

Summary
T lymphocytes regulate nutrient uptake to meet the metabolic demands of immune activation. The present study shows that the intracellular supply of large neutral amino acids (LNAAs) in T cells is regulated by pathogen and the T cell antigen receptor (TCR). A single System L transporter, Slc7a5, mediated LNAA uptake in activated T cells. Slc7a5-null T cells could not metabolically reprogram in response to antigen and failed clonal expansion and effector differentiation. The metabolic catastrophe caused by Slc7a5 loss reflects the requirement for sustained uptake of the LNAA leucine for activation of mammalian target of rapamycin complex 1 (mTORC1) and for expression of c-myc. Pathogen control of System L transporters is thus a critical metabolic checkpoint for T cells.
Introduction
T lymphocytes respond to antigen by proliferating and differentiating to effector subpopulations that mediate the adaptive immune response. Activated T cells must modulate their metabolic programs to match the metabolic demands of participating in an immune response. For example, T cell activation strikingly increases protein synthesis and these cells need to accordingly increase cellular amino acid uptake. Importantly, amino acids are not only required for protein synthesis. Glutamine can thus be diverted into metabolic intermediates such as pyruvate and lactate via a metabolic process known as glutaminolysis1,2. It is also important that T cells control the intracellular supply of leucine because this controls the activity of the serine/threonine kinase complex Mammalian target of rapamycin complex 1 (mTORC1)3-6. This kinase regulates CD8 cytotoxic T cell (CTL) differentiation, memory and migratory capacity7,8,9,10. mTORC1 also promotes the differentiation of CD4+ TH1 and TH17 cells while suppressing the differentiation of FoxP3 expressing regulatory T cells6,11,12,13. The mechanisms used by mTORC1 to control T cell differentiation are not fully understood although it is known that this kinase complex is required for the glycolytic reprograming that accompanies effector T cell differentiation5,14,15,16.
The transport of leucine, a large neutral amino acid (LNAA), is predominantly mediated by system L1 and system y+L amino acid transporters. These comprise a heterodimer of CD98 (Slc3a2)17-19 and either Slc7a5 (also known as LAT1), Slc7a8 (LAT2), Slc7a7 (y+LAT1) and Slc7a6 (y+LAT2)20. CD98 deletion or mutation results in early embryonic lethality21,22, and selective deletion of CD98 has shown that CD98 is critical for T and B cell proliferative expansion23,24. Moreover, mice with a T cell-specific deficiency of CD98 can accept a full major histocompatibility complex-mismatched cardiac allograft25. However, these studies do not inform about the relevance of System L amino transporters in lymphocytes because CD98 not only complexes with amino acid transporters but also forms complexes with integrins. Moreover, reconstitution experiments have shown that the integrin-binding domain of CD98 is important for lymphocyte proliferation whereas the amino acid-transport function was dispensable. Hence, CD98 is proposed to regulate lymphocytes because it amplifies integrin signals and not because it forms complexes with amino acid transporter light chains23,24.
The relevant LNAA transporters in T lymphocytes are thus not known and indeed, it is not known whether LNAA transport activity is coupled to T cell activation. Evidence exists, however, showing that pharmacological blockade of System L transport function blocks T cell proliferation5,26. Accordingly, the focus of the present study was to examine the dynamics and relevance of System L transport activity in immune activated T cells. We show that pathogen and T cell antigen receptor (TCR) triggering induced a striking increase in the ability of T cells to transport LNAAs via System L transporters. Moreover, System L mediated transport of leucine was essential for mTORC1 activity in T cells. System L transport activity in T cells required sustained immune activation via the T cell antigen receptor (TCR) or inflammatory cytokines such as interleukin 2 (IL-2). Transcriptional profiling of activated T cells identified Slc7a5 as a candidate for the TCR regulated System L amino acid transporter. Accordingly, we examined the consequences of the loss of Slc7a5 for T cell development and peripheral T cell function. Slc7a5 was shown to be the main LNAA transporter in antigen receptor activated T cells. Moreover, the loss of Slc7a5 prevented the proliferation and differentiation of CD4+ and CD8+ T cells although the capacity of CD4+ T cells to differentiate to regulatory T cells was unimpaired. Unexpectedly, antigen receptor activated Slc7a5-null T cells have complex metabolic defects beyond a simple loss of LNAA uptake. TCR-triggered Slc7a5-null T cells failed to increase glutamine, transferrin and glucose uptake and failed to undergo a metabolic switch to glycolysis. The catastrophic impact of Slc7a5 loss is explained in part by the requirement for sustained leucine uptake for mTORC1 activity, but also reflects that Slc7a5-null T cells can only respond to T cell activation to increase mRNA but not protein encoding c-Myc and key nutrient transporters. These data demonstrate that TCR and cytokine regulated amino acid transport controls mTORC1 activity in T cells. Moreover, the regulated expression of Slc7a5 is a key event for T cell metabolic reprogramming.
RESULTS
Antigen and IL-2 regulate System L amino acid transport
Naïve CD8+ T cells did not effectively take up 3H-labeled phenylalanine, a large neutral amino acid that is transported via System L amino acid transporters (Fig. 1a). However, TCR triggering of CD8+ T cells with cognate peptide induced a substantial increase in phenylalanine transport (Fig. 1a). Effector CD8+ T cells from mice immunized with Listeria also showed enhanced phenylalanine transport compared to naïve T cells (Fig. 1b). TCR-primed CD8+ T cells cultured in IL-2 clonally expand and differentiate to cytotoxic T cells (CTLs). CTLs cultured in IL-2 had enhanced phenylalanine uptake; the removal of IL-2 or the exposure of cells to limiting IL-2 concentrations caused CTLs to decrease phenylalanine uptake (Fig. 1c). 3H-phenylalanine uptake by CTLs can be competed by unlabeled leucine but not by basic amino acids such as lysine or by the acidic amino acid aspartic acid (Fig. 1d). Moreover, treatment of CTLs with 2-aminobicyclo-(2,2,1)-heptane-2-carboxylic acid (BCH), an inhibitor of System L amino acid transporters, blocked the influx of phenylalanine in CTLs, whereas treatment with the MeIAB, a System A amino acid transport inhibitor, did not (Fig. 1d). Thus, activated effector T cells increase System L amino acid transporter activity.
Figure 1. System L amino acid transport in CD8+ T cells.

The data show 3H-phenylalanine uptake (c.p.m.) per 106 cells in (a) OT-I TCR transgenic lymph node cells cultured with IL-7 or stimulated with SIINFEKL for 4h (p=0.0147 )or 20 h p<0.0001. (b) Purified CD8+ T cells from Listeria-infected mice, 3 days post infection, and from uninfected mice. (c) Effector CTLs exposed to 20 ng/ml IL-2, 1.25 ng/ml IL-2 or medium alone for 20 h. (d) Phenylalanine flux (rate of Phe uptake) of IL-2 maintained CTLs in the presence of 10 mM cold amino acids; Phe, Lys, Asp or Leu, or in the presence of 10 mM of BCH or MeAIB. Data is from a minimum of 3 experiments done in triplicates (a, c and d), data in b is from 6 mice, 1 experiment.
Amino acid deprivation resulted in rapid inactivation of mTORC1 in CTLs as judged by the loss of phosphorylation of the mTORC1 substrate sequence in p70 S6-Kinase 1 (S6K1-threonine389) and the p70 S6-Kinase substrate sequences in the S6 ribosomal subunit (pS6-serines235/6) (Fig. 2a,b). mTORC1 activity was rapidly restored after adding back exogenous leucine to the cells (Fig. 2b). Furthermore, when replete media lacking glutamine alone was added back after amino acid depletion, S6 phosphorylationwas recovered, whereas when media lacking leucine was used then S6 phosphorylation was not restored (Fig. 2c). CTLs treated with BCH, the selective inhibitor of System L transport, also rapidly lost mTORC1 activity comparable to treatment with the mTORC1 inhibitor rapamycin (Fig. 2d). T cells deprived of leucine alone also failed to sustain mTORC1 activity (Fig. 2e). Previous studies have indicated that glutamine availability also regulates mTORC1 activity. The molecular basis for this glutamine sensitivity has been proposed to be due to the actions of Slc7a5-Slc3a2 operating as a bidirectional transporter regulating the simultaneous efflux of glutamine and influx of leucine27. Removal of glutamine for 1 hour also resulted in a loss of mTORC1 activity (Fig. 2e), whereas the addition of glutamine alone to amino acid-deprived CTLs without the presence of leucine did not restore mTORC1 activity (Fig. 2b,c). Collectively these data show that the intracellular supply of the branched-chain amino acid leucine via System L transporters is important to sustain mTORC1 activity in T cells.
Figure 2. Amino acids and mTORC1 in CD8+ T cells.

(a) Flow cytometric analysis with antibodies to phospho-Ser235/236 of ribosomal S6 protein (p-S6) in CTLs in amino acid replete RPMI (left) or switched to amino acid-free HBSS (right) for 15 min +/− 20 nM rapamycin (Rap) for 15 min. (b) Immunoblot analysis with phospho-p70S6K(T389) (p-70S6K) or pan-p70S6K antibodies of CTLs deprived of amino acids for 30 mins then re-fed complete amino acids (RPMI), gln (2 mM), leu (0.4 mM), leu and gln for 30 min +/− Rapamcyin as indicated. (c) Flow cytometric analysis of p-S6 in CTLs deprived of amino acids in HBSS for 30 min, then re-fed with complete RPMI, glutamine-free RPMI or leucine-free RPMI for 30 min. (d) Immunoblot analysis (left) with phospho-70S6K(T389), pan-p70S6K or SMC1 antibodies in CTLs treated with inhibitors BCH (50 mM) or Rapamycin. Right, flow cytometric analysis with antibodies to p-S6 of the same samples. (e) Immunoblot analysis (left) phospho-p70S6KT389, pan-p70S6K or SMC1 antibodies in CTLs maintained in RPMI (AA) or switched to glutamine free RPMI, leucine free RPMI or treated with Rapamycin for 1 h. Right, flow cytometric analysis with p-S6 antibodies of the same samples. Data are representative of 3 experiments.
Slc7a5, the system L transporter in activated T cells
To identify candidate System L transporters in T cells we examined Affymetrix microarray data of naïve versus antigen receptor activated T cells28. These data revealed that the most highly TCR-induced System L transporter was Slc7a5 (LAT1) (Fig. 3a). TCR triggering also induced increases in mRNA encoding CD98 (Slc3a2), the heavy chain subunit of System L transporters. Quantitative PCR analysis confirmed that Slc7a5 mRNA abundance was increased in T cells activated with antigen for 4 hours or 20 hours (Fig. 3b). Slc7a5 protein was only detected in the T cells activated for 20 hours (Fig. 3b). Surface expression of CD98 was detected in T cells activated with antigen for 4 hours, yet more CD98 was expressed in T cells activated for 20 hours (Fig. 3b). 3H-phenylalanine uptake was also increased in 20-hour TCR-stimulated cells (Fig. 1a). CTLs cultured in IL-2 had abundant Slc7a5 mRNA and protein, but this expression was dependent on sustained exposure to cytokine (Fig. 3c). Transcriptional profiling also indicated that OT-I effector CD8+ T cells isolated from Listeria-infected mice selectively up-regulated Slc7a5 (data assembled by the ImmGen consortium)29. Thus, a strong correlation exists between the expression of Slc7a5 and System L transporter activity in peripheral T cells.
Figure 3. Regulation of System L amino acid transporters in T cells.

(a) Fold induction of mRNA expression of the indicated nutrient transporters in naïve P14 CD8+ T cells versus 4h TCR stimulated cells (3 mice per group). (b) Relative Slc7a5 mRNA levels from RTPCR analysis of OT-I lymph node cells stimulated with SIINFEKL for 4 or 20 h (left) immunoblot analysis of Slc7a5 protein expression (middle), and CD98 surface expression by flow cytometry (right) of the same samples. (c) Left, Relative Slc7a5 mRNA levels from RTPCR analysis of IL-2 maintained CTLs exposed to 20 or 1.25 ng/ml IL-2 or media alone for 20 h; immunoblot analysis of Slc7a5 protein expression (center) and CD98 surface expression by flow cytometry (right) of the same samples. (d,e) 3H-phenylalanine uptake by OT-I T cells TCR-stimulated 18 h +/− inhibitors PD184352 (2 μM), rapamycin (20 nM) or CyclosporinA (CsA) (100 nM) p=0.0242 (d) and (e) IL-2 (20 ng/ml) +/− CsA compared to IL-7–maintained cells p=0.0246. (f) RTPCR of Slc7a5 gene expression in OT-I T cells TCR-stimulated +/− CsA (100 nM) compared to IL-7–maintained OT-I T cells p=0.0070. (b,c) Data are representative of 3 experiments in triplicates; (d-f) 2 collated experiments. SMC1 is loading control for immunoblots.
We then addressed how TCR signaling induces Slc7a5 expression. TCR-regulated glucose and glutamine uptake is regulated by the MAP kinases Erk1/2 and mTORC1 (refs. 2,14,16 ). However, PD184352, which prevents ERK1/2 activation, and rapamycin, which inhibits the mTORC1 complex, had no impact on the ability of TCR triggering to induce System L transport activity (Fig. 3d). Triggering of the TCR complex is known to lead to an increase in intracellular calcium and sustained activation of the calcium-regulated phosphatase calcineurin. Cyclosporin A (CsA), which inhibits TCR-induced calcium-calcineurin–regulated signaling pathways, abrogated TCR-induced system L activity (Fig. 3d). CsA prevents the production of the cytokine IL-2 by immune-activated T cells. Nevertheless, the addition of exogenous IL-2 to the TCR-stimulated cultures did not restore System L activity to the CsA-treated TCR-activated T cells (Fig. 3e). CsA also prevented TCR induction of Slc7a5 mRNA expression (Fig. 3f). These results indicate that calcineurin-regulated signaling pathways mediate TCR control of Slc7a5.
Generation of Slc7a5-null T cells
Systemic deletion of Slc7a5 causes embryonic lethality (unpublished data). However, mice with a single functional Slc7a5 allele deleted are born at normal Mendelian frequency and have normal peripheral lymphocyte subpopulations (data not shown). T cells haplo-insufficient for Slc7a5 underwent normal blastogenesis and proliferation in response to TCR triggering and IL-2. Slc7a5+/− T cells showed the expected reduction in Slc7a5 mRNA and protein expression and also had a 50% reduction in System L transporter activity while maintaining normal glutamine uptake (Supplementary Fig. 1). To further probe the importance of Slc7a5 in T cells we generated Slc7a5fl/flCD4-Cre mice to delete the Slc7a5 gene in CD4+CD8+ double-positive (DP) thymocytes and all subsequent T cell populations. The deletion of Slc7a5 alleles in Slc7a5fl/flCD4-Cre T cells was confirmed by genomic PCR analysis (Fig. 4a). Slc7a5fl/flCD4-Cre mice had normal numbers and frequency of conventional αβ T cells and NKT cells in the thymus (Fig. 4b). Slc7a5 forms a heterodimer with CD98 and one question we wished to address was the impact of Slc7a5 deletion on CD98 expression. Slc7a5fl/flCD4-Cre DP and single-positive (SP) thymocytes expressed approximately 50% less CD98 as compared to wild-type cells (Fig. 4c), whereas CD98 levels were reduced 2 fold in Slc7a5 null peripheral T cells compared to control cells (Fig. 4d). Slc7a5fl/flCD4-Cre mice also had normal numbers and frequency of naïve CD4+ and CD8+ T cell subsets in the spleen and lymph node and a normal frequency of FoxP3 regulatory T cells (Fig. 4e). We also used a Vav-Cre transgene to delete Slc7a5 in hematopoietic progenitors in the bone marrow30. These Slc7a5fl/flVav-Cre mice had normal thymocyte numbers and a normal distribution of CD4 and CD8 double-negative, DP and SP subsets. They also had normal numbers and frequency of peripheral T lymphocyte subpopulations, B lymphocytes and NK cells (Supplementary Fig. 2).
Figure 4. Slc7a5fl/flCD4-Cre mice.

(a) PCR analysis of genomic DNA isolated from the indicate thymocytes showing Slc7a5 floxed (FL), wild-type (WT) and deleted (DEL) PCR products. (b) Thymocyte (left) and thymocyte subset (middle) numbers of Slc7a5fl/fl and Slc7a5fl/flCD4-Cre mice, the right panel shows flow cytometric analysis of NKT cells from the indicated mice (c) CD98 in thymic subsets. (d,e) Cellular analysis of spleen and lymph node from Slc7a5fl/fl and Slc7a5fl/flCD4-Cre mice. (d) Flow cytometry analysis of CD98, CD62L and CD44 levels in lymph node T cells. (e) Total numbers of spleen (left) and brachial lymph node cells (center left). Flow cytometry analysis of CD4 and CD8 expression by lymph node T cells (center right) or Foxp3 and CD25 expression by splenic T cells (right). (f) immunoblot analysis with Slc7a5 antibodies of naïve and 20 h TCR stimulated Slc7a5fl/fl and Slc7a5fl/flCD4-Cre CD8+ T cells. (g) 3H-phenylalanine uptake by Slc7a5fl/fl and Slc7a5fl/flCD4-Cre CD8+ TCR stimulated (20 h) T cells, p=0.0003. Uptake performed in the presence or absence of cold competitor 10 mM Leu to quench. (b-e). Representative data from 8-12 week mice, 3 mice per group; (f) representative of 3 experiments. SMC1 is loading control. (g) 3 mice per group.
The phenotype of Slc7a5fl/flCD4-Cre mice indicated that Slc7a5 was not required for T cell selection in the thymus or for homeostasis of the naïve T cell pool. This conclusion was confirmed when we backcrossed Slc7a5fl/flCD4-Cre mice to OT-I TCR transgenic mice. These express a Vα2Vβ5 TCR specific for the ovalbumin (OVA)-derived peptide SIINFEKL (OT-IxSlc7a5fl/flCD4-Cre). Thymocyte development and peripheral T cell numbers in OT-IxSlc7a5fl/flCD4-Cre mice were normal (data not shown). T cells that develop in Slc7a5fl/flCD4-Cre expressed undetectable amounts of Slc7a5 protein (Fig. 4f). Importantly, analysis of phenylalanine uptake revealed that Slc7a5-null TCR-activated T cells have no detectable System L transporter activity (Fig. 4g).
slc7a5 null T cells have defective immune responses
We next explored the response of Slc7a5fl/flCD4-Cre T cells to immune activation. Slc7a5-null CD4+ T cells could not respond to antigen receptor ligation and the appropriate polarizing cytokines to effectively produce TH1 or TH17 cells (Fig. 5a). Slc7a5-null CD8+ T cells also had a severe defect in their ability to respond to cognate antigen and IL-2 to produce cytotoxic effector cells (Fig. 5a). However Slc7a5-null CD4+ T cells could respond normally to transforming growth factor-β (TGF-β) and IL-2 to make Foxp3 positive induced regulatory T cells (Fig. 5a).
Figure 5. Effector cell differentiation of Slc7a5fl/flCD4-Cre T cells.

(a) Data show numbers of in vitro differentiated effector T cells from Slc7a5fl/fl and Slc7a5fl/flCD4-Cre spleen T cells: Th1 (left) p=0.0041, Th17 (center left) p=0.0044, inducible T regs (center right) or CTL (far right) p<0.0001. (b) Slc7a5fl/fl and Slc7a5fl/flCD4-Cre were immunized with T cell-dependent antigen NP-OVA. Serum antibody endpoint titers of NP-32 specific IgM antibodies (left) p=0.0684 and NP-2 specific IgG1 antibodies (right) p<0.0001 day 7 post-immunization. (c-e) OT-I and OT-IxSlc7a5fl/flCD4-Cre lymph node cells were mixed 1:1, CFSE-labelled and co-injected into C57BL/6Ly5.1 hosts. Mice were then immunized with LPS and SIINFEKL. (c) Analysis of transferred cells recovered from spleens 7 days after immunization. Left, ratios of OT-I CD8+ Vα2 cells, right panels show surface expression of activation markers on OT-I CD8+ Vα2 cells. (d) Analysis of CD25, CD69, CD44 and CFSE on OT-I CD8+ Vα2 cells recovered after 48 h, 2 independent experiments, 5 mice. (e) The ratios of OT-I CD8+ Vα2 cells recovered from lymph node p<0.0001 and spleen p=0.0003 after 48 h. (a) collated data from at least 3 experiments in triplicates; (b-e) each data point represents one mouse.
To explore the importance of Slc7a5 for T cell-mediated immune responses in vivo we looked at the ability of Slc7a5fl/flCD4-Cre mice to make a T-dependent antibody response to the model antigen nitrophenyl(NP)-OVA. T cell expression of Slc7a5 was not required for mice to respond to NP-OVA immunization to make IgM antibodies (Fig. 5b). However, affinity maturation of antibody production and the production of high-affinity NP-specific IgG1 responses was severely impaired in Slc7a5fl/flCD4-Cre mice. The generation of high-affinity antibody requires the collaboration of T follicular helper and B cells within germinal centers. T cell ‘help’ to B cells is thus dependent on T cell expression of Slc7a5.
To explore the role of Slc7a5 in CD8+ T cell function in vivo, wild-type and Slc7a5-nullOT-I CD8+ T cells were adoptively transferred to naïve recipient mice and their responses to immunization with cognate antigen and lipopolysaccharide were monitored. Slc7a5fl/flCD4-Cre OT-I T cells had a severe defect in their ability to produce effector CD8+ T cells in vivo (Fig. 5c). Very few Slc7a5-null OT-I T cells were present compared to control OT-I T cells in the spleens of 7 day immunized mice and the Slc7a5-null T cells were CD62Lhi compared to the CD62Llo phenotype of the control cells. We examined the response of wild-type and Slc7a5-null OT-I CD8+ T cells to antigen 2 days after immunization. Pathogen-activated Slc7a5-null T cells normally upregulated expression of the IL-2 receptor alpha chain CD25, CD69 and CD44, however, only the wild-type but not the Slc7a5-null OT-I CD8+ T cells could undergo a proliferative expansion in vivo (Fig. 5d,e).
In vitro experiments also examined the importance of Slc7a5 expression for T cells. Slc7a5-null CD8+ T cells responded to cognate antigen in vitro by increasing expression of CD69, CD44 and CD25 (Fig. 6a). They are also able to secrete normal amounts of IL-2 and produce interferon-γ, although at much lower concentrations than control TCR-activated T cells (Fig. 6b). Thus Slc7a5 is not required for the initial events of T cell activation. Flow cytometric FSC and SSC analysis of Slc7a5-null T cells revealed that these cells did not undergo normal blastogenesis (Fig. 6c). Hence TCR-activated Slc7a5-null T cells are much smaller than normal T lymphoblasts. In this respect, TCR-mediated activation of normal T cells induces an enhancement of ribosomal biogenesis as measured by the high expression of the S6 ribosomal subunits in TCR-activated T cells. In contrast, Slc7a5-null T cells did not upregulate S6 protein (Fig. 6d). Furthermore, Slc7a5-null T cells could not proliferate in response to TCR triggering in vitro. Wild-type but not Slc7a5-null OT-I CD8+ T cells undergo multiple cell divisions in response to the TCR ligand SIINFEKL (Fig. 6c). We also examined the ability of Slc7a5-null CD4+ and CD8+ T cells to undergo lymphopenia-induced proliferation in vivo. In these experiments control and Slc7a5-null T cells were adoptively transferred at a 1:1 ratio into Rag2−/− mice. After 14 days, the recovery of control and Slc7a5-null T cells from the recipient spleens showed that Slc7a5 was essential to allow both CD4+ and CD8+ T cells to undergo proliferative expansion in a lymphopenic environment (Fig. 6e). Slc7a5 is thus essential for CD4 and CD8 T cells to mediate adaptive immune responses.
Figure 6. Activation and proliferation of Slc7a5fl/flCD4-Cre T cells.
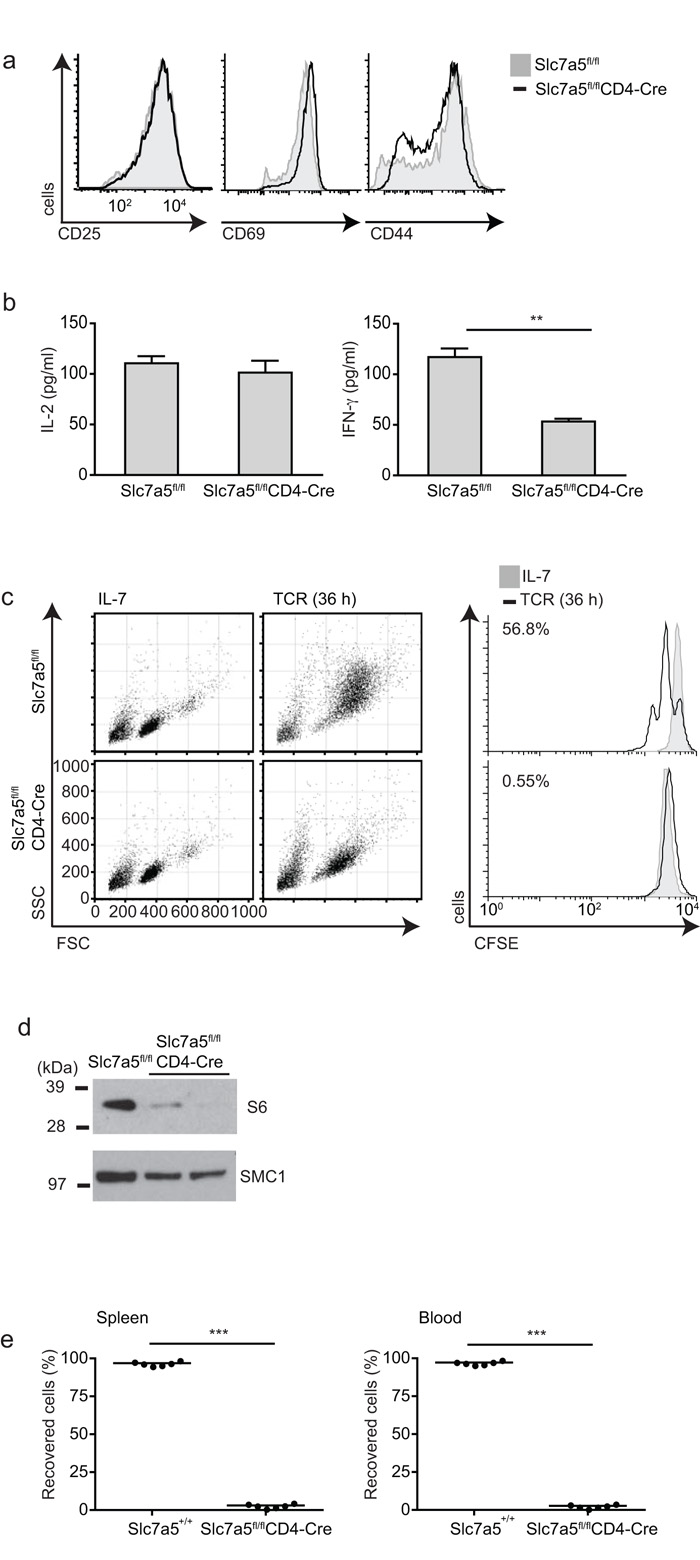
OT-I Slc7a5fl/fl and OT-IxSlc7a5fl/flCD4-Cre lymph node cells were stimulated through the TCR. (a) Cell surface expression of CD25, CD69 and CD44 on CD8+ T cells, and (b) the amount of IL-2 (left) and IFN-γ (right) produced after 20 h TCR stimulation. (c) Forward- and side-scatter profiles of CD8+ T cells after 36 h activation are shown compared to unstimulated cells maintained in IL-7 (left) and CFSE dilution (right). (d) Immunoblot analysis of total ribosomal S6 protein of CD8+ T cells after 20 h activation. (e) Slc7a5+/+ (CD45.1) and Slc7a5fl/flCD4-Cre (CD45.2) T cells were mixed at a ratio of 1:1 and adoptively transferred into Rag2−/− hosts. The graphs show percentage of recovered Slc7a5+/+ and Slc7a5fl/flCD4-Cre T cells in spleen and blood 14 days after adaptive transfer. Each data point represents data from one mouse. a, c, d data are representative of at least 3 experiments. (b) n = 3 mice per group, 1 independent experiment, triplicate samples, p=0.0019. (e) n=3 mice per group, 2 independent experiments, p<0.0001.
Slc7a5 is essential for T cell metabolic reprogramming
Why are Slc7a5-null T cells unable to differentiate or proliferate? This finding could not be solely due to loss of mTORC1 activity caused by failed leucine uptake because the phenotype of Slc7a5-null T cells was much more severe than the phenotypes caused by loss of mTORC1 activity; for example, inhibition of mTOR does not prevent T cell clonal expansion9,14. In this context, the cell growth and blastogenic defects of the Slc7a5-null T cells prompted analysis of the impact of Slc7a5 deletion on the ability of T cells to metabolically reprogram in response to immune activation. Previous studies have shown that glucose and glutamine metabolism in T cells is regulated by the transcription factor c-Myc that acts to control expression of glucose and glutamine transporters16. c-Myc is a protein with a very short half life31,32 and we hypothesized that the expression of c-Myc protein might be very sensitive to loss of a key amino acid transporter. Slc7a5fl/flCD4-Cre cells responded to immune activation and increased expression of c-Myc mRNA (Fig. 7a). However, they failed to increase expression of c-Myc protein. It is important to note that this effect of Slc7a5 loss on c-Myc expression was not explained by the role of amino acid uptake in controlling mTORC1 activity. Rapamycin treatment inhibited mTORC1 activity in T cells but did not prevent TCR-mediated increases in expression of c-Myc protein (Fig. 7b).
Figure 7. Metabolic consequences of Slc7a5 deletion in T cells.
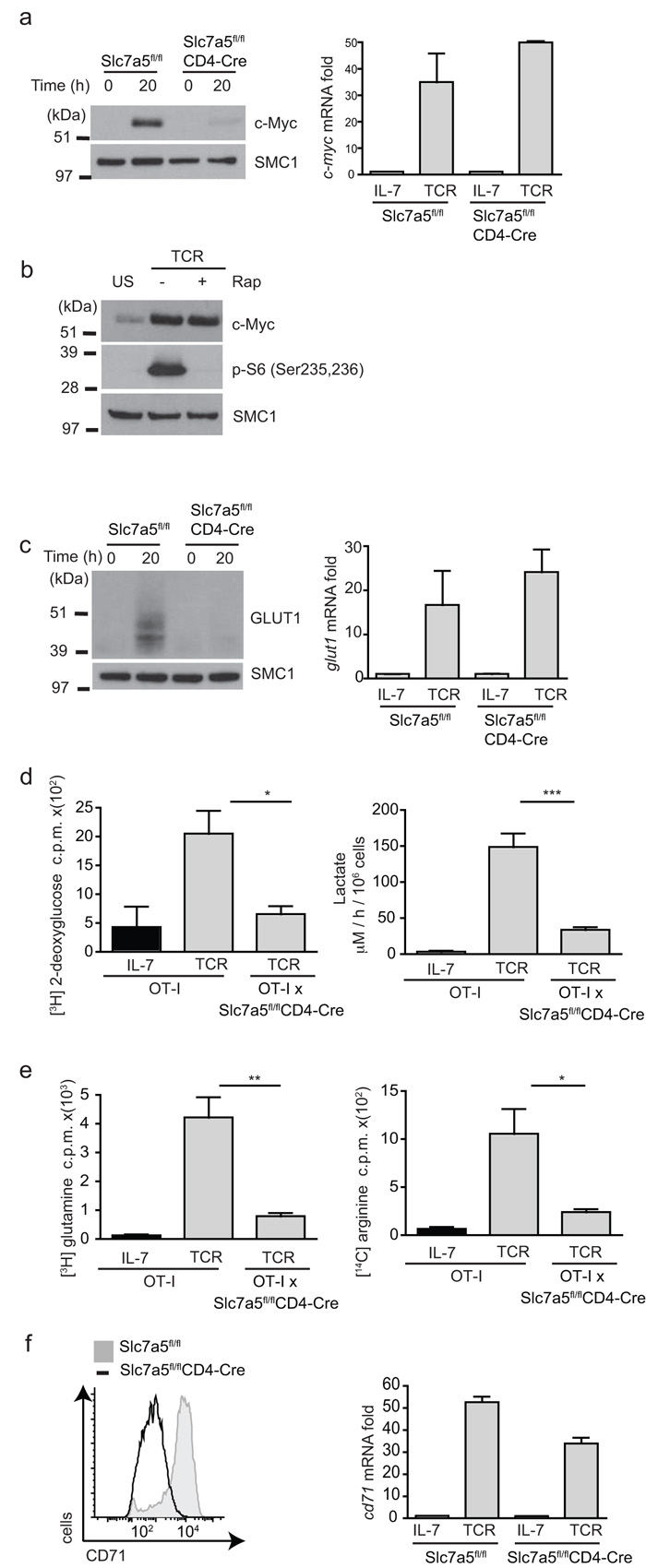
(a) Immunoblot analyses with c-Myc antbodies (left) and real-time PCR gene expression of c-Myc mRNA in OT-I and OT-Ix Slc7a5fl/flCD4-Cre CD8 T cells stimulated with SIINFEKL for 20h. (b) Immunoblot analyses with c-Myc and p-S6(Ser235,6) antisera in OT-I and OT-Ix Slc7a5fl/flCD4-Cre lymph node T cells stimulated with SIINFEKL for 20h with and without rapamycin (rap) treatment. (c) Immunoblot analysis with Glut1 antibodies (left) and Glut1 mRNA levels( right) in OT-I and OT-Ix Slc7a5fl/flCD4-Cre lymph node T cells stimulated with SIINFEKL for 20h. (d) 3H-2-deoxy-glucose uptake (left), p=0.0239 and lactate output (right), p=0.0002 in OT-I and OT-Ix Slc7a5fl/flCD4-Cre lymph node T cells. (e) 3H glutamine (left), p=0.0086 and 14C arginine (right) uptake, p=0.0358 in OT-I and OT-Ix Slc7a5fl/flCD4-Cre lymph node T cells stimulated with SIINFEKL for 20h. (f) Flow cytometry analysis with CD71 antibodies on CD8+ T cells (left) and CD71 mRNA expression in OT-I and OT-Ix Slc7a5fl/flCD4-Cre lymph node T cells stimulated with SIINFEKL for 20h (right). (a,b,e) data is representative of 3 experiments; (c,d) 3 mice per group. All samples done in triplicates. SMC1 is shown as a loading control.
The expression of c-Myc is required for the glycolytic switch and increase in glutaminolysis that accompanies T cell activation16. In this respect, Slc7a5-null T cells failed to increase expression of the glucose transporter Glut1 upon TCR stimulation (Fig. 7c) and have significantly reduced glucose uptake and lactate output in comparison to control TCR-activated cells (Fig. 7d). Furthermore, Slc7a5-null T cells also failed to increase glutamine and arginine uptake in response to T cell activation and they failed to express the transferrin receptor, CD71 (Fig. 7e,f). Note the failure of Slc7a5-null T cells to express CD71 and Glut1 protein did not lie in the inability of these cells to increase mRNA abundance for these proteins. There was no detectable difference in CD71 and Glut1 mRNA abundance between TCR-activated control and Slc7a5-null T cells, only a difference in protein expression (Fig. 7 c,f). The expression of Slc7a5 is thus required for T cells to sustain c-Myc expression and to sustain expression of proteins encoding key nutrient transporters.
DISCUSSION
The present study shows that the System L amino acid transport activity in peripheral T lymphocytes is linked to the immune activation of these cells by pathogens. Triggering of the T cell antigen receptor markedly increases in the ability of T cells to transport large neutral amino acids across their plasma membranes. Moreover, System L transport activity is not autonomous in antigen-primed T cells but requires sustained immune activation via the TCR or inflammatory cytokines such as Interleukin 2 (IL-2). The ability of the TCR to control System L transporter activity indicates that mechanisms have evolved to link T cell responses to pathogens with the ability of T cells to control intracellular pools of amino acids. In this context, it was noteworthy that TCR induction of Slc7a5 expression and System L transport activity was prevented by inhibition of calcineurin-mediated signaling pathways by CsA. The latter is a powerful immunosuppressive drug and its ability to prevent Slc7a5 expression affords some novel insight that part of its mechanism of action may be to prevent TCR-induced reprogramming of amino acid metabolism.
At least 4 genes encoding System L1 and System y+L transporters have been described20 but is striking that a single System L transporter, Slc7a5, was responsible for mediating LNAA uptake in immune-activated T cells. Slc7a5 is thus the dominant LNAA transporter in immune-activated T cells, and no redundancy exists between other System L1 or y+L transporters. In this respect it was intriguing that Slc7a5 was not apparently needed for T cell development in the thymus. T cell progenitors in the thymus proliferate rapidly following TCR beta chain selection and this would be a metabolically demanding process where one would expect expression of LNAA transporters to be essential. The failure to see defects in thymic development in Slc7a5fl/flCD4-Cre mice could reflect that the CD4-Cre transgene deletes Slc7a5 after the cells have passed the biosynthetically demanding stage of pre-T cell proliferation. However, we have also used a Vav-Cre transgene to delete Slc7a5 in hematopoietic progenitors in the bone marrow30 and these Slc7a5fl/flVav-Cre mice also had no defects in thymus development. There must therefore be other LNAA transporters that can substitute for Slc7a5 in T cell progenitors. In this context, transcriptional profiling indicates that T cell progenitors express abundant Slc7a6 (y+LAT2) and Slc7a5 (LAT1) (data assembled by the ImmGen consortium)29. The T cell dependency on Slc7a5 is thus restricted to effector T cells undergoing proliferative expansion and differentiation in response to pathogens. Slc7a5 is not required for the maintenance of naïve T cells. It was also noteworthy that Foxp3 expressing natural and induced regulatory T cells were not dependent on Slc7a5 expression.
Why would effector T cells be so dependent on Slc7a5? We show that System L mediated intracellular transport of leucine is essential for mTORC1 activity in activated T cells. The inability of Slc7a5-null T cells to sustain mTORC1 activity would thus cause defects in CD4+ and CD8+ T cell differentiation6,13,33. Note, the ability of mTORC1 to sense leucine uptake allows this kinase to modulate cellular responses to nutrient availability. The present data afford the insight that the TCR has assumed control of this evolutionarily conserved pathway and controls mTORC1 activity by controlling the expression of leucine transporters. The rate of intracellular leucine uptake in T cells is thus not determined by the extracellular leucine concentration but by TCR control of leucine transporter expression. How does leucine control mTORC1 activity in mammalian cells? This process is not fully understood although it is proposed that amino acid sensing for mTORC1 activity in mammalian cells is initiated within lysosomes and involves amino acid-dependent activation of the guanine nucleotide exchange activity of the Ragulator complex34. This results in the accumulation of active GTP-bound RagA GTPases, which then recruits mTORC1 to the lysosomal surface34 where it interacts with the small GTPase Rheb, a potent stimulator of mTORC1 kinase activity in T cells13. One candidate for the direct amino acid sensor involved in regulating the activity of the Rag GTPases is leucyl-tRNA synthetase35,36.
The role of leucine in controlling mTORC1 activity does not fully explain the functional defects of Slc7a5-null T cells. For example, Slc7a5 deletion, but not mTORC1 inhibition by rapamycin, prevents T cell clonal expansion. However, there are selective and critical protein synthesis defects in activated Slc7a5-null T cells that have a catastrophic impact on the ability of Slc7a5-null T cells to reprogram metabolism. Immune-activated Slc7a5-null T cells fail to increase glucose transport because they only increase expression of mRNA encoding the glucose transporter Glut1 but they cannot express Glut1 protein. Slc7a5-null T cells fail to express the transferrin receptor CD71 and they also fail to increase glutamine uptake in response to TCR triggering. In this context, the Slc7a5-null T cells closely phenocopy c-Myc-null T cells in being unable to upregulate glucose and glutamine metabolism in response to immune activation16. The explanation for this similarity is that Slc7a5-null T cells are effectively c-Myc null. They can respond to immune activation to increase expression of mRNA encoding c-Myc but they do not express c-Myc protein. Accordingly, Slc7a5-null T cells are unable to upregulate the “metabolic machinery” required to permit T cell proliferation and differentiation. It should be emphasized that Slc7a5-null T cells did not have global defects in protein synthesis and could respond to TCR triggering to up-regulate expression of the IL-2 receptor, CD69 and CD44. TCR-activated Slc7a5-null T cells could also secret normal amounts of IL-2 and reasonable, albeit reduced amounts of interferon-γ. Why the selectivity? One insight is that c-Myc has very short half-life and needs to be constantly resynthesized. For example the estimated half life of c-Myc is 15 minutes whereas the half life of CD25 is >18 hours31,37. Proteins with a short half-life that need to be continually resynthesized will be much more dependent on sustained amino acid uptake.
In summary, the present study highlights that inducing LNAA transport through controlling expression of Slc7a5 is a primary function of the TCR. The directed transport of leucine through System L transporters controls mTORC1 activity in T cells and is also required to sustain c-Myc expression in activated T cells. The failure of TCR-activated Slc7a5-null T cells to express c-Myc cannot be explained by the loss of mTORC1 signaling as rapamycin inhibition of mTORC1 does not ablate c-Myc expression14 . However the inability of immune-activated Slc7a5-null T cells to sustain c-Myc expression causes multiple metabolic defects. The loss of c-Myc and mTORC1 activity is thus sufficient to explain the phenotype of Slc7a5-null T cells although we would not exclude that Slc7a5 expression also controls the expression of other critical signaling molecules. Nevertheless our data show that antigen receptor and pathogen control of Slc7a5 expression and amino acid uptake is a critical switch for the metabolic reprogramming that allows immune-activated T cells to mediate adaptive immune responses.
METHODS
Mice and cells
C57BL/6, Rag2null, Slc7a5fl/fl, CD4Cre, VavCre, OT-I TCR38 and P14 LCMV TCR39 transgenic mice were maintained in the WTB/RUTG, University of Dundee in compliance with UK Home Office Animals (Scientific Procedures) Act 1986 guidelines. The University Ethical Review Committee approved the procedures.
Slc7a5fl/fl genotyping was carried out by PCR of genomic DNA using primers 5′-GGCTCCTGGACTTATCTTGACCAA-3′ (forward) and 5′-GTGGTGCTTTGCTGAAGGCAGGG-3′ (reverse), producing products of LAT1-fl (359 bp), WT (271 bp) and LAT1-deleted (253 bp).
Primary T cells were activated to generate CTLs as described previously14. To generate TH1, TH17 and Treg cells, CD8+ T cells were depleted from lymph node preparations using CD8 depletion kit (Miltenyi Biotech). CD4 cells and APCs were cultured at 3 × 105 cells/ml for 5 days in the presence of anti-CD3 (2 μg/ml) and anti-CD28 (3 μg/ml) and cytokines; TH1: IL-12 (20 ng/ml) and IL-2 (20 ng/ml), TH17: IL-6 (50 ng/ml), IL-1β(10 ng/ml), TGF-β(3 ng/ml) and FICZ (300 nM), Treg: TGF-β1 (20 ng/ml) and IL-2 (20 ng/ml).
Inhibitor treatment: mTORC1 inhibitor, rapamycin (20 nM) (Calbiochem), MEK inhibitor PD184352 (2 μM) (synthesized in-house by the DSTT, Dundee), System L inhibitor BCH (50 mM) (Sigma), cyclosporin A (100 nM) (Sigma).
Flow cytometry
For cell surface staining, fluorochrome-conjugated antibodies (BD Pharmigen or eBioscience) were used to detect: CD98 (RC388), CD4 (RM4-5), CD8 (53-6.7), TCRβ (H57-597), B220 (RA3-6B2) CD25 (PC61), CD71 (C2F2), CD44 (IM7), CD62L (MEL-14), CD45.1 (104), CD45.2 (A20), CD24 (M1/69), TCR Vα2 (B20.1) and Fc Block (2.4G2). Live cells were gated according to their forward scatter and side scatter. Data were acquired on FACSCalibur or LSR Fortessa (Becton Dickinson) and analyzed using FlowJo software (TreeStar). For NKT staining, CD1d-DimerX (BD Pharmingen) was loaded with the α-galactoceramide (α-GalCer) synthetic analog KRN7000 (Enzo Life Sciences) and used according to manufacturer’s instructions. Intracellular Foxp3 staining was performed using a Foxp3 intracellular staining kit (eBioscience). Intracellular phospho-S6 staining was performed using phospho-S6 ribosomal protein (Ser235/236) antibody (Cell Signaling Technology) and secondary goat-anti rabbit PE (Jackson ImmunoResearch Labs) or Alexa 647 phospho-S6(Ser235/236) (Cell Signaling Technology).
Affymetrix GeneChip mouse genome array
Lymph node T cells from P14 TCR transgenic mice were cultured in vitro for 4 h +/− the LCMV gp33 peptide (3 μM). RNA was extracted using the RNeasy Mini kit (Qiagen) and microarray analysis from three independent replicate samples was performed by the Finnish DNA Microarray Centre at the Centre for Biotechnology, Turku, Finland, using a GeneChip mouse genome 430_2.0 array (Affymetrix). Affymetrix Expression Console v1.1 (Affymetrix) was used to normalize the data.
Immunoblotting
Immunoblotting was done as described previously14. The Slc7a5 antisera was generated “in house”, and Glut1 antibody was a kind gift from G. Holman, University of Bath40. Structural maintenance of chromosomes 1 (SMC1) antibody (Bethyl) was used as a loading control.
Nutrient measurements
Briefly, nutrient uptake was carried out using 1×106 cells resuspended in 0.4 ml uptake medium. Phenylalanine uptake was carried out in HBSS (GIBCO) containing [3H] L-phenylalanine (0.5 μCi/ml) and a final extracellular L-leucine concentration of 5 μM. Uptake assay was performed in presence of 10 mM cold leucine to quench System L specific activity. 2-deoxyglucose uptake was carried out in glucose-free RPMI (GIBCO) containing [3H] 2-deoxyglucose (0.5 μCi/ml). Glutamine and arginine uptake was carried out in HBSS (GIBCO) containing 0.5 μCi/ml [3H] L-Glutamine and [14C] L-Arginine. 4 minute uptake assays were carried out layered over 0.5 ml of 1:1 silicone oil (Dow Corning 550 (BDH silicone products); specific density, 1.07 g/ml):dibutyl phthalate (Fluka). Cells were pelleted below the oil, the aqueous supernatant solution, followed by the silicon oil/dibutyl phthalate mixture was aspirated, and the cell pellet underneath resuspended in 200 μl NaOH (1M) and β-radioactivity measured by liquid scintillation counting in a Beckman LS 6500 Multi-Purpose Scintillation Counter (Beckman Coulter). Lactate production by TCR-triggered CD8+ T cells was measured as described 14.
Proliferation assays
Cells were labeled with 10 μM CFSE at 37 °C for 20 min in RPMI 1640 Cells were then activated and proliferation was assessed by flow cytometric analysis of dilution of CFSE labeling.
IL-2 and IFN-γ ELISA
IL-2 and IFN-γ production by activated T cells was measured using Mouse IL-2 Ready-Set-Go or IFN-γ ‘Femto-HS’ High Sensitivity ELISA Ready-Set-Go (eBioscience) as per manufacturers instructions.
Quantitative real-time PCR
RNA was purified using the RNeasy RNA purification Mini Kit (Qiagen). Genomic DNA was digested with RNase-free DNase (Qiagen) following manufacturer instructions and reverse-transcribed using the iScript cDNA synthesis kit (BioRad). Quantitative PCR was performed in 96-well plate format using iQ SYBR Green based detection (BioRad) on a BioRad iCycler.
QPCR Primers
slc7a5: Forward: 5′-CTGGATCGAGCTGCTCATC-3′
Reverse: 5′-GTTCACAGCTGTGAGGAGC-3′
c-myc: Forward: 5′-CCACCAGCAGCGACTCTG-3′
Reverse: 5′-GAGATGAGCCCGACTCCG-3′
Glut1: Forward: 5′-CCAGCAGCAAGAAGGTGAC-3′
Reverse: 5′-ATGTTTGATTGTAGAACTCCTC-3′
CD71: Forward: 5′-GACGCTTTGGTGCTGGTGTTG-3′
Reverse: 5′-GCCTGCAGTCCAGCTGGC-3′
Listeria monocytogenes infection
106 OT-I cells were injected i.p. into C57BL/6 Ly5.1 recipient mice, followed by i.v. injection of 5×106 colony forming units of the attenuated ActA-deleted OVA-expressing Listeria monocytogenes strain (provided by H. Shen, University of Pennsylvania)41. After 3 days of infection, the mice were culled and CD8+ T cells isolated from spleens using AUTOMACS.
Immunization and antibody responses
Mice were immunized i.p. with 100 μg of NP(13)OVA (Biosearch Technologies) adsorbed to alum and serum collected day 7. For ELISA quantification of antibody responses, Immunolon plates (Thermo Fisher) were coated with NP(32)BSA or for high affinity measurement NP(2)BSA. Serum samples were serially diluted and bound Ig was detected using biotinylated anti-IgM or IgG1 (BD Bioscience), followed by horseradish peroxidase-conjugated streptavidin (eBioscience) and TMB substrate (eBioscience). Endpoint titers were calculated to give a measure of relative antibody concentration.
Adoptive transfers
For in vivo activation and proliferation, OT-I (CD45.1/2) and OT-IxSlc7a5fl/flCD4-Cre (CD45.2) lymph node cells were mixed 1:1, labeled with CFSE and co-injected into C57BL/6 (CD45.1) hosts. Transferred cells were identified and analyzed for activation and proliferation at 48 h and 1 week after activation.
For lymphopenia-induced proliferation, C57BL/6 (CD45.1) and Slc7a5fl/flCD4-Cre (CD45.2) lymph node cells were mixed 1:1 and co-injected into RAG2null hosts. The transferred T cells were quantified 2 weeks after transfer.
Statistical Analyses
Statistical analyses were performed using GraphPad Prism 4.00.. Unpaired t-test was used to calculate P-values, error bars on graphs show the s.e.m.
Supplementary Material
Acknowledgements
We wish to thanks and acknowledge L. Chen, an undergraduate student involved in initial characterizing of amino acid transporters expressed by CTLs. We thank members of the Biological Services Unit; R. Clarke of the Flow Cytometry Facility; and members of the D.A.C. laboratory and Simon Arthur for critical reading of the manuscript. This work was supported by a Welcome Trust Principal Research Fellowship and Program Grant 065975/Z/01/A and 097418/Z/11Z to D.A.C. and by Wellcome Trust Grant WT094226 to P.T. This research was also supported in part by the Intramural Research Program of NICHD, NIH.
Footnotes
Accession codes The data have been deposited in NCBI’s Gene Expression Omnibus and are accessible through GEO Series accession number GSE33942 28.
Author Contributions L.V.S. all in vitro and most in vivo experiments; J.R. in vivo NP-ova immunisation and Listeria infections; E.E. genomic PCR of Slc7a5fl/flCD4-Cre mice; Y-B.S. generation of Slc7a5fl/fl mice; P.M.T conceptual design and D.A.C conceptual design and manuscript authorship.
The authors have no conflicting financial interests.
References
- 1.Wang R, Green DR. Metabolic checkpoints in activated T cells. Nat. Immunol. 2012;13:907–915. doi: 10.1038/ni.2386. [DOI] [PubMed] [Google Scholar]
- 2.Carr EL, et al. Glutamine uptake and metabolism are coordinately regulated by ERK/MAPK during T lymphocyte activation. J. Immunol. 2010;185:1037–1044. doi: 10.4049/jimmunol.0903586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beugnet A, Tee AR, Taylor PM, Proud CG. Regulation of targets of mTOR (mammalian target of rapamycin) signalling by intracellular amino acid availability. Biochem. J. 2003;372:555. doi: 10.1042/BJ20021266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schriever SC, Deutsch MJ, Adamski J, Roscher AA, Ensenauer R. Cellular signaling of amino acids towards mTORC1 activation in impaired human leucine catabolism. The Journal of Nutritional Biochemistry. 2012 doi: 10.1016/j.jnutbio.2012.04.018. doi:10.1016/j.jnutbio.2012.04.018. [DOI] [PubMed] [Google Scholar]
- 5.Zheng Y, Delgoffe GM, Meyer CF, Chan W, Powell JD. Anergic T Cells Are Metabolically Anergic. J. Immunol. 2009;183:6095–6101. doi: 10.4049/jimmunol.0803510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Powell JD, Delgoffe GM. The Mammalian Target of Rapamycin: Linking T Cell Differentiation, Function, and Metabolism. Immunity. 2010;33:301–311. doi: 10.1016/j.immuni.2010.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rao RR, Li Q, Odunsi K, Shrikant PA. The mTOR kinase determines effector versus memory CD8+ T cell fate by regulating the expression of transcription factors T-bet and Eomesodermin. Immunity. 2010;32:67–78. doi: 10.1016/j.immuni.2009.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sinclair LV, et al. Phosphatidylinositol-3-OH kinase and nutrient-sensing mTOR pathways control T lymphocyte trafficking. Nat. Immunol. 2008;9:513–521. doi: 10.1038/ni.1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Araki K, et al. mTOR regulates memory CD8 T-cell differentiation. Nature. 2009;460:108–112. doi: 10.1038/nature08155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pearce EL, et al. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature. 2009;460:103–107. doi: 10.1038/nature08097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kopf H, la Rosa, de GM, Howard OMZ, Chen X. Rapamycin inhibits differentiation of Th17 cells and promotes generation of FoxP3+ T regulatory cells. International Immunopharmacology. 2007;7:1819–1824. doi: 10.1016/j.intimp.2007.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sauer S, et al. T cell receptor signaling controls Foxp3 expression via PI3K, Akt, and mTOR. Proc. Natl. Acad. Sci. U.S.A. 2008;105:7797–7802. doi: 10.1073/pnas.0800928105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Delgoffe GM, et al. The kinase mTOR regulates the differentiation of helper T cells through the selective activation of signaling by mTORC1 and mTORC2. Nat. Immunol. 2011;12:295–303. doi: 10.1038/ni.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Finlay DK, et al. PDK1/mTOR and Hypoxia-inducible factor 1 integrate metabolism and migration of CD8 T cells. Journal of Experimental Medicine. 2012;209:2441–2453. doi: 10.1084/jem.20112607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shi LZ, et al. HIF1 -dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. Journal of Experimental Medicine. 2011;208:1367–1376. doi: 10.1084/jem.20110278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang R, et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity. 2011;35:871–882. doi: 10.1016/j.immuni.2011.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gottesdiener KM, et al. Isolation and structural characterization of the human 4F2 heavy-chain gene, an inducible gene involved in T-lymphocyte activation. Mol. Cell. Biol. 1988;8:3809–3819. doi: 10.1128/mcb.8.9.3809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lindsten T, June CH, Thompson CB, Leiden JM. Regulation of 4F2 heavy-chain gene expression during normal human T-cell activation can be mediated by multiple distinct molecular mechanisms. Mol. Cell. Biol. 1988;8:3820–3826. doi: 10.1128/mcb.8.9.3820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Parmacek MS, Karpinski BA, Gottesdiener KM, Thompson CB, Leiden JM. Structure, expression and regulation of the murine 4F2 heavy chain. Nucleic acids research. 1989;17:1915–1931. doi: 10.1093/nar/17.5.1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Verrey FO, et al. CATs and HATs: the SLC7 family of amino acid transporters. Pflugers Archiv European Journal of Physiology. 2004;447:532–542. doi: 10.1007/s00424-003-1086-z. [DOI] [PubMed] [Google Scholar]
- 21.Tsumura H, et al. The targeted disruption of the CD98 gene results in embryonic lethality. Biochemical and Biophysical Research Communications. 2003;308:847–851. doi: 10.1016/s0006-291x(03)01473-6. [DOI] [PubMed] [Google Scholar]
- 22.Sato Y, Heimeier RA, Li C, Deng C, Shi Y-B. Extracellular domain of CD98hc is required for early murine development. Cell Biosci. 2011;1:7. doi: 10.1186/2045-3701-1-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cantor J, et al. CD98hc facilitates B cell proliferation and adaptive humoral immunity. Nat. Immunol. 2009;10:412–419. doi: 10.1038/ni.1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cantor J, Slepak M, Ege N, Chang JT, Ginsberg MH. Loss of T Cell CD98 H Chain Specifically Ablates T Cell Clonal Expansion and Protects from Autoimmunity. J. Immunol. 2011;187:851–860. doi: 10.4049/jimmunol.1100002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu Z, et al. Deletion of CD98 Heavy Chain in T Cells Results in Cardiac Allograft Acceptance by Increasing Regulatory T Cells. Transplantation Journal. 2012;93:1116–1124. doi: 10.1097/TP.0b013e31824fd7cd. [DOI] [PubMed] [Google Scholar]
- 26.Usui T, et al. Brasilicardin A, a Natural Immunosuppressant, Targets Amino Acid Transport System L. Chemistry & Biology. 2006;13:1153–1160. doi: 10.1016/j.chembiol.2006.09.006. [DOI] [PubMed] [Google Scholar]
- 27.Nicklin P, et al. Bidirectional Transport of Amino Acids Regulates mTOR and Autophagy. Cell. 2009;136:521–534. doi: 10.1016/j.cell.2008.11.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Navarro MN, et al. Protein kinase D2 has a restricted but critical role in T-cell antigen receptor signalling in mature T-cells. Biochem. J. 2012;442:649–659. doi: 10.1042/BJ20111700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Heng TSP, et al. The Immunological Genome Project: networks of gene expression in immune cells. Nat. Immunol. 2008;9:1091–1094. doi: 10.1038/ni1008-1091. [DOI] [PubMed] [Google Scholar]
- 30.Ogilvy S, et al. Promoter elements of vav drive transgene expression in vivo throughout the hematopoietic compartment. Blood. 1999;94:1855–1863. [PubMed] [Google Scholar]
- 31.Hann SR, Eisenman RN. Proteins encoded by the human c-myc oncogene: differential expression in neoplastic cells. Mol. Cell. Biol. 1984;4:2486–2497. doi: 10.1128/mcb.4.11.2486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hann SR, Abrams HD, Rohrschneider LR, Eisenman RN. Proteins encoded by v-myc and c-myc oncogenes: identification and localization in acute leukemia virus transformants and bursal lymphoma cell lines. Cell. 1983;34:789–798. doi: 10.1016/0092-8674(83)90535-4. [DOI] [PubMed] [Google Scholar]
- 33.Delgoffe GM, et al. The mTOR Kinase Differentially Regulates Effector and Regulatory T Cell Lineage Commitment. Immunity. 2009;30:832–844. doi: 10.1016/j.immuni.2009.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bar-Peled L, Schweitzer LD, Zoncu R, Sabatini DM. Ragulator is a GEF for the rag GTPases that signal amino acid levels to mTORC1. Cell. 2012;150:1196–1208. doi: 10.1016/j.cell.2012.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Han JM, et al. Leucyl-tRNA synthetase is an intracellular leucine sensor for the mTORC1-signaling pathway. Cell. 2012;149:410–424. doi: 10.1016/j.cell.2012.02.044. [DOI] [PubMed] [Google Scholar]
- 36.Bonfils G, et al. Leucyl-tRNA synthetase controls TORC1 via the EGO complex. Mol. Cell. 2012;46:105–110. doi: 10.1016/j.molcel.2012.02.009. [DOI] [PubMed] [Google Scholar]
- 37.Smith KA, Cantrell DA. Interleukin 2 regulates its own receptors. Proc. Natl. Acad. Sci. U.S.A. 1985;82:864–868. doi: 10.1073/pnas.82.3.864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kurts C, et al. Constitutive class I-restricted exogenous presentation of self antigens in vivo. Journal of Experimental Medicine. 1996;184:923–930. doi: 10.1084/jem.184.3.923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pircher H, Bürki K, Lang R, Hengartner H, Zinkernagel RM. Tolerance induction in double specific T-cell receptor transgenic mice varies with antigen. Nature. 1989;342:559–561. doi: 10.1038/342559a0. [DOI] [PubMed] [Google Scholar]
- 40.Holman GD, et al. Cell surface labeling of glucose transporter isoform GLUT4 by bis-mannose photolabel. Correlation with stimulation of glucose transport in rat adipose cells by insulin and phorbol ester. J. Biol. Chem. 1990;265:18172–18179. [PubMed] [Google Scholar]
- 41.Pearce EL, Shen H. Generation of CD8 T cell memory is regulated by IL-12. J. Immunol. 2007;179:2074–2081. doi: 10.4049/jimmunol.179.4.2074. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.