folded according to the ‘RNAstructures’ webserver.
Thereafter, we analysed in more detail the potential for secondary structure within the ESAG9-EQ 3′UTR in the presence or absence of the regulatory region. This was carried out by comparing the minimum folding energy (MFE) of the wild-type or eΔ sequences against 50–100 copies of each sequence permuted in base order, in each case these being constrained in order to maintain the dinucleotide frequencies (supplementary material Table S1). Thereafter, analysis of the difference in MFE between the sequences (MFED) and the position of the native sequence in the distribution of control MFE values (Z-score) provided an indication of its sequence-order-dependent structure and its dependence on the presence of the regulatory element. This analysis revealed that the ESAG9-EQ 3′UTR shows evidence for secondary structure (with an MFED value of around 20% and a Z-score of <−2 (P<0.05), whereas deletion of the regulatory element abolished this (supplementary material Table S1). This highlighted that it was only this sequence within the 3′UTR that provided support for a predicted secondary structure. Confirming the sequence specificity of the predicted structure in the ESAG9-EQ 3′UTR, a control analysis using antisense copies of sequences (and exploiting G–U asymmetry), showed no evidence for sequence-order-dependent folding.
Although this analysis provided support for a structural element coinciding with the ESAG9-EQ regulatory sequence, using the same analysis for all other ESAG9 family 3′UTRs failed to provide a MFED score consistent with the existence of a conserved secondary structural element (supplementary material Table S1). Therefore, we reanalysed the ESAG9 family 3′UTRs for common secondary structures using multiple structural comparisons through the ‘RNAstructures’ webserver (http://rna.urmc.rochester.edu/RNAstructureWeb/Servers/Predict3/Predict3.html). Analysis of the structures with the greatest conservation between the multiple structural predictions of the ESAG9 family revealed that the regulatory element sequence had a predicted propensity to fold into one or two stem loop structures terminated by a common AAU sequence element at the base of the distal stem (Fig. 7B, components of the ascending stem and descending stem are highlighted in yellow and purple, respectively). Hence, consensus folding analysis has provided evidence for a structural component to the regulatory element in ESAG9-EQ, although independent analyses of predicted pseudoknots and tertiary structural elements via other algorithms did not provide further support for a generally conserved structure (P. S., unpublished observation).
Discussion
To date, the study of most regulated gene expression in trypanosomes has focussed on the differentiation between bloodstream and tsetse midgut procyclic forms. During this transition, several examples where 3′UTRs are important to lifecycle stage-dependent control of expression have been identified (reviewed by Schwede et al., 2011). The most extensively characterised 3′UTRs are those of the procyclin genes. For example, the 3′UTR of EP procyclin contains three regulatory elements; two positive elements (loops I and III) and a negative element (loop II) (Furger et al., 1997). The EP procyclin gene is positively regulated by a small RNA binding protein, TbZFP3; when overexpressed TbZFP3 promotes EP procyclin expression, this being dependent upon sequences in the loop II element, such that TbZFP3 has been proposed to displace a negative regulatory protein (Walrad et al., 2009). Another procyclin isoform, GPEET procyclin, is regulated by glycerol and hypoxia (Vassella et al., 2000), this being mediated by a glycerol response element in its 3′UTR that is absent in EP procyclin mRNAs. Other developmentally regulated mRNAs are also regulated via their 3′UTR, though conserved elements responsible for widespread developmental regulation have proved difficult to identify. For example, the functionally and coordinately regulated nuclear encoded components of the cytochrome oxidase complex are all upregulated upon differentiation to procyclic forms, but show different emphasis on either mRNA stability or translational control, and little evidence for identifiable common regulatory motifs between the distinct mRNAs (Mayho et al., 2006). Overall, therefore, the signals governing developmental expression are largely 3′UTR directed, but consensus regulatory motifs and regulatory proteins have proved elusive.
Here we focussed on a family of co-regulated genes that are developmentally enriched in bloodstream stumpy forms, thereby providing a route to identify regulatory signals important for this transition. Moreover, we anticipated that analysis of the conservation of identified regulatory regions within the co-regulated family members would help to decipher cryptic signals and structures that control expression in a developmental regulon.
Dissection of the 3′UTR of ESAG9-EQ identified a highly defined regulatory region of only 34 nt that governed the developmental expression of this gene. Interestingly, the element exhibited both negative and positive regulatory potential, depending on the developmental stage of the parasite. Hence, in slender forms, the element contributed to the repression of ESAG9-EQ gene expression, whereas upon development to stumpy forms, the element enabled the strong activation of ESAG9-EQ expression. This identified the 34 nt region as a tightly defined bifunctional regulatory element, among the shortest such regions identified to date in trypanosome gene expression. In Fig. 8 we present a model for how this regulatory element might function. In the simplest scenario, the element might be bound by a negative regulator in slender forms, this being displaced by a positive regulator in stumpy forms. This scenario would resemble the regulation of EP procyclin by the small RNA binding protein TbZFP3, where TbZFP3 apparently counteracts a negative regulator operating upon loop II of the EP procyclin 3′UTR (Walrad et al., 2009). Although the regulatory element within the 3′UTR affects both mRNA levels and protein levels, insertion of the element in isolation into the 3′UTR of a constitutively expressed gene in pleomorphic cells repressed mRNA levels but did not strongly affect protein levels. Hence, the element apparently influences gene expression at more than one level, this being influenced by the context in which it is placed. Other sequences in the ESAG9 3′UTR might, therefore, contribute to translation of the mRNA in the presence of the 34 nt element, for example by engaging or stabilising the association of positive regulators of translation. Since the protein level expression in stumpy forms was considerably higher than in monomorphic forms induced to become stumpy-like by the cell permeable cAMP analogue, we propose that either monomorphic cells have lost such factors, or that cell permeable cAMP analogues drive only an incomplete development to cells with only some stumpy characteristics.
Fig. 8.
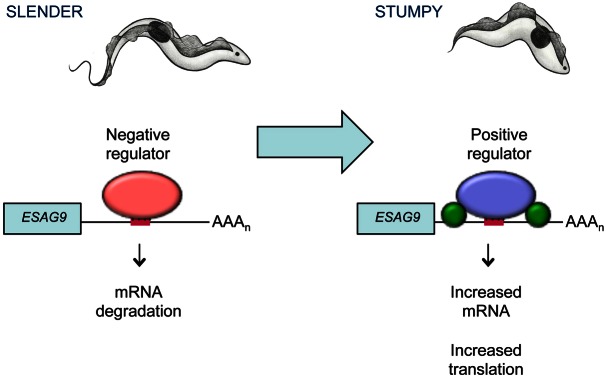
Proposed model for the mechanisms of regulation of ESAG9-EQ expression in slender and stumpy forms. In slender forms (left-hand side), a negative regulator (red oval) binds to the element sequence (red bar) in the ESAG9-EQ 3′UTR and causes mRNA degradation of the transcript, repressing the expression of ESAG9-EQ. In stumpy forms (right-hand side), a positive regulator (purple oval) binds to the element sequence, increasing its mRNA abundance. Translation of the transcript also increases, potentially through the association or stabilisation of additional regulators (green circles) that require other sequences in the ESAG9 3′UTR outside the 34 nt element. Combined, these result in the release of gene expression repression in the stumpy lifecycle stage. AAAn: poly(A) tail.
Although the regulatory element was identified through analysis of reporter assays in monomorphic forms, the effects of its deletion, and insertion into other 3′UTRs were quite limited. In contrast, when the element was analysed in pleomorphic slender and stumpy forms, dramatic changes in gene expression were conferred by the presence of the regulatory element, such that almost 10-fold changes in reporter protein expression were observed. This scenario matches the analysis for another stumpy enriched gene, PAD1, where the regulation was more pronounced in true stumpy forms than monomorphic cells- even when the latter were induced to undergo stumpy-like formation with cell permeable cAMP analogues (MacGregor and Matthews, 2012). This reemphasises the distinction between monomorphic trypanosomes and pleomorphic trypanosomes (Matthews et al., 2004) and highlights that monomorphic cells provide a poor model for developmental events in the mammalian bloodstream even when artificially induced to generate cells with stumpy characteristics by chemical treatment. Clearly, developmental events are more dramatic and robust in cells naturally capable of stumpy formation, and such cells are necessary to provide an accurate picture of the regulatory events that prepare the cell for transmission in the mammalian bloodstream.
The developmental co-regulation of ESAG9 transcripts suggested that common regulatory sequences would be uncovered by the dissection of the 3′UTR of one family member. However, linear alignment of all ESAG9 gene 3′UTRs did not reveal stringent identity of the different family members over the key regulatory region identified in the ESAG9-EQ 3′UTR, limiting the value of more detailed mutational analysis. Rather, this suggests that higher order structures contribute to the specific recognition of mRNAs of this gene family. While Sfold comparisons and multiple RNA fold alignments highlighted that a potential stem loop region is contained within the 34 nt regulatory section, a strikingly conserved structural pattern was not observed. This highlights that predictive approaches to RNA structure are currently insufficient to uncover the conservation within genes that are highly likely to share interactions with regulatory RNA binding proteins. Nonetheless, the identification of a short and dominant regulatory motif in the ESAG9-EQ 3′UTR provides a powerful ligand to select such regulatory RNA binding proteins from slender and stumpy forms and their identification will allow mapping of the contact sites on the ESAG9 3′UTR from different family members. For this reason, the precise identification of the regulatory element in ESAG9-EQ 3′UTR provides an invaluable reagent to further understanding of developmental gene expression in trypanosomes.
In summary, the regulation of ESAG9-EQ has uncovered the most highly defined regulatory motif yet identified as being important in the developmental control of trypanosome gene expression in the mammalian bloodstream. This element acts as both a negative and positive regulator, dependent on the developmental context of the parasite, indicating that it acts as a downstream target of the quorum-sensing pathway. As signalling pathways in trypanosomes become better characterised, the components of the regulatory network that interact to govern development of the parasite in its mammalian host can be connected. Breaking these networks pharmacologically has obvious potential in manipulating the virulence and transmission potential of the parasite in a disease relevant stage of its life cycle.
Materials and Methods
Trypanosomes
Monomorphic bloodstream form T. b. brucei strain Lister 427 were maintained at 37°C with 5% CO2 in HMI-9 medium (Hirumi and Hirumi, 1989) containing 20% foetal calf serum (FCS; Fetal Bovine Serum Gold, PAA Laboratories Ltd, Yeovil, Somerset, UK) and penicillin-streptomycin (100 Units/ml penicillin, 100 µg/ml streptomycin; Invitrogen, Life technologies, Paisley, Scotland).
Mice (male, MF1 strain) were infected with pleomorphic trypanosomes (T. brucei AnTat1.1 90∶13 strain) by intraperitonial (IP) injection. If infected for the harvest of a stumpy form population, the mouse was injected (IP) with 250 µl cyclophosphamide (25 mg/ml) the day prior to infection. Infected blood was collected by cardiac puncture and allowed to pass through a diethylaminoethyl (DEAE) cellulose (DE-52; GE Healthcare, Little Chalfont, Buckinghamshire) anion exchange column (Lanham and Godfrey, 1970) equilibrated with PSG (pH 7.8). All animal experiments were performed according to UK Home Office approved guidelines as specified under Project licence no. 60/4373.
Subcloning
Reporter constructs used the previously described CAT449 plasmid (Biebinger et al., 1997) containing the chloramphenicol acetyltransferase (CAT) coding region (Mayho et al., 2006). Digestion with NotI results in linearisation of the CAT449 vector in the tubulin targeting sequence. Consequently, integration in to the T. brucei genome is through the β-tubulin locus and the CAT reporter gene is transcribed by read-through Pol II transcription of this locus. Investigation of experimental 3′UTRs was made by replacement of the truncated aldolase (ΔALD) 3′UTR present in the CAT449 vector through BamHI and BbsI restriction sites. This was achieved through PCR amplification of the 3′UTR sequence using genomic DNA from T. b. brucei as template (primer 1: ESAG9-EQ FL forward; 5′-cccggatccaaatcattaccgcaattcc-3′; primer 2: ESAG9-EQ FL reverse; 5′-cccgaagaccttgctgttatcacgttccc-3′). The element deletion (eΔ) 3′UTR sequence was generated by two PCR reactions performed using the full-length 3′UTR sequence PCR product as template; one reaction with primers 1 and 6 (primer 6; element deletion R: 5′-ggggtctagagttccatccacatgatcttcattg-3′) to amplify the region of the full-length 3′UTR sequence upstream of the element sequence, the other with primers 2 and 7 (primer 7; element deletion F: 5′-ggggtctagaactaatgtaatcataagtagcgtacatgag-3′) to amplify the region of the full-length 3′UTR sequence downstream of the element sequence. To adjoin the two PCR amplicons, XbaI restriction enzyme sites where included in primers 6 and 7. The two PCR amplicons were used in a single ligation reaction with BamHI- and BbsI-digested CAT449 vector. The element insertion (eI) 3′UTR sequence was synthesised by GENEART AG (Invitrogen, Paisley, UK), in their pMA-T vector with flanking BamHI and BbsI restriction enzyme sites for ligation in to the CAT449 vector. Each amplicon was initially subcloned in to pGEM T-easy vector (Promega UK, Southampton, UK) for sequence verification. The deletion series was created using primer 2 in combination with primer 3 (primer 3: ESAG9-EQ 3′UTR 350 nt F; 5′-ggccggatccaacaccgcatgatg-3′; 350 nt construct); primer 2 with primer 4 (primer 4; ESAG9-EQ 3′UTR 300 nt F; 5′-ggcccggatccctgctttcaatgaag-3′; 300 nt construct) or primer 2 with primer 5 (primer 5: ESAG9-EQ 3′UTR 250 nt F; 5′-ggcgcggatccagaggatcgaataac-3′; 250 nt construct).
Transfection
Transfection of monomorphic bloodstream form cells was performed as previously (MacGregor and Matthews, 2012) described with 15 µg of NotI-linearised DNA. 24 hours post-transfection, selection was made with 0.5–2.5 µg/ml phleomycin on serial dilutions of transfected cells to allow clonal selection, with a control of drug-treated parental (non-transfected) cells to confirm drug sensitivity. Transfection of pleomorphic cells was as described previously (MacGregor and Matthews, 2012).
CAT ELISA assay
To determine the level of CAT protein derived from each construct, CAT ELISA assays (Roche Products Ltd, Welwyn Garden City, UK) were used according to the manufacturer's instructions. Absorbance values at 405 nm were measured using a BioTek ELx808 ELISA microplate reader and for each assay a CAT standard curve was calculated. Each assay included the same independently derived clone of the unmodified vector, which was used for comparison between assays. This clone was representative of several independently derived cell lines for the same construct (supplementary material Fig. S1).
Northern blot analysis
RNA extraction was performed using an RNeasy RNA purification kit (Qiagen Ltd, Crawley, UK) according to the manufacturer's instructions for ‘animal cell spin’ RNA purification. RNA (typically 2.0 µg per sample) was resolved on formaldehyde-agarose gels, transferred to nylon membranes using the upward capillary transfer method and cross-linked on to the membrane at 0.12 joules using a UV Crosslinker (Uvitec). Riboprobes were generated using the DIG RNA labeling kit (Roche Products Ltd, Welwyn Garden City, UK) according to the manufacturer's instructions. For transcript detection CDP-star (Roche Products Ltd, Welwyn Garden City, UK) was used and the signal identified by exposure to X-ray film or using the Chemiluminescence function of a GBOX (Syngene, Cambridge, UK) for transcript quantification by normalisation to rRNA level.
8-pCPT-2′-O-Me-cAMP treatment
8-(4-Chlorophenylthio)-2′-O-methyladenosine-3′,5′-cyclic monophosphate (8-pCPT-2′-O-Me-cAMP; Biolog Life Science Institute, Bremen, Germany) dissolved in DMSO (DMSO) to a concentration of 10 mM was added to the cell culture (∼5×105 cells/ml concentration) to a final concentration of 100 µM. As a control, to an identical cell culture, the same volume of DMSO alone was added. Following incubation at 37°C for 48 hours, 5 ml of each culture was used to generate cell lysate samples for use in a CAT ELISA assay and the remaining culture used for RNA extraction and then northern blot analysis (as described above).
Bioinformatics and RNA structure prediction
For basic prediction of RNA secondary structure the Srna application module of the freely available web-based software package Sfold (Ding et al., 2004) was used with the default settings (http://sfold.wadsworth.org/cgi-bin/index.pl). Multi-alignment of DNA sequences was performed using ClustalW2 (Multiple Sequence Alignment tool, version 2.1) or Clustal Omega with the default settings (Larkin et al., 2007). For more in-depth analysis, mean folding energies (MFEs) were calculated for 250 base segments of the 3′UTR from each ESAG9 3′UTR using UNAfold (Markham and Zuker, 2005) within the SSE package (Simmonds, 2012). For each sequence, MFE differences from the null expectation (MFEDs) and Z-scores (Workman and Krogh, 1999) were calculated by parallel submission of 100 control sequences scrambled using the NDR algorithm in SSE that retains biases in dinucleotide frequencies (Simmonds et al., 2004). Results were expressed as MFE differences {MFEDs; i.e. the percentage difference in MFEs of native and scrambled sequence, calculated as [(MFENATIVE/MFESCRAMBLED)−1]×100, and as Z-scores (the position of the native sequence in the distribution of control values (Workman and Krogh, 1999)}. Alternative approaches for RNA structure prediction in nucleotide sequences aligned by the program MUSCLE (Edgar, 2004) used PFOLD, a stochastic context-free grammar method to identify phylogenetically conserved co-variant sites supportive of an RNA structure model (Knudsen and Hein, 1999), by STRUCTUREDIST in the SSE package that identifies conserved paired and unpaired bases in minimum energy fold (Simmonds, 2012), and a combined minimum energy/covariant site detection method implemented in ALIFOLD (Gruber et al., 2008).
Supplementary Material
Acknowledgments
We thank Julie Wilson for generating stumpy form parasites and for technical assistance, Dr Paula MacGregor for advice and the pleomorphic ΔALD cell line and Dr Margo Chase-Topping for statistical analysis.
Footnotes
Author contributions
S.M. performed the experiments; S.M. and K.M. analysed the data; P.S., S.M. and K.M. carried out RNA structural analyses; S.M., P.S. and K.M. wrote the manuscript.
Funding
This work was supported by a Wellcome Trust Programme grant [grant number 088293MA to K.M.]; a Strategic Award to support the Centre for Immunity, Infection and Evolution from the Wellcome Trust [grant number 095831MA to K.M. and P.S.]; and a studentship from the Medical Research Council (UK) [grant number G0700029 to S.M.]. Deposited in PMC for release after 6 months.
Supplementary material available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.126011/-/DC1
References
- Barnwell E. M., van Deursen F. J., Jeacock L., Smith K. A., Maizels R. M., Acosta-Serrano A., Matthews K. (2010). Developmental regulation and extracellular release of a VSG expression-site-associated gene product from Trypanosoma brucei bloodstream forms. J. Cell Sci. 123, 3401–3411 10.1242/jcs.068684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biebinger S., Wirtz L. E., Lorenz P., Clayton C. (1997). Vectors for inducible expression of toxic gene products in bloodstream and procyclic Trypanosoma brucei. Mol. Biochem. Parasitol. 85, 99–112 10.1016/S0166-6851(96)02815-0 [DOI] [PubMed] [Google Scholar]
- Borst P. (2002). Antigenic variation and allelic exclusion. Cell 109, 5–8 10.1016/S0092-8674(02)00711-0 [DOI] [PubMed] [Google Scholar]
- Brecht M., Parsons M. (1998). Changes in polysome profiles accompany trypanosome development. Mol. Biochem. Parasitol. 97, 189–198 10.1016/S0166-6851(98)00151-0 [DOI] [PubMed] [Google Scholar]
- Clayton C. E. (2002). Life without transcriptional control? From fly to man and back again. EMBO J. 21, 1881–1888 10.1093/emboj/21.8.1881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cully D. F., Ip H. S., Cross G. A. (1985). Coordinate transcription of variant surface glycoprotein genes and an expression site associated gene family in Trypanosoma brucei. Cell 42, 173–182 10.1016/S0092-8674(85)80113-6 [DOI] [PubMed] [Google Scholar]
- Dean S. D., Marchetti R., Kirk K., Matthews K. R. (2009). A surface transporter family conveys the trypanosome differentiation signal. Nature 459, 213–217 10.1038/nature07997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Y., Chan C. Y., Lawrence C. E. (2004). Sfold web server for statistical folding and rational design of nucleic acids. Nucleic Acids Res. 32, W135–W141 10.1093/nar/gkh449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 10.1093/nar/gkh340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florent I. C., Raibaud A., Eisen H. (1991). A family of genes related to a new expression site-associated gene in Trypanosoma equiperdum. Mol. Cell. Biol. 11, 2180–2188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furger A., Schürch N., Kurath U., Roditi I. (1997). Elements in the 3′ untranslated region of procyclin mRNA regulate expression in insect forms of Trypanosoma brucei by modulating RNA stability and translation. Mol. Cell. Biol. 17, 4372–4380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruber A. R., Lorenz R., Bernhart S. H., Neuböck R., Hofacker I. L. (2008). The Vienna RNA websuite. Nucleic Acids Res. 36, W70–W74 10.1093/nar/gkn188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Günzl A., Bruderer T., Laufer G., Schimanski B., Tu L. C., Chung H. M., Lee P. T., Lee M. G. (2003). RNA polymerase I transcribes procyclin genes and variant surface glycoprotein gene expression sites in Trypanosoma brucei. Eukaryot. Cell 2, 542–551 10.1128/EC.2.3.542-551.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertz-Fowler C., Figueiredo L. M., Quail M. A., Becker M., Jackson A., Bason N., Brooks K., Churcher C., Fahkro S., Goodhead I. et al. (2008). Telomeric expression sites are highly conserved in Trypanosoma brucei. PLoS ONE 3, e3527 10.1371/journal.pone.0003527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirumi H., Hirumi K. (1989). Continuous cultivation of Trypanosoma brucei blood stream forms in a medium containing a low concentration of serum protein without feeder cell layers. J. Parasitol. 75, 985–989 10.2307/3282883 [DOI] [PubMed] [Google Scholar]
- Jensen B. C., Sivam D., Kifer C. T., Myler P. J., Parsons M. (2009). Widespread variation in transcript abundance within and across developmental stages of Trypanosoma brucei. BMC Genomics 10, 482 10.1186/1471-2164-10-482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knudsen B., Hein J. (1999). RNA secondary structure prediction using stochastic context-free grammars and evolutionary history. Bioinformatics 15, 446–454 10.1093/bioinformatics/15.6.446 [DOI] [PubMed] [Google Scholar]
- Kooter J. M., van der Spek H. J., Wagter R., d'Oliveira C. E., van der Hoeven F., Johnson P. J., Borst P. (1987). The anatomy and transcription of a telomeric expression site for variant-specific surface antigens in T. brucei. Cell 51, 261–272 10.1016/0092-8674(87)90153-X [DOI] [PubMed] [Google Scholar]
- Landeira D., Navarro M. (2007). Nuclear repositioning of the VSG promoter during developmental silencing in Trypanosoma brucei. J. Cell Biol. 176, 133–139 10.1083/jcb.200607174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanham S. M., Godfrey D. G. (1970). Isolation of salivarian trypanosomes from man and other mammals using DEAE-cellulose. Exp. Parasitol. 28, 521–534 10.1016/0014-4894(70)90120-7 [DOI] [PubMed] [Google Scholar]
- Larkin M. A., Blackshields G., Brown N. P., Chenna R., McGettigan P. A., McWilliam H., Valentin F., Wallace I. M., Wilm A., Lopez R. et al. (2007). Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948 10.1093/bioinformatics/btm404 [DOI] [PubMed] [Google Scholar]
- Laxman S., Riechers A., Sadilek M., Schwede F., Beavo J. A. (2006). Hydrolysis products of cAMP analogs cause transformation of Trypanosoma brucei from slender to stumpy-like forms. Proc. Natl. Acad. Sci. USA 103, 19194–19199 10.1073/pnas.0608971103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacGregor P., Matthews K. R. (2012). Identification of the regulatory elements controlling the transmission stage-specific gene expression of PAD1 in Trypanosoma brucei. Nucleic Acids Res 40, 7705–7717 10.1093/nar/gks533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacGregor P., Savill N. J., Hall D., Matthews K. R. (2011). Transmission stages dominate trypanosome within-host dynamics during chronic infections. Cell Host Microbe 9, 310–318 10.1016/j.chom.2011.03.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacGregor P., Szöőr B., Savill N. J., Matthews K. R. (2012). Trypanosomal immune evasion, chronicity and transmission: an elegant balancing act. Nat. Rev. Microbiol. 10, 431–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markham N. R., Zuker M. (2005). DINAMelt web server for nucleic acid melting prediction. Nucleic Acids Res. 33, W577–W581 10.1093/nar/gki591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews K. R., Ellis J. R., Paterou A. (2004). Molecular regulation of the life cycle of African trypanosomes. Trends Parasitol. 20, 40–47 10.1016/j.pt.2003.10.016 [DOI] [PubMed] [Google Scholar]
- Mayho M., Fenn K., Craddy P., Crosthwaite S., Matthews K. (2006). Post-transcriptional control of nuclear-encoded cytochrome oxidase subunits in Trypanosoma brucei: evidence for genome-wide conservation of life-cycle stage-specific regulatory elements. Nucleic Acids Res. 34, 5312–5324 10.1093/nar/gkl598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro M., Gull K. (2001). A pol I transcriptional body associated with VSG mono-allelic expression in Trypanosoma brucei. Nature 414, 759–763 10.1038/414759a [DOI] [PubMed] [Google Scholar]
- Navarro M., Peñate X., Landeira D. (2007). Nuclear architecture underlying gene expression in Trypanosoma brucei. Trends Microbiol. 15, 263–270 10.1016/j.tim.2007.04.004 [DOI] [PubMed] [Google Scholar]
- Paindavoine P., Rolin S., Van Assel S., Geuskens M., Jauniaux J. C., Dinsart C., Huet G., Pays E. (1992). A gene from the variant surface glycoprotein expression site encodes one of several transmembrane adenylate cyclases located on the flagellum of Trypanosoma brucei. Mol. Cell. Biol. 12, 1218–1225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pays E., Tebabi P., Pays A., Coquelet H., Revelard P., Salmon D., Steinert M. (1989). The genes and transcripts of an antigen gene expression site from T. brucei. Cell 57, 835–845 10.1016/0092-8674(89)90798-8 [DOI] [PubMed] [Google Scholar]
- Pays E., Lips S., Nolan D., Vanhamme L., Pérez-Morga D. (2001). The VSG expression sites of Trypanosoma brucei: multipurpose tools for the adaptation of the parasite to mammalian hosts. Mol. Biochem. Parasitol. 114, 1–16 10.1016/S0166-6851(01)00242-0 [DOI] [PubMed] [Google Scholar]
- Reuner B., Vassella E., Yutzy B., Boshart M. (1997). Cell density triggers slender to stumpy differentiation of Trypanosoma brucei bloodstream forms in culture. Mol. Biochem. Parasitol. 90, 269–280 10.1016/S0166-6851(97)00160-6 [DOI] [PubMed] [Google Scholar]
- Rudenko G. (2011). African trypanosomes: the genome and adaptations for immune evasion. Essays Biochem. 51, 47–62 [DOI] [PubMed] [Google Scholar]
- Rudenko G., Bishop D., Gottesdiener K., Van der Ploeg L. H. (1989). Alpha-amanitin resistant transcription of protein coding genes in insect and bloodstream form Trypanosoma brucei. EMBO J. 8, 4259–4263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmon D., Geuskens M., Hanocq F., Hanocq-Quertier J., Nolan D., Ruben L., Pays E. (1994). A novel heterodimeric transferrin receptor encoded by a pair of VSG expression site-associated genes in T. brucei. Cell 78, 75–86 10.1016/0092-8674(94)90574-6 [DOI] [PubMed] [Google Scholar]
- Salmon D., Bachmaier S., Krumbholz C., Kador M., Gossmann J. A., Uzureau P., Pays E., Boshart M. (2012a). Cytokinesis of Trypanosoma brucei bloodstream forms depends on expression of adenylyl cyclases of the ESAG4 or ESAG4-like subfamily. Mol. Microbiol. 84, 225–242 10.1111/j.1365-2958.2012.08013.x [DOI] [PubMed] [Google Scholar]
- Salmon D., Vanwalleghem G., Morias Y., Denoeud J., Krumbholz C., Lhommé F., Bachmaier S., Kador M., Gossmann J., Dias F. B. et al. (2012b). Adenylate cyclases of Trypanosoma brucei inhibit the innate immune response of the host. Science 337, 463–466 10.1126/science.1222753 [DOI] [PubMed] [Google Scholar]
- Schwede A., Kramer S., Carrington M. (2011). How do trypanosomes change gene expression in response to the environment? Protoplasma 249, 223–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmonds P. (2012). SSE: a nucleotide and amino acid sequence analysis platform. BMC Res. Notes 5, 50 10.1186/1756-0500-5-50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmonds P., Tuplin A., Evans D. J. (2004). Detection of genome-scale ordered RNA structure (GORS) in genomes of positive-stranded RNA viruses: Implications for virus evolution and host persistence. RNA 10, 1337–1351 10.1261/rna.7640104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassella E., Reuner B., Yutzy B., Boshart M. (1997). Differentiation of African trypanosomes is controlled by a density sensing mechanism which signals cell cycle arrest via the cAMP pathway. J. Cell Sci. 110, 2661–2671 [DOI] [PubMed] [Google Scholar]
- Vassella E., Den Abbeele J. V., Bütikofer P., Renggli C. K., Furger A., Brun R., Roditi I. (2000). A major surface glycoprotein of trypanosoma brucei is expressed transiently during development and can be regulated post-transcriptionally by glycerol or hypoxia. Genes Dev. 14, 615–626 [PMC free article] [PubMed] [Google Scholar]
- Walrad P., Paterou A., Acosta-Serrano A., Matthews K. R. (2009). Differential trypanosome surface coat regulation by a CCCH protein that co-associates with procyclin mRNA cis-elements. PLoS Pathog. 5, e1000317 10.1371/journal.ppat.1000317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Workman C., Krogh A. (1999). No evidence that mRNAs have lower folding free energies than random sequences with the same dinucleotide distribution. Nucleic Acids Res. 27, 4816–4822 10.1093/nar/27.24.4816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xong H. V., Vanhamme L., Chamekh M., Chimfwembe C. E., Van Den Abbeele J., Pays A., Van Meirvenne N., Hamers R., De Baetselier P., Pays E. (1998). A VSG expression site-associated gene confers resistance to human serum in Trypanosoma rhodesiense. Cell 95, 839–846 10.1016/S0092-8674(00)81706-7 [DOI] [PubMed] [Google Scholar]
- Young R., Taylor J. E., Kurioka A., Becker M., Louis E. J., Rudenko G. (2008). Isolation and analysis of the genetic diversity of repertoires of VSG expression site containing telomeres from Trypanosoma brucei gambiense, T. b. brucei and T. equiperdum. BMC Genomics 9, 385 10.1186/1471-2164-9-385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zomerdijk J. C., Kieft R., Borst P. (1991). Efficient production of functional mRNA mediated by RNA polymerase I in Trypanosoma brucei. Nature 353, 772–775 10.1038/353772a0 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.