

Abstract
The synthesis of the pharmaceutical (R)-tolterodine is reported using lithiation/borylation–protodeboronation of a homoallyl carbamate as the key step. This step was tested with two permutations: an electron-neutral aryl Li-carbamate reacting with an electron-rich boronic ester and an electron-rich aryl Li-carbamate reacting with an electron-neutral boronic ester. It was found that the latter arrangement was considerably better than the former. Further improvements were achieved using magnesium bromide in methanol leading to a process that gave high yield and high enantioselectivity in the lithiation/borylation reaction. The key step was used in an efficient synthesis of (R)-tolterodine in a total of eight steps in a 30% overall yield and 90% ee.
Keywords: asymmetric synthesis, boronic esters, gem-diarylalkyl, lithiation/borylation reaction, tolterodine
Introduction
The enantioselective synthesis of pharmaceuticals and biologically active natural products is a major theme for synthetic chemists. In particular, the formation of chiral gem-diarylalkyl compounds, a motif that is present in many therapeutically important molecules, is especially challenging owing to the lack of nearby functional groups that might be enlisted to enable the formation of C–C bonds.1 Examples of these compounds include (R)-tolterodine (1, Detrol), (+)-sertraline (2, Zoloft), and (+)-indatraline (3) (Fig. 1).
Fig. 1.

Pharmaceutically important molecules containing the gem-diaryl substructure.
(R)-Tolterodine (1) is a potent competitive muscarine receptor antagonist for the treatment of urinary incontinence and other symptoms related to an overactive bladder,2 a compound that has achieved blockbuster status ($1.21 billion in sales in 20083). Because of its importance and challenging gem-diarylalkyl stereocenter, it has become a popular target for asymmetric synthesis. Whereas the original route relied on optical resolution to separate (R)-tolterodine from the racemate,4 initial asymmetric routes used chiral auxiliaries5 or asymmetric Me-CBS (Corey–Bakshi–Shibata) reduction6 to introduce the stereogenic center. More recently, asymmetric strategies have utilized transition-metal-catalyzed processes including hydrogenation,7 hydroformylation,8 or arylboration.9
Recently, we reported the highly enantioselective syntheses of (+)-sertraline (2) and (+)-indatraline (3) using a lithiation/borylation10–protodeboronation11 strategy (Scheme 1). (+)-Sertraline (2), a potent competitive selective serotonin reuptake inhibitor (SSRI), is a commonly prescribed therapeutic for the treatment of depression and other anxiety-related disorders and has also achieved blockbuster status12 ($3.36 billion in sales in 200413). The related molecule, (+)-indatraline (3), is a potent psychoactive compound that acts as a monoamine reuptake inhibitor14 and has the potential to be used in maintenance therapy to treat cocaine abuse.15 A lithiation/borylation reaction and subsequent protodeboronation gave access to both 2 and 3 in high yield and excellent enantioselectivity. In contrast with other substrates devoid of the alkene function, for homoallylic carbamate 4 it was found necessary to add a crown ether or to carry out a solvent exchange to CHCl3 to achieve 1,2-metalate rearrangement of the intermediate ate complex.11b The requirement for such unusual conditions has made us probe this type of rearrangement further with a view to applying it in the asymmetric synthesis of (R)-tolterodine. Once again, we found that modifications of the improved conditions for this class of substrates were required to promote 1,2-metallate rearrangement, but this did ultimately enable us to complete a successful synthesis of (R)-tolterodine using our lithiation/borylation–protodeboronation strategy.
Scheme 1.

Enantioselective syntheses of (+)-sertraline (2) and (+)-indatraline (3) using a lithiation/borylation–protodeboronation strategy.
Results and discussion
Encouraged by the results for the synthesis of (+)-sertraline and (+)-indatraline, we applied a similar strategy for the synthesis of (R)-tolterodine (1). The retrosynthetic analysis for 1 (Scheme 2) involves, as a key step, the lithiation/borylation of benzylic carbamate 4 with boronic ester 7 to form 8. Subsequent protodeboronation of tertiary boronic ester 8 would lead to olefin 9. Both the lithiation/borylation10 and protodeboronation11 have been found to be highly stereoselective, occurring with retention of the stereochemistry. Functional group manipulation of the key intermediate 9 would then provide (R)-tolterodine (1).
Scheme 2.

Retrosynthetic scheme for the synthesis of (R)-tolterodine (1).
However, attempts to realize this in practice were not straightforward: when we carried out the lithiation/borylation of carbamate 4 with boronic ester 7, using 12-crown-4, trimethylsilyl chloride (TMSCl), and H2O as additives, no product was formed (Scheme 3). A solvent switch after ate complex formation from diethyl ether to CHCl3 gave only trace amounts of tertiary boronic ester 8. Both sets of conditions had been successfully employed in the synthesis of sertraline; the only difference here was the use of 2-methoxy-5-methylphenyl boronic ester in place of 3,4-dichlorophenyl boronic ester. Monitoring the reaction by 11B NMR revealed that an intermediate boron–ate complex (A) formed (~5 ppm), but instead of a 1,2-migration to the product, ate complex A decomposed over time returning some starting material.
Scheme 3.
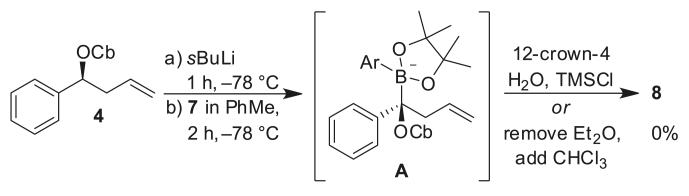
Lithiation/borylation of carbamate 4 and boronic ester 7.
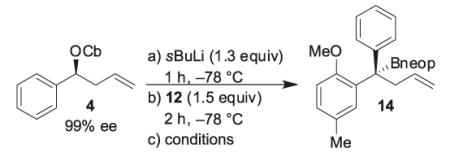
Previous experiments in the syntheses of 2 and 3 revealed that the double bond can interfere in the lithiation/borylation process, presumably by formation of a Li–π complex, as the saturated compound underwent smooth lithiation/borylation under standard conditions (either (i) no additives were required, simply warming to room temperature (RT) or (ii) the addition of MgBr2/MeOH prior to warming to RT).11b To determine whether the problem was related to the double bond, propylcarbamate 10 was tested in the lithiation/borylation reaction with pinacol boronic ester 7 (Scheme 4). The formation of a boron–ate complex was confirmed by 11B NMR, but again no product could be isolated. We believed that the extra steric hindrance of the aryl boronic ester (bearing an ortho substituent) was perhaps responsible for the low reactivity. We therefore investigated the use of the less-hindered neopentyl boronic ester 12, instead of the pinacol boronic ester 7, under the same reaction conditions. Utilizing carbamate 10 and neopentyl boronic ester 12 in the lithiation/borylation reaction gave tertiary boronic ester 13 in 62% isolated yield after column chromatography. To our delight, lithiation/borylation of homoallylic carbamate 4 (99% enantiomeric excess (ee)) and neopentyl boronic ester 12 gave tertiary boronic ester 14 in good yield and high enantioselectivity (Table 1, entry 1). This showed that the presence of the double bond was not interfering in the reaction. However, attempts to improve both yield and enantioselectivity by the addition of additives after ate complex formation were not successful (Table 1, entries 2–8).
Scheme 4.

Lithiation/borylation of carbamate 10, pinacol boronic ester 7, and neopentyl boronic ester 12.
Table 1.
Optimization of the lithiation/borylation reaction conditions for carbamate 4.
| Entry | Conditions | Yield (%) | es (%)a |
|---|---|---|---|
| 1 | No additives | 52 | 94 |
| 2 | Solvent exchange to CHCl3 | 10 | 90 |
| 3 | Reaction in toluene | 19 | 45 |
| 4 | 12-Crown-4 | 32 | 53 |
| 5 | TMSCl | 8 | 93 |
| 6 | 12-Crown-4, TMSCl | 1 | 93 |
| 7 | MgBr2/MeOH | 1 | 96 |
| 8 | MgBr2·Et2O | 50 | 13 |
Note: es, enantiospecifity. Reaction conditions: 4 (0.3 mol/L in diethyl ether), s-BuLi (1.3 mol/L in cyclohexane/hexane, 98:2), 12 (0.9 mol/L in diethyl ether). Where applicable, 1.2 equiv of additive were used. The enantiomeric excess (ee) was determined after oxidation of an aliquot of the tertiary boronic ester by chiral HPLC.
es = (product ee/starting material ee) × 100.
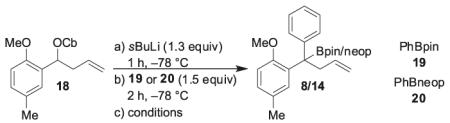
We therefore considered an alternative protocol. Instead of using carbamate 4 with 2-methoxy-5-methylphenyl boronic ester we decided to switch the two aryl groups. In other words, we considered the possibility of the reaction of homoallylic carbamate 18 with phenyl boronic ester in a lithiation/borylation reaction. Although the two ate complexes have similar steric properties, we believed that the presence of the ortho-methoxy group might assist in the expulsion of the carbamate, thereby promoting the 1,2-metallate rearrangement.
Homoallylic carbamate 18 was synthesized in a three-step sequence starting from the commercially available aldehyde 15 (Scheme 5). The phenolic hydroxyl group was protected as the methyl ether to give aldehyde 16. Subsequent, Brown-mediated asymmetric allylation using (+)-B-allyl (diisopinocampheyl)borane ((+)-Ipc2Ball) gave alcohol 17 in 92% ee,16 which was followed by carbamoylation furnishing the desired carbamate in 97% yield and 92% ee.
Scheme 5.

Synthesis of carbamate 18.
The lithiation/borylation reaction between racemic homoallylic carbamate 18 and phenyl pinacol boronic ester 19 now proceeded well to give tertiary boronic ester 8 in 40% yield (Table 2, entry 1). In contrast with our earlier work, the reaction gave only a poor yield when neopentyl boronic ester 20 was used (Table 2, entry 2). Therefore, an optimization of the lithiation/borylation reaction of carbamate 18 with phenyl pinacol boronic ester 19 was carried out. Better results were obtained with the Lewis acidic additives TMSCl or MgBr2 in anhydrous MeOH (Table 2, entries 6 and 8). The optimum conditions/additives of the lithiation/borylation reaction (MgBr2/MeOH; Table 2, entry 8) were applied to the enantioenriched carbamate 18 and we were pleased to find that the reaction proceeded with 98% es (Scheme 6). Hence, we used these optimized conditions to complete the synthesis of (R)-tolterodine.
Table 2.
Optimization of the lithiation/borylation reaction conditions for carbamate 18.
| Entry | Boronic ester | Conditions | Yield (%) |
|---|---|---|---|
| 1 | 19 | No additives | 40 |
| 2 | 20 | No additives | 13 |
| 3 | 19 | Solvent exchange to CHCl3 | 44 |
| 4 | 19 | Reaction in toluene | 16 |
| 5 | 19 | 12-Crown-4 | 33 |
| 6 | 19 | TMSCl | 51 |
| 7 | 19 | 12-Crown-4, TMSCl | 17 |
| 8 | 19 | MgBr2/MeOH | 74 |
| 9 | 19 | MgBr2·Et2O | 15 |
Note: Reaction conditions: 18 (0.3 mol/L in diethyl ether), s-BuLi (1.3 mol/L in cyclohexane/hexane, 98:2), 19 or 20 (0.9 mol/L in diethyl ether). Where applicable, 1.2 equiv of additive were used.
Scheme 6.

Synthesis of (R)-tolterodine (1).
Interestingly, when we reinvestigated the lithiation/borylation reaction as applied in the synthesis of sertraline, the reaction with an electron-rich aryl group associated with carbamate 4 and an electron-deficient aryl group (3,4-Cl2C6H4) on boronic ester 21 worked much better than having an electron-deficient aryl group associated with carbamate 22 and an electron-rich aryl group on boronic ester 19 (Scheme 7). This suggests that this arrangement of electronic substitution of the aryl rings is likely to be general.
Scheme 7.

Lithiation/borylation of carbamates 4 and 22.
With the lithiation/borylation problem solved, we focused on the next step: protodeboronation of tertiary boronic ester 8. Unfortunately, our standard procedure for the protodeboronation of diarylalkyl boronic esters (1.5 equiv CsF, 2.5 equiv H2O, 1,4-dioxane)11 proceeded very sluggishly and it took 48 h at 80 °C until full conversion of 8 was achieved. This is probably due to the electron-rich aryl ring, which makes deboronation more difficult. In the case of protodeboronation of aryldialkyl boronic esters, we also found that CsF/H2O was very slow, but that tetrabutylammonium fluoride trihydrate (TBAF·3H2O) was considerably superior.11a Using these previously reported conditions (1.5 equiv TBAF·3H2O, toluene), tertiary boronic ester 8 was smoothly protodeboronated at ambient temperature within 90 min in excellent yield and enantioselectivity to give key intermediate 9 (Scheme 6).
The remaining synthesis was completed in a three-step sequence (Scheme 6). The double bond in olefin 9 was successfully cleaved by a Lemieux–Johnson oxidation,17 with in situ generated osmium tetroxide, to the corresponding aldehyde 23. Subsequent reductive amination with diisopropylamine and sodium triacetoxyborohydride gave the protected tolterodine precursor 24 in a good yield. Finally, removal of the methyl group by acidic hydrolysis resulted in the formation of (R)-tolterodine (1) in an excellent yield. The R configuration was confirmed by comparison of the optical rotations, +28.0 (c 0.50, MeOH) for 1 with that reported in the literature ( +32.5 (c 0.89, MeOH)).7e Thus, using our lithiation/borylation–protodeboronation methodology as a key step, (R)-tolterodine was synthesized through an eight-step sequence in an overall yield of 30%.
Conclusion
In conclusion, lithiation/borylation and subsequent protodeboronation has been used in the generation of gem-diarylalkyl stereocenters in both high yield and high enantioselectivity. More importantly, we have found that the optimum arrangement of groups is to have an electron-rich aryl group associated with the carbamate to promote the 1,2-metallate rearrangement, otherwise, reversion to starting materials tends to dominate. This methodology has been applied to a highly enantioselective synthesis of (R)-tolterodine.
Experimental
Reaction mixtures were stirred magnetically. Air- and moisture-sensitive reactions were carried out in flame-dried glassware under argon atmosphere using standard Schlenk manifold technique. All required fine chemicals were purchased from Acros Organics, Alfa Aesar, Inochem-Frontier Scientific, or Sigma-Aldrich and used as received unless otherwise mentioned. Anhydrous solvents were prepared using anhydrous solvent drying columns.18 Microwave reactions were carried out in a Biotage Initiator EXP EU microwave synthesizer. Analytical thinlayer chromatography (TLC) was performed on aluminum-backed silica plates (Merck, silica gel 60 F254, 0.25 mm). Compounds were visualized by exposure to UV light or by staining the plates with a 5% solution of phosphomolybdic acid (H3PMo12O40) in EtOH followed by heating. Flash column chromatography was performed on silica gel (Sigma-Aldrich, silica gel 60, 40–63 μm). 1H NMR spectra were recorded in CDCl3 at 400 or 500 MHz on a Joel ECP 400, a Varian 400, or a Varian 500 Fourier transform (FT) spectrometer. Chemical shifts (δH) are quoted in parts per million (ppm) and referred to CHCl3 (7.27 ppm). 1H NMR coupling constants are reported in hertz and refer to apparent multiplicities. Data are reported as follows: chemical shift, multiplicity (s = singlet, br s = broad singlet, d = doublet, t = triplet, m = multiplet, dd = doublet of doublet, etc.), coupling constant, integration, and assignment. 13C NMR spectra were recorded at 101 or 126 MHz on a Jeol ECP 400, a Varian 400, or a Varian 500 instrument. Chemical shifts (δC) are quoted in ppm and referenced to CHCl3 (77.0 ppm). 11B NMR spectra were measured using quartz NMR tubes at 96 or 128 MHz on a Jeol Lambda 300, a Joel ECP (Eclipse) 300, or a Varian 400 with complete proton decoupling using BF3·Et2O (0.0 ppm) as an external standard. Mass spectra were recorded by the University of Bristol, School of Chemistry departmental mass spectrometry service using chemical ionization (CI) or electrospray ionization (ESI) techniques for low- and high-resolution mass spectra (LR-MS and HR-MS, respectively). All IR spectra were recorded on the neat compounds using a PerkinElmer Spectrum One FT-IR spectrometer. Optical rotations were obtained using a Bellingham + Stanley Ltd. ADP220 polarimeter. Melting points were measured with a Reichert hot stage apparatus and are uncorrected. Chiral HPLC separations were performed on an Agilent 1100 Series HPLC unit equipped with a UV–vis diode array detector using a Daicel Chiralpak IB column (4.6 × 250 mm2, 5 μm) fitted with a guard (4 × 10 mm2). (Chiral HPLC traces and 1H and 13C NMR spectra can be found in the Supplementary data).
2-(2-Methoxy-5-methylphenyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (7)
This compound was synthesized following a literature procedure performed on a different substrate.19 Boronic ester 7 (2.02 g, 8.14 mmol, >99%) was obtained as a white solid, which was used without further purification; mp 75–76 °C (diethyl ether). IR (neat, cm−1) νmax: 2981, 1605, 1585, 1243. 1H NMR (400 MHz, CDCl3, ppm) δH: 7.49 (d, J = 2.3 Hz, 1H, HAr), 7.20 (dd, J = 8.4, 2.3 Hz, 1H, HAr), 6.77 (d, J = 8.4 Hz, 1H, HAr), 3.81 (s, 3H, OCH3), 2.29 (s, 3H, CArCH3), 1.36 (s, 12H, CH3). 13C NMR (101 MHz, CDCl3, ppm) δC: 162.2 (COCH3), 137.1 (CH), 132.9 (CH), 129.2 (C), 110.5 (CH), 83.4 (OC(CH3)2), 56.0 (OCH3), 24.8 (CH3), 20.2 (CArCH3). 11B NMR (96 MHz, CDCl3, ppm) δB: 30.0 (br s). MS m/z (%) (CI+): 249 ([M + H]+, 100), 248 ([M]+, 60), 247 (18), 145 (15), 123 ([Ar + H]+, 15), 101 (24). HR-MS (CI+) calcd for C14H22O311B [M + H]+: 249.1662; found: 249.1664.
2-(2-Methoxy-5-methylphenyl)-5,5-dimethyl-1,3,2-dioxaborinane (12)
This compound was synthesized following a literature procedure performed on a different substrate.19 Boronic ester 12 (3.51 g, 14.4 mmol, 96%) was obtained as a white solid, which required no further purification; mp 59–61 °C (diethyl ether). IR (neat, cm−1) νmax: 2936, 1603, 1584, 1241. 1H NMR (400 MHz, CDCl3, ppm) δH: 7.47 (d, J = 2.2 Hz, 1H, HAr), 7.17 (dd, J = 8.4, 2.2 Hz, 1H, HAr), 6.78 (d, J = 8.4 Hz, 1H, HAr), 3.82 (s, 3H, OCH3), 3.80 (s, 4H, OCH2), 2.29 (s, 3H, CArCH3), 1.05 (s, 6H, CH3). 13C NMR (101 MHz, CDCl3, ppm) δC: 161.7 (COCH3), 136.2 (CH), 132.0 (CH), 129.1 (C), 110.4 (CH), 72.4 (OCH2), 55.9 (OCH3), 31.7 (C(CH3)2), 21.9 (CH3), 20.3 (CArCH3). 11B NMR (96 MHz, CDCl3, ppm) δB: 26.3 (br s). MS m/z (%) (CI+): 235 ([M + H]+, 100), 234 ([M]+, 53), 233 (19), 123 ([Ar + H]+, 31), 75 (53). HR-MS (CI+) calcd. for C13H20O311B [M + H]+: 235.1506; found: 235.1500.
1-Phenylbutyl diisopropylcarbamate (10)
To a suspension of sodium hydride (60% dispersion in mineral oil, 300 mg, 7.50 mmol, 1.5 equiv) in anhydrous tetrahydrofuran (THF; 20 mL), 1-phenylbutan-1-ol (751 mg, 5.00 mmol, 1.0 equiv) was added dropwise and the mixture was stirred for 75 min at RT. A solution of N,N-diisopropylcarbamoyl chloride (982 mg, 6.00 mmol, 1.2 equiv) in anhydrous THF (5.0 mL) was added and the reaction mixture was heated under reflux for 44 h. The solvent was removed in vacuo and the residue was portioned between water and diethyl ether. The phases were separated and the aqueous layer was reextracted with diethyl ether. The combined organic layers were dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure. The residue was subjected to flash chromatography (SiO2, pentane/EtOAc, 6:1) to obtain carbamate 10 (1.13 g, 4.07 mmol, 81%) as a colourless oil. TLC (pentane/EtOAc, 6:1): 0.46. IR (neat, cm−1) νmax: 2962, 1687. 1H NMR (400 MHz, CDCl3, ppm) δH: 7.37–7.29 (m, 4H, HAr), 7.29–7.23 (m, 1H, HAr), 5.71 (t, J = 6.9 Hz, 1H, CHOCb), 4.04 (br m, 1H, CH(CH3)2), 3.84 (br m, 1H, CH(CH3)2), 1.94 (m, 1H, CHHCHO), 1.76 (m, 1H, CHH-CHO), 1.44–1.27 (m, 2H, CH2CH3), 1.22 (br m, 12H, CH(CH3)2), 0.92 (t, J = 7.4 Hz, 3H, CH2CH3). 13C NMR (101 MHz, CDCl3, ppm) δC: 155.1 (NCO), 141.8 (C), 128.2 (CH), 127.3 (CH), 126.5 (CH), 76.4 (CHOCb), 46.1 (br, CH(CH3)2), 39.0 (CH2CHO), 21.4 (br, CH(CH3)2), 18.9 (CH2CH3), 13.9 (CH2CH3). MS m/z (%) (CI+): 278 ([M + H]+, 77), 236 (11), 218 (10), 146 ([CbOH + H]+, 100), 133 ([Ph(CH2)4]+, 57), 128 (16), 102 (12), 91 ([PhCH2]+, 30). HR-MS (CI+) calcd for C17H28NO2 [M + H]+: 278.2120; found: 278.2116.
2-(1-(2-Methoxy-5-methylphenyl)-1-phenylbutyl)-5,5-dimethyl-1,3,2-dioxaborinane (13)
1-Phenylbutyl diisopropylcarbamate (10; 139 mg, 0.50 mmol, 1.0 equiv) was dissolved in anhydrous diethyl ether (1.5 mL) and chilled to −78 °C. s-BuLi (500 μL, 1.3 mol/L solution in cyclohexane/hexane, 98:2, 0.65 mmol, 1.3 equiv) was added dropwise and the mixture was stirred at this temperature for 1 h. A solution of neopentyl ester 12 (176 mg, 0.75 mmol, 1.5 equiv) in anhydrous diethyl ether (0.5 mL) was added dropwise and the mixture was stirred for 2 h at −78 °C. The cooling bath was removed and stirring was continued at RT overnight (15 h). The reaction mixture was cooled to 0 °C and 1 mol/L KH2PO4(aq) (2.0 mL) was added slowly. After stirring for 10 min at RT, the phases were separated and the aqueous phase was extracted with diethyl ether. The combined organic phases were dried over anhydrous MgSO4, filtered, and the solvent was removed in vacuo. The crude product was purified by column chromatography (SiO2, pentane/EtOAc, 30:1) to give tertiary boronic ester 13 (114 mg, 0.31 mmol, 62% or 92% based on recovered starting material (BRSM)) as a colourless oil, which crystallized upon standing to white cubes; mp 86–87 °C (pentane/EtOAc). TLC (pentane/EtOAc, 30:1): 0.22. IR (neat, cm−1) νmax: 2955, 2873, 1494, 1244. 1H NMR (400 MHz, CDCl3, ppm) δH: 7.42–7.38 (m, 2H, HAr), 7.28–7.23 (m, 2H, HAr), 7.16–7.11 (m, 1H, HAr), 6.97 (ddd, J = 8.2, 2.2, 0.6 Hz, 1H, HAr), 6.82–6.79 (m, 2H, HAr), 3.85 (s, 3H, OCH3), 3.53 (s, 4H, OCH2), 2.25–2.17 (m, 1H, CHHCH2), 2.19 (s, 3H, CArCH3), 2.02–1.93 (m, 1H, CHHCH2), 1.31–1.21 (m, 1H, CHHCH3), 0.87–0.78 (m, 4H, CH2CH3, CHHCH3), 0.65 (s, 6H, CH3). 13C NMR (101 MHz, CDCl3, ppm) δC: 154.9 (C), 144.0 (C), 136.3 (C), 130.9 (CH), 129.4 (CH), 129.3 (C), 127.2 (CH), 126.7 (CH), 124.5 (CH), 110.4 (CH), 72.3 (OCH2), 56.1 (OCH3), 34.8 (CH2CH2), 31.6 (C(CH3)2), 21.5 (CH3), 20.8 (CArCH3), 17.9 (CH2CH3), 14.8 (CH2CH3). 11B NMR (96 MHz, CDCl3, ppm) δB: 27.9 (br s). MS m/z (%) (CI+): 367 ([M + H]+, 16), 366 ([M]+, 30), 351 ([M – Me]+, 14), 323 ([M – n-Pr]+, 5), 289 ([M – Ph]+, 30), 253 ([M – Bneop]+, 7), 245 ([M – Ar]+, 100), 203 (28), 177 (16), 149 (12), 131 (34), 123 (46), 105 (15), 93 (99). HR-MS (CI+) calcd for C23H32O311B [M + H]+: 367.2445; found: 367.2457.
2-(1-(2-Methoxy-5-methylphenyl)-1-phenylbut-3-enyl)-5,5-dimethyl-1,3,2-dioxaborinane (14)
Following the experimental procedure for tertiary boronic ester 13, 1-phenylbut-3-enyl diisopropylcarbamate (4) (139 mg, 0.50 mmol, 1.0 equiv) in anhydrous diethyl ether (1.5 mL), s-BuLi (570 μL, 1.14 mol/L solution in cyclohexane/hexane, 98:2, 0.65 mmol, 1.3 equiv), and neopentyl ester 12 (176 mg, 0.75 mmol, 1.0 equiv) in anhydrous diethyl ether (0.75 mL) gave, after purification by column chromatography (SiO2, pentane/EtOAc, 30:1), tertiary boronic ester 14 (94 mg, 0.26 mmol, 52% or 97% BRSM) as a colourless oil. TLC (pentane/EtOAc, 30:1): 0.12. IR (neat, cm−1) νmax: 2928, 1495, 1245. 1H NMR (400 MHz, CDCl3, ppm) δH: 7.49–7.42 (m, 2H, HAr), 7.30–7.23 (m, 2H, HAr), 7.18–7.13 (m, 1H, HAr), 6.98 (ddd, J = 8.1, 2.2, 0.6 Hz, 1H, HAr), 6.85–6.78 (m, 2H, HAr), 5.68 (dddd, J = 17.3, 10.2, 7.4, 6.2 Hz, 1H, CHCH2), 4.97–4.84 (m, 2H, CHCH2), 3.86 (s, 3H, OCH3), 3.56 (s, 4H, OCH2), 3.13 (ddt, J = 15.4, 7.4, 1.2 Hz, 1H, CHH), 2.84 (ddt, J = 15.4, 6.2, 1.6 Hz, 1H, CHH), 2.20 (s, 3H, CArCH3), 0.71 (s, 6H, CH3). 13C NMR (101 MHz, CDCl3, ppm) δC: 154.8 (C), 143.7 (C), 137.1 (CHCH2), 135.8 (C), 130.7 (CH), 129.6 (CH), 129.4 (C), 127.3 (CH), 126.9 (CH), 124.8 (CH), 115.6 (CHCH2), 110.4 (CH), 72.3 (OCH2), 56.1 (OCH3), 37.5 (CH2), 31.6 (C(CH3)2), 21.6 (CH3), 20.8 (CArCH3). 11B NMR (96 MHz, CDCl3, ppm) δB: 28.1 (br s). MS m/z (%) (CI+): 365 ([M + H]+, 89), 364 ([M]+, 75), 349 ([M – Me]+, 49), 323 ([M – allyl]+, 49), 287 ([M – Ph]+, 32), 243 ([M – Ar]+, 100), 203 (12), 149 (36), 131 (33), 123 (19), 105 (24), 93 (77). HR-MS (CI+) calcd for C23H30O311B [M + H]+: 365.2288; found: 365.2278.
2-Methoxy-5-methylbenzaldehyde (16)
This compound was synthesized following a literature procedure20 and yielded a pale yellow oil (4.44 g, 29.6 mmol, 99%), which was used without further purification. IR (neat, cm−1) νmax : 2863, 1676, 1494, 1250. 1H NMR (400 MHz, CDCl3, ppm) δH: 10.45 (s, 1H, CHO), 7.64 (d, J = 2.5 Hz, 1H, HAr), 7.36 (dd, J = 8.6, 2.5 Hz, 1H, HAr), 6.90 (d, J = 8.6 Hz, 1H, HAr), 3.91 (s, 3H, OCH3), 2.32 (s, 3H, CH3). 13C NMR (101 MHz, CDCl3, ppm) δC: 190.0 (CHO), 160.0 (C), 136.6 (CH), 130.0 (C), 128.6 (CH), 124.5 (C), 111.6 (CH), 55.7 (OCH3), 20.2 (CH3). MS m/z (%) (CI+): 151 ([M + H]+ 100), 123 ([Ar + H]+, 26), 89 (68). HR-MS (CI+) calcd for C9H11O2 [M + H] : 151.0759; found: 151.0756.
(R)-1-(2-Methoxy-5-methylphenyl)but-3-en-1-ol (17)
This compound was synthesized following a literature procedure performed on a different substrate.16 Flash chromatography (SiO2, pentane/diethyl ether, 9:1) gave alcohol 17 (74 mg, 0.38 mmol, 77%) as white needles; mp 44–45 °C (pentane/diethyl ether). +56.3 (c 1.79, CHCl3, for 98% ee). TLC (pentane/diethyl ether, 9:1): 0.08. IR (neat, cm−1) νmax: 3361, 2922, 1498, 1243. 1H NMR (400 MHz, CDCl3, ppm) δH: 7.16 (d, J = 2.0 Hz, 1H, HAr), 7.05 (dd, J = 8.2, 2.0 Hz, 1H, HAr), 6.79 (d, J = 8.2 Hz, 1H, HAr), 5.93–5.81 (ddt, J = 16.9, 10.3, 6.9 Hz, 1H, CHCH2), 5.19–5.10 (m, 2H, CHCH2), 4.93 (dt, J = 8.1, 5.3 Hz, 1H, CHOH), 3.84 (s, 3H, OCH3), 2.62–2.47 (m, 3H, CH2, OH), 2.31 (s, 3H, CH3). 13C NMR (101 MHz, CDCl3, ppm) δC: 154.3 (C), 135.3 (CHCH2), 131.4 (C), 129.9 (C), 128.5 (CH), 127.5 (CH), 117.5 (CHCH2), 110.4 (CH), 69.8 (CHOH), 55.4 (OCH3), 42.0 (CH2), 20.6 (CH3). MS m/z (%) (ESI+): 215 ([M + Na]+, 100), 175 ([M – allyl + Na]+, 10), 107 ([Ar – Me]+, 5). HR-MS (ESI+) calcd. for C12H16O2Na [M + Na]+: 215.1043; found: 215.1038. HPLC: Chiralpak IB, 20 °C, 0.5% i-PrOH in hexane, 0.7 mL/min, UV detection at 210.8 nm, retention times: (S)-1-(2-methoxy-5-methylphenyl)but-3-en-1-ol, 27.6 min; (R)-1-(2-methoxy-5-methylphenyl)but-3-en-1-ol, 30.1 min; 92% ee.
(R)-1-(2-Methoxy-5-methylphenyl)but-3-enyl diisopropylcarbamate (18)
Alcohol 17 (1.32 g, 6.84 mmol, 1.0 equiv), N,N-diisopropylcarbamoyl chloride (1.34 g, 8.21 mmol, 1.2 equiv), and Et3N (1.23 mL, 8.89 mmol, 1.3 equiv) were dissolved in anhydrous toluene (7.0 mL) in a microwave vial. The vial was sealed and heated for 2 h at 150 °C. After cooling to ambient temperature, the salts were removed by filtration through a plug of silica and the solids were thoroughly washed with diethyl ether. The solvent was removed in vacuo and the residue was subjected to flash chromatography (SiO2, pentane/EtOAc, 9:1) to obtain 2.11 g (R)-carbamate 18 (6.6 mmol, 97%) as a pale yellow oil. −1.0 (c 1.00, CHCl3). TLC (pentane/EtOAc, 9:1): 0.27. IR (neat, cm−1) νmax: 2969, 1686, 1249. 1H NMR (400 MHz, CDCl3, ppm) δH: 7.09 (d, J = 2.2 Hz, 1H, HAr), 7.02 (dd, J = 8.2, 2.2 Hz, 1H, HAr), 6.76 (d, J = 8.2 Hz, 1H, HAr), 6.15 (dd, J = 7.1, 5.3 Hz, 1H, CHOCb), 5.80 (ddt, J = 17.1, 10.1, 7.0 Hz, 1H, CHCH2), 5.08–4.99 (m, 2H, CHCH2), 4.01 (br m, 1H, CH(CH3)2), 3.90 (br m, 1H, CH(CH3)2), 3.81 (s, 3H, OCH3), 2.65–2.53 (m, 2H, CH2), 2.28 (s, 3H, CArCH3), 1.24 (br m, 12H, CH3). 13C NMR (101 MHz, CDCl3, ppm) δC: 154.9 (NCO), 154.0 (C), 134.7 (CHCH2), 129.6 (C), 129.3 (C), 128.5 (CH), 127.3 (CH), 116.9 (CHCH2), 110.5 (CH), 70.8 (CHOCb), 55.5 (OCH3), 45.7 (br, CH(CH3)2), 40.1 (CH2), 21.1 (br, CH3), 20.7 (CArCH3). MS m/z (%) (ESI+): 342 ([M + Na]+, 100), 220 ([M – Ar + Na]+, 4), 175 ([M – OCb]+, 7), 114 (4). HR-MS (ESI+) calcd for C19H29O3NaN [M + Na] : 342.2040; found: 342.2033. HPLC: Chiralpak IB, 0 °C, 0.2% i-PrOH in hexane, 0.5 mL/min, UV detection at 210.8 nm, retention times: (S)-1-(2-methoxy-5-methylphenyl)but-3-enyl diisopropylcarbamate, 36.3 min; (R)-1-(2-methoxy-5-methylphenyl)but-3-enyl diisopropylcarbamate, 40.9 min; 92% ee.
(R)-2-(1-(2-Methoxy-5-methylphenyl)-1-phenylbut-3-enyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (8)
(R)-Carbamate 18 (515 mg, 1.61 mmol, 1.0 equiv) was dissolved in anhydrous diethyl ether (5.0 mL) and chilled to −78 °C. s-BuLi (1.41 mL, 1.49 mol/L solution in cyclohexane/hexane, 98:2, 2.10 mmol, 1.3 equiv) was added dropwise and the reaction mixture was stirred at this temperature for 1 h. A solution of PhBpin (4,4,5,5-tetramethyl-2-phenyl-1,3,2-dioxaborolane, 490 mg, 2.42 mmol, 1.5 equiv) in anhy-drous diethyl ether (2.5 mL) was added dropwise and the mixture was stirred for 2 h at −78 °C. Afterwards, a 1.0 mol/L solution of MgBr2 in anhydrous MeOH10b (1.93 mL, 1.93 mmol, 1.2 equiv) was added slowly. After 5 min, the cooling bath was removed and stirring was continued at RT overnight (16 h). Then, the reaction mixture was cooled to 0 °C and 1 mol/L KH2PO4(aq) (2.0 mL) was added slowly. After stirring for 10 min at RT, the phases were separated and the aqueous phase was extracted with diethyl ether. The combined organic phases were dried over anhydrous MgSO4, filtered, and the solvent was removed in vacuo. The crude product was purified by column chromatography (SiO2, pentane/EtOAc, 30:1) to give tertiary boronic ester 8 (449 mg, 1.19 mmol, 74%) as a white solid; mp 134–136 °C (pentane/EtOAc). –88.0 (c 1.00, CHCl3, for 90% ee). TLC (pentane/EtOAc, 30:1): 0.10. IR (neat, cm−1) νmax: 2974, 1497, 1240. 1H NMR (400 MHz, CDCl3, ppm) δH: 7.46 (d, J = 7.8 Hz, 2H, HAr), 7.26 (t, J = 7.8 Hz, 2H, HAr), 7.15 (t, J = 7.8 Hz, 1H, HAr), 6.96 (dd, J = 8.2, 1.7 Hz, 1H, HAr), 6.81 (d, J = 1.7 Hz, 1H, HAr), 6.76 (d, J = 8.2 Hz, 1H, HAr), 5.65 (ddt, J = 17.1, 10.2, 6.8 Hz, 1H, CHCH2), 4.95 (dd, J = 17.1, 1.4 Hz, 1H, CHCHtransH), 4.87 (dd, J = 10.2, 1.4 Hz, 1H, CHCHcisH), 3.77 (s, 3H, OCH3), 3.10 (dd, J = 15.2, 6.8 Hz, 1H, CHH), 2.94 (dd, J = 15.2, 6.8 Hz, 1H, CHH), 2.19 (s, 3H, CArCH3), 1.11 (s, 6H, CH3), 1.10 (s, 6H, CH3). 13C NMR (101 MHz, CDCl3, ppm) δC: 154.8 (C), 143.0 (C), 136.7 (CHCH2), 134.4 (C), 130.4 (CH), 129.9 (CH), 129.2 (C), 127.3 (CH), 127.1 (CH), 125.1 (CH), 115.7 (CHCH2), 110.1 (CH), 83.0 (C), 55.1 (OCH3), 38.8 (br, CB), 37.8 (CH2), 24.6 (CH3), 24.4 (CH3), 20.8 (CArCH3). 11B NMR (96 MHz, CDCl3, ppm) δB: 30.2 (br s). MS m/z (%) (ESI+): 401 ([M + Na]+, 100), 279 (9), 223 (4), 134 (5). HR-MS (ESI+) calcd for C24H31O3Na11B [M + Na]+: 401.2258; found: 401.2245. The enantiomeric excess of the chiral boronic ester was determined by HPLC analysis of an aliquot oxidized to 25.
(S)-1-(2-Methoxy-5-methylphenyl)-1-phenylbut-3-en-1-ol (25)
A solution of tertiary boronic ester 8 (38 mg, 0.10 mmol, 1.0 equiv) in THF (5.0 mL) was cooled to 0 °C and a mixture of 2 mol/L NaOH(aq) (2.0 mL) and 30% H2O2 (1.0 mL) was added slowly under vigorous stirring. The reaction mixture was stirred at RT for 2 h. The solvent was removed under reduced pressure and the residue was portioned between water and diethyl ether. The phases were separated and the aqueous layer was extracted with diethyl ether. The combined organic layers were washed with brine, dried over anhydrous MgSO4, filtered, and the solvent was removed in vacuo. The residue was purified by flash chromatography (SiO2, pentane/EtOAc, 9:1) to give tertiary alcohol 25 (21 mg, 79 μmol, 79%) as a colourless oil, which crystallized on standing to form a white solid; mp 52–53 °C (pentane/EtOAc). +100.2 (c 1.57, CHCl3, for 94% ee). TLC (pentane/EtOAc, 9:1): 0.32. IR (neat, cm−1) νmax: 3517, 2923, 1497, 1237. 1H NMR (400 MHz, CDCl3, ppm) δH: 7.35–7.31 (m, 2H, HAr), 7.30–7.24 (m, 3H, HAr), 7.22–7.17 (m, 1H, HAr), 7.08 (ddd, J = 8.2, 2.2, 0.6 Hz, 1H, HAr), 6.78 (d, J = 8.2 Hz, 1H, HAr), 5.86 (ddt, J = 17.2, 10.3, 6.8 Hz, 1H, CHCH2), 5.15–5.05 (m, 2H, CHCH2), 4.64 (br s, 1H, OH), 3.56 (s, 3H, OCH3), 3.14 (ddd, J = 14.5, 6.8, 1.1 Hz, 1H, CHH), 2.89 (ddd, J = 14.5, 6.8, 1.1 Hz, 1H, CHH), 2.35 (s, 3H, CH3). 13C NMR (101 MHz, CDCl3, ppm) δC: 155.0 (C), 147.7 (C), 134.5 (CHCH2), 134.0 (C), 129.9 (C), 128.8 (CH), 128.1 (CH), 127.5 (CH), 126.3 (CH), 125.6 (CH), 117.6 (CHCH2), 112.5 (CH), 77.5 (COH), 55.8 (OCH3), 45.8 (CH2), 20.9 (CH3). MS m/z (%) (CI+): 269 ([M + H]+, 4), 251 ([M – OH]+, 100), 227 ([M – allyl]+, 85), 149 [(M – Ar + H]+, 30), 135 (20), 105 (34). HR-MS (CI+) calcd for C18H21O2 [M + H] : 269.1542; found: 269.1545. HPLC: Chiralpak IB, 20 °C, 3.0% i-PrOH in hexane, 0.7 mL/min, UV detection at 210.8 nm, retention times: (R)-1-(2-methoxy-5-methylphenyl)-1-phenylbut-3-en-1-ol, 8.6 min; (S)-1-(2-methoxy-5-methylphenyl)-1-phenylbut-3-en-1-ol, 9.7 min; 90% ee.
(R)-1-Methoxy-4-methyl-2-(1-phenylbut-3-enyl)benzene (9)
A solution of tertiary boronic ester 8 (328 mg, 0.87 mmol, 1.0 equiv) and TBAF·3H2O (411 mg, 1.30 mmol, 1.5 equiv) in anhydrous toluene (9.0 mL) was stirred for 1.5 h at RT. Afterwards, the reaction mixture was washed with 0.5 mol/L NaOH(aq), water, and brine. The organic phase was dried over anhydrous MgSO4, filtered, and the solvent was removed under reduced pressure to give olefin 9 (207 mg, 0.82 mmol, 95%) as a colourless oil, which required no purification. −10.0 (c 1.00, CHCl3, for 90% ee). IR (neat, cm−1) νmax: 2922, 1498, 1240. 1H NMR (400 MHz, CDCl3, ppm) δH: 7.30–7.25 (m, 4H, HAr), 7.19–7.12 (m, 1H, HAr), 7.02 (d, J = 2.2 Hz, 1H, HAr), 6.96 (dd, J = 8.3, 2.2 Hz, 1H, HAr), 6.74 (d, J = 8.3 Hz, 1H, HAr), 5.75 (ddt, J = 17.1, 10.2, 6.8 Hz, 1H, CHCH2), 5.03 (ddt, J = 17.1, 2.8, 1.3 Hz, 1H, CHCHtransH), 4.93 (ddt, J = 10.2, 2.8, 1.3 Hz, 1H, CHCHcisH), 4.47 (t, J = 7.9 Hz, 1H, CH), 3.75 (s, 3H, OCH3), 2.78 (ddt, J = 7.9, 6.8, 1.3 Hz, 2H, CH2), 2.27 (s, 3H, CH3). 13C NMR (101 MHz, CDCl3, ppm) δC: 154.9 (C), 144.5 (C), 137.3 (CHCH2), 132.8 (C), 129.5 (C), 128.5 (CH), 128.2 (CH), 128.1 (CH), 127.4 (CH), 125.8 (CH), 115.8 (CHCH2), 110.8 (CH), 55.6 (OCH3), 43.3 (CH), 39.1 (CH2), 20.7 (CH3). MS m/z (%) (ESI+): 275 ([M + Na]+, 100), 270 ([M + NH4]+, 18), 211 ([M – allyl]+, 22). HR-MS (ESI+) calcd for C18H20ONa [M + Na]+: 275.1406; found: 275.1413. The enantiomeric excess of the chiral olefin was determined by HPLC analysis of an aliquot, which has been hydroborated and oxidized to 26.
(R)-4-(2-Methoxy-5-methylphenyl)-4-phenylbutan-1-ol (26)
A solution of 9-borabicyclo[3.3.1]nonane (9-BBN; 390 μL, 0.5 mol/L in THF, 0.20 mmol, 1.75 equiv) was added to a solution of olefin 9 (29 mg, 0.11 mmol, 1.0 equiv) in anhydrous THF (1.0 mL) and stirred for 2 h at ambient temperature. The reaction mixture was cooled to 0 °C and a mixture of 2 mol/L NaOH(aq) (1.0 mL) and 30% H2O2 (0.5 mL) was added slowly. The reaction mixture was stirred vigorously for 2 h at RT. The solution was diluted with water and extracted with diethyl ether. The combined organic layers were dried over anhydrous MgSO4, filtered, concentrated under reduced pressure, and the residue was purified by column chromatography (SiO2, pentane/EtOAc, 4:1) to give alcohol 26 (23 mg, 85 μmol, 75%) as a colourless oil. +6.5 (c 0.62, CHCl3, for 90% ee). TLC (pentane/EtOAc, 4:1): 0.10. IR (neat, cm−1) νmax: 3334, 2937, 1498, 1243. 1H NMR (400 MHz, CDCl3, ppm) δH: 7.31–7.24 (m, 4H, HAr), 7.19–7.12 (m, 1H, HAr), 7.03–7.00 (m, 1H, HAr), 6.96 (dd, J = 8.2, 2.2 Hz, 1H, HAr), 6.74 (d, J = 8.2 Hz, 1H, HAr), 4.39 (t, J = 7.8 Hz, 1H, CH), 3.76 (s, 3H, OCH3), 3.67 (t, J = 6.6 Hz, 2H, CH2OH), 2.27 (s, 3H, CH3), 2.14–2.00 (m, 2H, CH2CH), 1.60–1.51 (dq, J = 8.1, 6.6 Hz, 2H, CH2CH2OH), 1.35 (br s, 1H, OH). 13C NMR (101 MHz, CDCl3, ppm) δC: 154.8 (C), 144.8 (C), 133.2 (C), 129.6 (C), 128.3 (CH), 128.1 (CH), 128.1 (CH), 127.3 (CH), 125.8 (CH), 110.7 (CH), 62.9 (CH2OH), 55.7 (OCH3), 42.7 (CH), 31.1 (CH2), 31.0 (CH2), 20.7 (CH3). MS m/z (%) (ESI+): 293 ([M + Na]+, 100), 290 (13), 282 (2), 211 ([M – (CH2)OH)]+ , 2). HR-MS (ESI+) calcd for C18H22ONa [M + Na]+: 293.1512; found: 293.1517. HPLC: Chiralpak IB, 20 °C, 1.5% i-PrOH in hexane, 0.7 mL/min, UV detection at 210.8 nm, retention times: (S)-1-(2-methoxy-5-methylphenyl)-1-phenylbut-3-en-1-ol, 48.8 min; (R)-4-(2-methoxy-5-methylphenyl)-4-phenylbutan-1-ol, 50.2 min; 90% ee.
(R)-3-(2-Methoxy-5-methylphenyl)-3-phenylpropanal (23)
This compound was synthesized following a literature procedure performed on a different substrate.17 The crude material was filtered through a plug of silica (pentane/EtOAc, 1:1), and the solvent was removed in vacuo to afford aldehyde 23 (163 mg, 0.64 mmol, 97%) as a pale yellow oil, which required no further purification. +14.0 (c 1.00, CHCl3) (lit.21 value −16.2 (c 1.4, CHCl3, for 99% ee of the (S)-isomer)). IR (neat, cm−1) νmax: 2919, 1721, 1498, 1241. 1H NMR (400 MHz, CDCl3, ppm) δH: 9.70 (t, J = 2.2 Hz, 1H, CHO), 7.32–7.23 (m, 4H, HAr), 7.22–7.16 (m, 1H, HAr), 6.98 (dd, J = 8.3, 2.0 Hz, 1H, HAr), 6.87 (d, J = 2.0 Hz, 1H, HAr), 6.75 (d, J = 8.3 Hz, 1H, HAr), 5.00 (t, J = 7.8 Hz, 1H, CH), 3.77 (s, 3H, OCH3), 3.09 (dd, J = 7.8, 2.2 Hz, 2H, CH2), 2.23 (s, 3H, CH3). 13C NMR (126 MHz, CDCl3, ppm) δC: 201.9 (CHO), 154.5 (C), 142.9 (C), 131.3 (C), 129.8 (C), 128.8 (CH), 128.4 (CH), 128.0 (CH), 128.0 (CH), 126.3 (CH), 110.8 (CH), 55.5 (OCH3), 48.5 (CH2), 38.3 (CH), 20.6 (CH3). MS m/z (%) (ESI+): 277 ([M + Na]+, 38), 237 (11), 211 ([M –CH2CHO], 100). HR-MS (ESI+) calcd for C17H18O2Na [M + Na]+: 277.1199; found: 277.1204. HPLC: Chiralpak IB, 0 °C, 2.0% i-PrOH in hexane, 0.5 mL/min, UV detection at 210.8 nm, retention times: (S)-3-(2-methoxy-5-methylphenyl)-3-phenylpropanal, 24.7 min; (R)-3-(2-methoxy-5-methylphenyl)-3-phenylpropanal, 27.6 min; 90% ee.
(R)-N,N-Diisopropyl-3-(2-methoxy-5-methylphenyl)-3-phenylpropan-1-amine (24)
A mixture of aldehyde 23 (25 mg, 98 μmol, 1.0 equiv), diisopropylamine (28 μL, 0.20 mmol, 2.0 equiv), and NaBH(OAc)3 (42 mg, 0.20 mmol, 2.0 equiv) in anhydrous THF (1.0 mL) was stirred for 18 h at ambient temperature. Then, saturated NaHCO3(aq) solution (0.5 mL) was added, the phases were separated, and the aqueous layer was extracted with EtOAc. The combined organic layers were washed with 3.0 mol/L HCl(aq) and brine, dried over anhydrous Na2SO4, filtered, and the solvent was removed under reduced pressure. Purification of the residue by column chromatography (SiO2, pentane/EtOAc/Et3N, 75:24:1) afforded tertiary amine 24 (23 mg, 68 μmol, 69%) as a pale yellow oil. −11.0 (c 1.00, CHCl3) (lit.22 value [α]D −6.14 (c 0.95, CHCl3, for 81% ee). TLC (pentane/EtOAc/Et3N, 75:24:1): 0.16. IR (neat, cm–1) νmax: 2962, 1497, 1239. 1H NMR (400 MHz, CDCl3, ppm) δH: 7.31–7.22 (m, 4H, HAr), 7.16–7.12 (m, 1H, HAr), 7.07 (d, J = 2.2 Hz, 1H, HAr), 6.94 (dd, J = 8.2, 2.2 Hz, 1H, HAr), 6.72 (d, J = 8.2 Hz, 1H, HAr), 4.35 (t, J = 7.7 Hz, 1H, CH), 3.75 (s, 3 H, OCH3), 2.98 (sept, J = 6.5 Hz, 2H, CH(CH3)2), 2.37–2.30 (m, 2H, CHHN + CHHCH), 2.26 (s, 3H, CArCH3), 2.16–2.09 (m, 2H, CHHN + CHHCH), 0.94 (d, J = 6.5 Hz, 6H, CH3), 0.93 (d, J = 6.5 Hz, 6H, CH3). 13C NMR (126 MHz, CDCl3, ppm) δC: 154.9 (C), 145.1 (C), 133.5 (C), 129.5 (C), 128.3 (CH), 128.2 (CH), 128.0 (CH), 127.1 (CH), 125.6 (CH), 110.6 (CH), 55.6 (OCH3), 48.7 (CH(CH3)2), 44.1 (CH2N), 41.3 (CArCH), 37.0 (CH2CH), 20.7 (CH3), 20.5 (CArCH3). MS m/z (%) (ESI+): 340 ([M + H]+, 100), 298 ([M – i-Pr]+, 15), 211 ([M – (CH)2N(i-Pr)2]+, 7), 113 (4). HR-MS (ESI+) calcd for C23H34ON [M + H]+: 340.2635; found: 340.2625.
(R)-Tolterodine (1)
This compound was synthesized following a literature procedure performed on a different substrate.7e Flash chromatography (SiO2, pentane/EtOAc, 70:30, 2% Et3N) yielded a pale yellow oil, which crystallized upon standing. (R)-tolterodine (1; 91 mg, 0.28 mmol, 87%) was obtained as an off-white solid; mp 78–79 °C (pentane/EtOAc/Et3N). +28.0 (c 0.50, MeOH) (lit.7e value [α]D +32.5 (c 0.89, MeOH, for 99% ee). TLC (pentane/EtOAc, 70:30, 2% Et3N): 0.18. IR (neat, cm−1) νmax: 3249, 2971, 2927, 1493. 1H NMR (500 MHz, CDCl3, ppm) δH: 10.27 (br s, 1H, CArOH), 7.36–7.32 (m, 4H, HAr), 7.26–7.22 (m, 1H, HAr), 6.87 (dd, J = 8.2, 2.0 Hz, 1H, HAr), 6.82 (dd, J = 8.2, 1.5 Hz, 1H, HAr), 6.57 (s, 1H, HAr), 4.50 (dd, J = 10.8, 3.2 Hz, 1H, CH), 3.25 (sept, J = 6.7 Hz, 2H, CH(CH3)), 2.79–2.70 (m, 1H, CHHN), 2.46–2.33 (m, 2H, CHHN + CHHCH), 2.14 (s, 3H, CArCH3), 2.13–2.04 (m, 1H, CHHCH), 1.15 (d, J = 6.7 Hz, 6H, CH3), 1.10 (d, J = 6.7 Hz, 6H, CH3). 13C NMR (126 MHz, CDCl3, ppm) δC: 153.2 (C), 144.7 (C), 132.3 (C), 129.3 (C), 128.6 (CH), 128.5 (CH), 128.3 (CH), 127.7 (CH), 126.1 (CH), 118.1 (CH), 48.0 (CH(CH3)2), 42.1 (CH2N), 39.4 (CArCH), 33.2 (CH2CH), 20.7 (CArCH3), 19.9 (CH3), 19.5 (CH3). MS m/z (%) (ESI+): 348 ([M + Na]+, 1), 326 ([M + H]+, 100), 284 ([M – i-Pr + H]+, 2), 197 ([M – (CH2)2N(i-Pr)2] , 1), 109 ([Ar]+, 5). HR-MS (ESI+) calcd for C22H32ON [M + H]+: 326.2478; found: 326.2466.
Supplementary Material
Acknowledgements
We wish to thank the Engineering and Physical Sciences Research Council (EPSRC) and the European Research Council (ERC: FP7/2007–2013, ERC grant No. 246785) for financial support. V.K.A. thanks the Royal Society for a Wolfson Research Merit Award and EPSRC for a Senior Research Fellowship. We thank Inochem-Frontier Scientific for the generous donation of boronic acids and boronic esters.
Footnotes
Supplementary data Supplementary data are available with the article through the journal Web site http://nrcresearchpress.com/doi/supp/10.1139/v2012-069.
References
- (1)(a).Prat L, Mojovic L, Levacher V, Dupas G, Quéguiner G, Bourguignon J. Tetrahedron Asymmetry. 1998;9(14):2509. doi:10.1016/S0957-4166(98)00228-6. [Google Scholar]; (b) Selim KB, Matsumoto Y, Yamada K, Tomioka K. Angew. Chem. Int. Ed. 2009;48(46):8733. doi: 10.1002/anie.200904676. and references therein. doi:10.1002/ anie.200904676. [DOI] [PubMed] [Google Scholar]
- (2)(a).Nilvebrant L, Andersson K-E, Gillberg P-G, Stahl M, Sparf B. Eur. J. Pharmacol. 1997;327(2–3):195. doi: 10.1016/s0014-2999(97)89661-6. doi:10.1016/S0014-2999(97)89661-6. [DOI] [PubMed] [Google Scholar]; (b) Hills CJ, Winter SA, Balfour JA. Drugs. 1998;55(6):813. doi: 10.2165/00003495-199855060-00008. doi:10.2165/00003495-1998 55060-00008. [DOI] [PubMed] [Google Scholar]
- (3).Pfizer Annual Report to Shareholders. 2008.
- (4).Jönsson NA, Sparf BA, Mikiver L, Moses P, Nilvebrant L, Glas G. EP 0325571. 1989. p. A1.
- (5).Andersson PG, Schink HE, Österlund K. J. Org. Chem. 1998;63(22):8067. doi:10.1021/jo981259r. [Google Scholar]
- (6).Hedberg C, Andersson PG. Adv. Synth. Catal. 2005;347(5):662. doi:10.1002/adsc.200404234. [Google Scholar]
- (7)(a).Ulgheri F, Marchetti M, Piccolo O. J. Org. Chem. 2007;72(16):6056. doi: 10.1021/jo0705667. doi:10.1021/jo0705667. [DOI] [PubMed] [Google Scholar]; (b) Jagdale AR, Sudalai A. Tetrahedron Lett. 2008;49(23):3790. doi:10.1016/j.tetlet.2008.04.007. [Google Scholar]; (c) Gallagher BD, Taft BR, Lipshutz BH. Org. Lett. 2009;11(23):5374. doi: 10.1021/ol9020404. doi:10.1021/ol9020404. [DOI] [PubMed] [Google Scholar]; (d) Yoo K, Kim H, Yun J. J. Org. Chem. 2009;74(11):4232. doi: 10.1021/jo900530s. doi:10.1021/jo900530s. [DOI] [PubMed] [Google Scholar]; (e) Wang X, Guram A, Caille S, Hu J, Preston JP, Ronk M, Walker S. Org. Lett. 2011;13(7):1881. doi: 10.1021/ol200422p. doi:10.1021/ol200422p. [DOI] [PubMed] [Google Scholar]
- (8).Botteghi C, Corrias T, Marchetti M, Paganelli S, Piccolo O. Org. Process Res. Dev. 2002;6(4):379. doi:10.1021/op020014k. [Google Scholar]
- (9)(a).Chen G, Tokunaga N, Hayashi T. Org. Lett. 2005;7(11):2285. doi: 10.1021/ol0507367. doi:10.1021/ol0507367. [DOI] [PubMed] [Google Scholar]; (b) Sörgel S, Tokunaga N, Sasaki K, Okamoto K, Hayashi T. Org. Lett. 2008;10(4):589. doi: 10.1021/ol702879u. doi:10.1021/ol702879u. [DOI] [PubMed] [Google Scholar]; (c) Kobayashi K, Nishikata T, Yamamoto Y, Miyaura N. Bull. Chem. Soc. Jpn. 2008;81(8):1019. doi:10.1246/bcsj.81.1019. [Google Scholar]
- (10)(a).Stymiest JL, Bagutski V, French RM, Aggarwal VK. Nature. 2008;456(7223):778. doi: 10.1038/nature07592. doi:10.1038/nature07592. [DOI] [PubMed] [Google Scholar]; ; ; (b) Bagutski V, French RM, Aggarwal VK. Angew. Chem. Int. Ed. 2010;49(30):5142. doi: 10.1002/anie.201001371. doi:10.1002/anie.201001371. [DOI] [PubMed] [Google Scholar]; ; These reactions are based on the seminal studies by Hoppe on the lithiation and trapping of secondary benzylic carbamates. See ; (c) Hoppe D, Carstens A, Krämer T. Angew. Chem. Int. Ed. Engl. 1990;29(12):1424. doi:10.1002/anie.199014241. [Google Scholar]; ; ; (d) Carstens A, Hoppe D. Tetrahedron. 1994;50(20):6097. doi:10.1016/S0040-4020(01)90461-2. [Google Scholar]
- (11)(a).Nave S, Sonawane RP, Elford TG, Aggarwal VK. J. Am. Chem. Soc. 2010;132(48):17096. doi: 10.1021/ja1084207. doi:10.1021/ja1084207. [DOI] [PubMed] [Google Scholar]; (b) Roesner S, Casatejada JM, Elford TG, Sonawane RP, Aggarwal VK. Org. Lett. 2011;13(21):5740. doi: 10.1021/ol202251p. doi:10.1021/ol202251p. [DOI] [PubMed] [Google Scholar]; (c) Elford TG, Nave S, Sonawane RP, Aggarwal VK. J. Am. Chem. Soc. 2011;133(42):16798. doi: 10.1021/ja207869f. doi:10.1021/ja207869f. [DOI] [PubMed] [Google Scholar]; (d) Aggarwal VK, Ball LT, Carobene S, Connelly RL, Hesse MJ, Partridge BM, Roth P, Thomas SP, Webster MP. Chem. Commun. 2012;48:9230. doi: 10.1039/c2cc32176a. doi:10.1039/C2CC32176A. [DOI] [PubMed] [Google Scholar]
- (12)(a).Koe BK, Weissman A, Welch WM, Browne RG. J. Pharmacol. Exp. Ther. 1983;226:686. [PubMed] [Google Scholar]; (b) Welch WM, Kraska AR, Sarges R, Koe BK. J. Med. Chem. 1984;27(11):1508. doi: 10.1021/jm00377a021. doi:10.1021/jm00377a021. [DOI] [PubMed] [Google Scholar]
- (13).Maggon K. Drug Discov. Today. 2005;10(11):739. doi: 10.1016/S1359-6446(05)03468-9. doi: 10.1016/S1359-6446(05)03468-9. [DOI] [PubMed] [Google Scholar]
- (14)(a).Bøgesø KP, Christensen AV, Hyttel J, Liljefors T. J. Med. Chem. 1985;28(12):1817. doi: 10.1021/jm00150a012. doi:10.1021/jm00150a012. [DOI] [PubMed] [Google Scholar]; (b) Hyttel J, Larsen JJ. J. Neurochem. 1985;44(5):1615. doi: 10.1111/j.1471-4159.1985.tb08803.x. doi:10.1111/j.1471-4159.1985.tb08803.x. [DOI] [PubMed] [Google Scholar]
- (15).Negus SS, Brandt MR, Mello NK. J. Pharmacol. Exp. Ther. 1999;291:60. [PubMed] [Google Scholar]
- (16).Jadhav PK, Bhat KS, Perumal PT, Brown HC. J. Org. Chem. 1986;51(4):432. doi:10.1021/jo00354a003. [Google Scholar]
- (17).Yu W, Mei Y, Kang Y, Hua Z, Jin Z. Org. Lett. 2004;6(19):3217. doi: 10.1021/ol0400342. doi:10.1021/ol0400342. [DOI] [PubMed] [Google Scholar]
- (18).Pangborn AB, Giardello MA, Grubbs RH, Rosen RK, Timmers FJ. Organometallics. 1996;15(5):1518. doi: 10.1021/om9503712. [Google Scholar]
- (19).Roush WR, Walts AE, Hoong LK. J. Am. Chem. Soc. 1985;107(26):8186. doi:10.1021/ja00312a062. [Google Scholar]
- (20).Kahnberg P, Lager E, Rosenberg C, Schougaard J, Camet L, Sterner O, Nielsen EØ, Nielsen M, Liljefors T. J. Med. Chem. 2002;45(19):4188. doi: 10.1021/jm020839k. doi:10.1021/jm020839k. [DOI] [PubMed] [Google Scholar]
- (21).Tokunaga N, Hayashi T. Tetrahedron Asymmetry. 2006;17(4):607. doi:10.1016/j.tetasy.2006.01.036. [Google Scholar]
- (22).Paras NA, Simmons B, MacMillan DWC. Tetrahedron. 2009;65(16):3232. doi:10.1016/j.tet.2008.12.054. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.