

Abstract
ARAP3, a GTPase activating protein for Rho and Arf family GTPases, is one of many PI3K effectors. Here we investigate the regulatory input of PI3K upstream of ARAP3, by analysing neutrophils from an ARAP3 PH domain point mutation knock-in mouse (R302, 303A), in which ARAP3 is uncoupled from activation by PI3K. ARAP3 PH domain point mutant neutrophils are characterised by disturbed responses linked to stimulation by either integrin ligands or immobilised immune complexes. These cells exhibit increased β2 integrin inside-out signalling (binding affinity and avidity), and our work suggests the disturbed responses to immobilised immune complexes are secondary to this. In vitro, neutrophil chemotaxis is affected in the mutant. In vivo, ARAP3 PH domain point mutant bone marrow chimeras exhibit reduced neutrophil recruitment to the peritoneum on induction of sterile peritonitis and also reduced inflammation in a model for rheumatoid arthritis. The present work suggests a dramatic regulatory input of PI3K into the regulation of β2 integrin activity, and processes dependent on this, by signalling through its effector ARAP3.
Keywords: PI3K, GTPase activating protein, neutrophil, integrin, Fcγ receptor, signalling, adhesion
Introduction
Neutrophils are highly specialised cells that form part of the innate immune system (1). These terminally differentiated, short-lived cells are continuously being generated in the bone marrow. Mature neutrophils reside in a quiescent state in the blood stream. On stimulation they follow a well-defined activation pathway which includes rolling along the vessel wall, followed by weak and then firm adhesion. Neutrophils finally extravasate and chemotax to sites of damage and/or infection, to phagocytose pathogens. Insufficient neutrophil activation leaves individuals susceptible to recurrent infections, as exemplified in leukocyte adhesion deficiency patients, where β2 integrin function is affected. On the flip side, neutrophils contribute to inflammation by producing reactive oxygen species (ROS) and cytotoxic enzymes. IgG-coated microorganisms drive the activation of neutrophil Fcγ receptors (FcγR), triggering neutrophil activation, normally at a site of infection. Immobilised immune complexes formed by autoantibodies reacting with their (host) antigens also trigger FcγR mediated neutrophil activation targeted against host tissue, leading to exacerbated autoimmune inflammatory responses. Neutrophil activity therefore needs to be tightly controlled to ensure an adequate innate immune response, whilst avoiding exaggerated inflammatory responses. On a molecular level, this control is exerted by many regulators, including agonist activated (class I) phosphoinositide 3-OH kinases (PI3Ks) and their effectors.
Agonist-activated PI3Ks are heterodimeric proteins that are activated downstream of tyrosine kinase coupled receptors (class IA) and G protein coupled receptors (class IB), respectively, as well as Ras. PI3Ks are responsible for the generation of the lipid second messengers phosphatidylinositol-(3,4,5)-trisphosphate [PtdIns(3,4,5)P3] and PtdIns(3,4)P2 in the plasma membrane (2). This drives the activation, mediated often at least in part by recruitment to the plasma membrane, of PI3K effectors. Although most attention has been placed on Akt / PKB signalling downstream of PI3K, there are also many other PI3K effectors; it is estimated that most cell types express in excess of 25 different PI3K effectors, which all mediate downstream effects. A large number of PI3K effectors isolated to date are regulators of small GTPases.
ARAP3 is a PI3K effector that was identified from neutrophils in a screen for PtdIns(3,4,5)P3 binding proteins (3). ARAP3 is a GTPase activating protein (GAP) for the small GTPases RhoA and Arf6, and it is regulated by PI3K and Rap-GTP. PtdIns(3,4,5)P3 regulates ARAP3 catalytically as an Arf GAP and causes ARAP3’s recruitment to the plasma membrane, thereby bringing it into vicinity of its substrates and of Rap-GTP, which activates ARAP3 as a Rho GAP (3, 4). We previously showed that ARAP3 is a regulator of integrin-dependent neutrophil functions (5).
To understand the regulatory input of PI3K into ARAP3 in the neutrophil, we here report the analysis of neutrophils from knock-in mice in which ARAP3 carries a double point mutation in the most N-terminal PH domain (R302, 303A). This double point mutation renders ARAP3 unable to interact with, and be activated by PtdIns(3,4,5)P3 (3, 6). For ease of reading, we refer here to this PH domain point mutation knock-in as PH*. Like ARAP3-deficient neutrophils, Arap3PH*/PH* cells exhibited perturbed responses on adhesion-dependent stimulation, and showed increased β2 integrin affinity and clustering. Arap3PH*/PH* neutrophils were also hyperactive when plated onto immobilised immune complexes, but not on cross-linking FcγR in solution. In vitro chemotaxis assays showed a directionality defect of Arap3PH*/PH* neutrophils, whilst in vivo, Arap3PH*/PH* neutrophils were characterised by reduced recruitment to the peritoneum after induction of sterile peritonitis and by subtly reduced inflammation in an autoantibody-induced arthritis model.
Materials and Methods
Unless otherwise stated, materials were obtained from Sigma, Dorset, UK.
Antibodies
Mouse anti-BSA (clone BSA-33) was from Sigma (Dorset, UK); rat anti-FcγRII/III (CD16/32; clone 93) was from eBioscience (Hatfield, UK); hamster anti-FcγRIV (blocking antibody; clone 9E9) has been described (7) and was a kind gift from Falk Nimmerjahn (Erlangen University, Germany). Sheep anti-ARAP3 has been described (3) and mouse anti β-COP was a gift from Nick Ktistakis. Mouse anti RhoA, anti Rap1 and anti Arf6 were from Santa Cruz Biotechnology (Santa Cruz, California, USA). Rabbit anti phospho-Akt, anti phospho-p38 and anti phospho myosin light chain antibodies were from Cell Signaling Technology / New England Biolabs (Hitchin, UK). Goat anti-mouse HRP conjugate was obtained from BioRad (Hemel Hempstead, UK), and goat anti-rabbit and anti-sheep were from Santa Cruz Biotechnology. Alexafluor-conjugated, highly cross-absorbed secondary antibodies were from BD Invitrogen - Molecular probes (Paisley, UK). Goat anti-rat F(ab’)2 was from Jackson Immuno Research (via Stratech, Newmarket, UK); FITC-conjugated mouse anti-hamster IgG was from BD Pharmingen (San Diego, California, USA). Fluorescently-conjugated anti-GR1, anti-LFA1 and anti-Mac1 as well as all isotype controls were obtained from eBioscience.
Arap3PH* mouse model
Generation of the Arap3PH* mouse has been previously described. Experiments performed for this work were carried out with mice derived from the BB10 clone, which produces rare homozygous mutants (8). Since the frequency of Arap3PH*/PH* mice was very low, we generated cohorts of Arap3PH*/PH* and wild-type bone marrow chimeras. In vitro experiments were carried out with matched wild-type and Arap3PH*/PH* mice, and in parallel with bone marrow chimeras (using chimeras originating from different donors for duplicate experiments). The results obtained in this fashion were pooled. Since ARAP3 is known to be expressed in the murine vasculature, in vivo experiments were only carried out with bone marrow chimeras, ensuring only neutrophil-dependent defects were assessed. Animal work carried out at Babraham Institute was approved by United Kingdom Home Office Project licenses PPL08/175 and PPL80/2335. Experiments carried out at Semmelweiss University were approved by the Semmelweis University Animal Experimentation Review Board.
Reconstitutions
Cohorts of C57Bl/6 mice were lethally irradiated and subsequently reconstituted by tail vein injection with 4 × 106 bone marrow cells from Arap3PH*/PH* mice or from sex and age matched wild-type controls. Complete reconstitution of the hematopoietic system was confirmed by flow cytometry.
Neutrophil purification
Neutrophils were isolated from bone marrows of 12 to 14 week old sex- and age-matched mice using discontinuous Percoll gradients (GE Healthcare Amersham, Uppsala, Sweden) as described (9, 10), using endotoxin-free reagents throughout. Chimeric mice were used six to eight weeks after reconstitution.
Analysis of peripheral blood
Tail vein blood was collected in EDTA-coated microvettes (Sarstedt, Nümbrecht, Germany) for analysis using a Vetabc animal blood cell counter.
Immobilisation of adhesive proteins and immune complexes
Sheep fibrinogen (150μg/ml) or polyRGD (20μg/ml) were adsorbed onto tissue culture grade plastics overnight at 4°C (fibrinogen) or for 3 hours at room temperature (polyRGD). Surfaces were washed prior to performing any assays. For immune complexes, dishes were coated overnight at 4°C with essentially endotoxin and fatty acid free BSA in PBS (100μg/ml), blocked with 1% fat free milk in PBS for 1 hour, and incubated with mouse anti-BSA (1:10000) for 1 hour at room temperature.
FcγR cross-linking
FcγR cross-linking was performed as described elsewhere (11) using rat anti-FcγRII/III and goat anti-rat F(ab’)2.
ROS production assays
ROS production was measured by chemiluminescence employing a luminol-based assay in polystyrene 96-well plates (Berthold Technologies, Bad Wildbad, Germany) as described (12). 5×105 cells were incubated with luminol (150μM) and HRP (18.75 U/ml) for 10 minutes at 37°C. Where indicated, neutrophils were added manually to wells pre-coated with fibrinogen or polyRGD with or without murine TNFα (20ng/ml, R&D systems) or to immobilised immune complexes. For soluble agonist assays with fMLF (10μM final concentration), cells were primed for 1hr at 37°C in the absence (mock) or presence of mouse TNFα (1000U/ml) and GM-CSF (100ng/ml). For all assays, measurements were started immediately and light emission was recorded. Data output is relative light units per second (RLU/sec) or total RLU integrated over indicated measured periods of time.
Chemotaxis assays
For micropipette assays, neutrophils were resuspended in HBSS, 15mM Hepes pH 7.4, 0.05% BSA, and allowed to attach to a glass coverslip. Cells were stimulated at 37°C with a point source of fMLF delivered from a microinjection pipette (femtotip, Eppendorf, Cambridge, UK) using 50 mbar pressure as described (5) and monitored by time-lapse imaging for 30 minutes using an inverted Zeiss axiovert microscope (Welwynn Garden City, UK) and Axiovision software. Dunn chamber chemotaxis assays were carried out as described (10, 13), using fMLF or MIP2 as chemoattractant. Cells were tracked using the ‘manual tracking’ and tracks were analysed using the ‘Chemotaxis Tool’ plug-ins (Ibidi, Planegg, Germany) into Image J.
MAPK, Erk and PKB activation assays
These assays were carried out with cells plated onto polyRGD, BSA-antiBSA immune complex coated, or hiFCS-blocked dishes essentially as described (14) or cells were lysed at indicated timepoints after FcγRII/III cross-linking as described (11).
Degranulation assay
Release of gelatinase granules after plating onto a fibrinogen- or immune complex coated surface, or as induced by stimulation with fMLF and cytochalasin B was detected by zymography as described (15).
Analysis of β2 integrins
Surface integrins were visualized by FACS analysis of stained cell populations. Bone marrow cells were co-stained with anti-GR1 FITC and anti-integrin Cy5. GR1high cells were gated and analysed for level of integrin expression. Integrin clustering was analysed by confocal microscopy of unstimulated, stained cells that were kept in solution at the time of assaying and that were allowed to settle onto electrostatic slides (superfrost +; VWR, Lutterworth, UK) after fixation. To measure integrin binding affinity, unstimulated cells were allowed to bind to ICAM1-Fc chimera (R&D systems, Abingdon, UK) in solution; after fixation, bound ICAM1-Fc was stained using a highly cross-absorbed alexafluor-conjugated anti-human Fc antibody, and cells were allowed to settle onto electrostatic slides. Bound ICAM1 was measured using quantitative immunofluorescence as described (5).
K/BxN serum transfer arthritis model
The K/BxN serum transfer model was performed as described (16) using lethally irradiated recipients reconstituted with Arap3PH* or matched control bone marrows. Mice were injected with 150μl of arthritogenic or control serum and ankle thickness and clinical scores were measured for the following 14 days. Sample preparation for histology was as described (17).
Small GTPase activity assays
RhoA-GTP was determined by G-LISA assay (Cytoskeleton Inc, Denver, Colorado, USA); Arf6-GTP and Rap1-GTP were identified by pull-down assay as described (5).
Results
ARAP3 is a PI3K and Rap regulated dual Rho and Arf GAP that was isolated in a screen for PtdIns(3,4,5)P3 binding proteins from neutrophils. PtdIns(3,4,5)P3 binds ARAP3 in a pleckstrin homology (PH) domain dependent fashion, and regulates ARAP3 catalytically as well as causing its plasma membrane recruitment. ARAP3 can be uncoupled from activation by PI3K by insertion of a double point mutation in its most N-terminal PH domain (3, 6). We previously showed that introducing this PH domain mutation (R302, 303A; here termed PH*) into ARAP3 led to embryonic lethality (8). A small number of homozygous mutant pups from Arap3+/PH* intercrosses from mice derived from one of two independently targeted ES cell clones were viable and fertile [(8); Table 1]. Arap3PH*/PH* knock-ins which survived to adulthood were healthy and fertile. We analysed their neutrophils and those isolated from lethally irradiated recipient mice that had been reconstituted with bone marrow cells from Arap3PH*/PH* or matched wild-type control mice, to elucidate the regulatory input of PI3K into ARAP3 in the neutrophil.
Table I.
Litters from Arap3R302,3A intercrosses
|
Total | Arap3 +/+ | Arap3+/R302,3A | Arap3R302,3A/R302,3A |
|---|---|---|---|---|
|
Arap3+/R302,3A x
Arap3R302,3A/R302,3A |
90 | - | 80 | 10 |
|
Arap3+/R302,3A x
Arap3+/R302,3A |
264 | 85 | 174 | 5 |
Expression of ARAP3 protein was not affected by the presence of the PH domain point mutation in heterozygous or homozygous mutants (Fig. 1A). We detected no differences in expression of the ARAP family members ARAP1 and ARAP2 (not shown). Peripheral blood cell counts from chimeric mice reconstituted with control and Arap3PH*/PH* bone marrow cells were very similar (Fig. 1B).
Figure 1. Incorporating a PH domain point mutation does not affect ARAP3 expression.
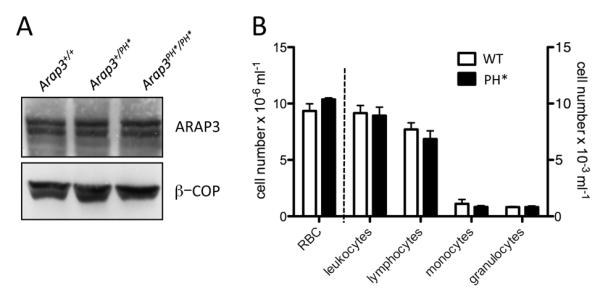
(A) Protein from 1×106 bone marrow derived neutrophils from control (Arap3+/+), heterozygous (Arap3+/PH*) or homozygous mutant (Arap3PH*/PH*) mice was immunoblotted for ARAP3 (top panel) or β-COP (bottom panel) as a loading control. Representative experiment shown from four independent experiments. (B) Peripheral blood cells from bone marrow chimeras reconstituted with Arap3PH*/PH* (PH*) or matched control (WT) hematopoietic stem cells were analysed using a Vet ABC animal blood cell counter. Data shown (means ±SEM) were obtained with 14 wild-type and 12 knock-in chimeras generated with three individual bone marrow donors per genotype.
Adhesion-dependent events are upregulated in Arap3PH*/PH* neutrophils
Neutrophils produce ROS in a well-characterised manner in response to a variety of stimuli. This allows the analysis of the machinery required for ROS production, and also of signalling pathways required for ROS production after a particular stimulus. ROS production of purified wild-type control and Arap3PH*/PH* neutrophils on stimulation with the soluble agonist fMLF (Fig. 2A,D), or with the non-physiological soluble stimulus PMA (not shown), was very similar, indicating ARAP3 is not required downstream of PI3K for ROS generation per se. However, when we assayed ROS production after plating neutrophils onto the physiological Mac1 ligand fibrinogen or onto the synthetic multivalent integrin ligand polyRGD, ROS production was significantly increased in Arap3PH*/PH* compared to the control neutrophils (Fig 2. B,C,E,F). For stimulation by fibrinogen neutrophils require a co-stimulatory event, for example by a pro-inflammatory cytokine like TNFα (18). Interestingly, plating Arap3PH*/PH* onto fibrinogen caused significantly increased ROS production even in the absence of any co-stimulation (Fig. 2B,E), suggesting that the knock-in cells were present in a pre-activated state.
Figure 2. Integrin-dependent events are upregulated in Arap3PH*/PH* neutrophils.

(A-F) Bone marrow-derived Arap3PH*/PH* (PH*) and matched wild-type control (WT) neutrophils were prepared, primed with TNFα and GM-CSF or mock primed (A,D) and (all) pre-incubated with luminol as described in materials and methods. 5×105 cells were plated into 96 well plates containing fMLF as a soluble stimulus (A,D) or that had been coated with fibrinogen (B,E) or polyRGD (C,F). Light emission was measured in a Berthold Microluminat Plus luminometer. Data were recorded in duplicate. Data (means ± range) from a representative experiment are shown in panels A-C whilst panels D-F represent accumulated light emissions (means ± SEM) from four separate, pooled experiments expressed as a percentage of the responses obtained with control neutrophils. (G-I) Neutrophils were allowed to adhere to heat inactivated serum-blocked (hiFCS) or polyRGD-coated plastic. Lysates were prepared and immunoblotted with antibodies specific for phospho-PKB (Ser 473) or phospho-p38 (T180, Y182) or β-COP as a loading control. A representative example is shown (G). Blots were quantitated using ImageJ software; integrated data obtained from five independent experiments are shown (H,I). (J,K) Gelatinase granule release was measured by zymography of supernatants of neutrophils that were allowed to adhere to hiFCS-blocked or fibrinogen-coated dishes. As a control, neutrophils were stimulated with fMLF in the presence of cytochalasin B (CB). A representative experiment is shown (J; the samples were run on two separate gels and are pasted next to one another for ease of viewing here, this is indicated by a dotted line). Integrated, quantitated data obtained from four independent experiments are plotted (K). (L-N) Neutrophils were allowed to adhere to pRGD-coated tissue culture dishes, washed and fixed. Numbers of attached cells (phase dark) per field of view, and the percentage of spread cells (phase light) were counted. Integrated data obtained from three separate experiments (L,M) and representative examples (N) are shown. (All) Raw data were analysed by T-test (Mann Whitney); * p<0.05; ** p<0.01; ***p<0.001.
We investigated whether signalling events known to lie downstream of β2 integrin ligation (outside-in signalling) were affected by uncoupling ARAP3 from PI3K. We assessed adhesion-dependent activation of PKB (also known as Akt) and p38 MAPK using phospho-specific antibodies and observed significantly enhanced PKB and p38 phosphorylation in Arap3PH*/PH* cells that had been plated onto polyRGD (Fig. 2G-I).
We next analysed the effect of uncoupling ARAP3 from PI3K on neutrophil degranulation by assaying gelatinase activity in supernatants of neutrophils that had or had not been stimulated. Like ROS production, this can be triggered by soluble stimuli, or by ligation of integrins. In-line with the results obtained with the ROS assays, Arap3PH*/PH* and control neutrophils released very similar amounts of gelatinase granules on stimulation with a soluble agonist (fMLF in the presence of cytochalasin B), whilst granule release in the PH domain point mutants was enhanced on being plated onto the integrin ligand fibrinogen in the presence of TNFα (Fig. 2J,K).
We finally compared the ability of control and Arap3PH*/PH* neutrophils to adhere and spread. Arap3PH*/PH* neutrophils plated onto polyRGD adhered and spread significantly more than controls (Fig. 2L-N; we observed the same pattern with fibrinogen in the presence of TNFα, not shown).
In summary, our results indicated that uncoupling ARAP3 from PI3K perturbed neutrophil responses on β2 integrin ligation, giving results typical for increased outside-in signalling.
β2 integrin inside-out signalling is increased in Arap3PH*/PH* neutrophils
To address why integrin ligation caused these enhanced responses with Arap3PH*/PH* neutrophils, we compared the major neutrophil β2 integrins, LFA1 and Mac1, of knock-in and control neutrophils. Surface expression of Mac1 and LFA1 was not affected by the presence of the PH domain point mutation (Fig. 3A).
Figure 3. β2 integrins on Arap3PH*/PH*neutrophils have higher affinity and avidity.

(A) Wild-type control and Arap3PH*/PH* bone marrow cells were or were not pre-incubated with 20ng/ml TNFα at 37°C before being labelled with PE-conjugated anti GR1, APC-conjugated anti Mac1 and FITC-conjugated anti LFA1. For FACS analysis, GR1-positive cells were gated and Mac1 and LFA1 staining was measured. Results were analysed with FlowJo V6.4.7 software. Cells from 15 knock-in and 11 control chimeras obtained from three separate reconstitutions were analysed in three experiments.
Representative traces are shown. black lines, wild-type; grey lines, Arap3PH*/PH* ; broken lines, no TNFα, full lines, with TNFα. (B) Unstimulated, bone marrow derived control (WT) and Arap3PH*/PH* (PH*) neutrophils were allowed to bind to ICAM1-Fc in solution in the absence of any stimulation. Cells were washed, fixed and bound ICAM1 was stained with a fluorescently conjugated secondary antibody. Washed, stained cells were allowed to settle on electrostatically coated slides and signal strength was measured by quantitative immunofluorescence. Mean fluorescence intensity obtained from 64 knock-in and 50 control cells in three independent experiments is plotted (mean ± SEM) and representative examples are shown. (C) Integrin clustering was analysed in unstimulated control and Arap3PH*/PH* neutrophils in solution. Bone marrow derived neutrophils were prepared and incubated at 37C in the presence of anti LFA1 or anti Mac1, washed, fixed, stained with a fluorescently conjugated secondary antibody and allowed to settle on electrostatically labelled slides. Distribution of fluorescence was assessed by confocal microscopy. Representative examples (stained for LFA1) are shown together with their corresponding heatmaps (obtained by analysis with ImageJ). Pooled results stem from four independent experiments, in each of which 25 cells per genotype were analysed are plotted (mean ± SEM). (B,C). Results obtained were analyed by paired T-tests. * p<0.05; ** p<0.01.
Integrins are regulated by inside-out signalling (affinity and avidity modulation) (19). Since we are not aware of any activation state-specific antibodies for murine β2 integrins, we analysed ligand binding affinity experimentally. We tested the ability of neutrophils to bind to ICAM1, a ligand for LFA1, in solution in the absence of any stimulation, using quantitative immunofluorescence as a read-out. Arap3PH*/PH* cells were able to bind significantly stronger to ICAM1 than wild-type controls (Fig. 3B), indicating that LFA1 on Arap3PH*/PH* neutrophils was present in a higher affinity status than those on wild-type cells.
We also microscopically analysed integrin lateral mobility (clustering), a read-out for integrin avidity, in unstimulated cells that were kept in suspension. In confocal images of unstimulated control cells, distribution of LFA1 and Mac1 resembled that of uniform beads on a string (as did the neutrophil surface marker GR1, not shown). In contrast, Arap3PH*/PH* neutrophils had a distinctively more clustered (patchy) LFA1 and Mac1 distribution (Fig. 3C). This suggested that the avidity of β2 integrins in Arap3PH*/PH* neutrophils was increased compared to controls. In summary, whilst the surface expression of Mac1 and LFA1 was unaltered by the PH domain point mutation in ARAP3, these integrins were characterised by increased inside-out signalling in Arap3PH*/PH* neutrophils.
Immobilised immune complex-dependent events are upregulated in Arap3PH*/PH* neutrophils
Immune complex binding induced cross-linking of FcγRs activates neutrophils, a function important for autoimmune responses. We assayed ROS production after plating neutrophils onto immobilised immune complexes, and found this to be significantly increased in Arap3PH*/PH* cells (Fig. 4A,B). In-line with this observation, we also observed significantly enhanced PKB and p38 phosphorylation, gelatinase granule release and cell spreading with Arap3PH*/PH* neutrophils that had been plated onto immobilised immune complexes (Fig. 4C-J). Therefore, responses induced by immobilised immune complexes were perturbed in Arap3PH*/PH* neutrophils.
Figure 4. Immune complex-induced events are upregulated in Arap3PH*/PH* neutrophils.

(A,B) Bone marrow-derived Arap3PH*/PH* (PH*) and matched wild-type control (WT) neutrophils were prepared and pre-incubated with luminol as described in materials and methods. 5×105 cells were plated into 96 well plates that had been coated with an immune-complex (BSA anti-BSA). Light emission was measured in a Berthold Microluminat Plus luminometer. Data were recorded in duplicate. Data (means ± range) from a representative experiment are shown in panel A whilst panel B represents accumulated light emissions (means ± SEM) from four independent experiments expressed as a percentage of the responses obtained with wild-type control neutrophils. (C-E) Neutrophils were allowed to adhere to heat inactivated BSA-blocked (BSA) or immune complex-coated (BSA-αBSA) plastic. Lysates were prepared and subjected to immunoblotting with antibodies specific for phospho-PKB (Ser 473) or phospho-p38 (T180, Y182) or β-COP as a loading control. A representative example is shown (C). Blots were quantitated using ImageJ software; integrated data obtained from five independent experiments are shown (D,E). (F,G) Gelatinase granule release was measured by zymography of supernatants of neutrophils that were allowed to adhere to BSA-blocked or immune complex-coated dishes. A representative experiment is shown (F; the samples were not in this order on the original gel and have been pasted next to one another for ease of viewing, this is indicated by a dotted line). Integrated, quantified data obtained from four independent experiments are plotted (G). (H-J) Neutrophils were allowed to adhere to immune complex-coated tissue culture dishes, washed and fixed. Numbers of attached cells (phase dark) per field of view, and the percentage of spread cells (phase light) were counted. Integrated data obtained from three separate experiments are plotted (I,J) and representative examples are shown (H). (All) Raw data were analysed by T-test (Mann Whitney). * p<0.05; ** p<0.01; ***p<0.001.
FcγR signalling is not directly affected in Arap3R302,3A/R302,3A neutrophils
To address why Arap3PH*/PH* neutrophils displayed these perturbed responses on being plated onto immobilised immune complexes, we analysed FcγR signalling further. We did not detect significantly different surface expression of FcγRII/III or of FcγRIV between control and Arap3PH*/PH* neutrophils (Fig. 5A). Using specific blocking antibodies in ROS assays with neutrophils plated onto immobilised immune complexes we found blocking FcγRII/III caused a significant reduction in both wild-type and Arap3PH*/PH* neutrophils, whilst blocking FcγRIV alone had very little effect. Using both blocking antibodies together virtually abolished the ROS production in both control and Arap3PH*/PH* neutrophils (Fig. 5B), in agreement with published work (15), and confirmed that immune complex induced ROS production was indeed FcγR dependent.
Figure 5. FcγR are not affected in Arap3PH*/PH* neutrophils.

(A) Surface distribution of FcγRII/III and FcγRIV was analysed by FACS analysis of bone marrow cell populations. Seven wild-type and seven knock-in chimeras obtained from three different pairs of donors were analysed in two independent experiments. A representative example is shown. Black lines represent control cells and grey lines Arap3PH*/PH*. FcγRII/III FACS: broken lines represent unstimulated and full lines TNFα-stimulated samples; FcγRIV FACS: dotted lines represent isotype control. (B) Immune complex induced ROS formation depends on signalling through FcγRs. Neutrophils were or were not pre-incubating with FcγR blocking antibodies and/or isotype controls as indicated, before being plated onto immobilised immune complexes for ROS assays. Pooled results from three independent experiments are plotted (mean ± SEM). Differences between wild-type and Arap3PH*/PH* were highly significant (p<0.001) for all conditions except anti-FcγRII/III + anti-FcγRIV (p>0.05), two-way ANOVA with Bonferroni post-test. (C) Cross-linking FcγRII/III does not cause increased ROS production in Arap3PH*/PH* neutrophils. Bone marrow derived neutrophils were prepared from control and knock-in mice and incubated with anti FcγRII/III with or without priming. After washing, ROS production was measured on cross-linking the FcγR with a F(ab’)2 fragment. A representative example is shown (left) and integrated results from five independent experiments are plotted (mean ± SEM). Differences did not reach statistical significance (p>0.1; Mann Whitney T-test). (D) Control and Arap3PH*/PH* neutrophils were incubated with FcγRII/III antibody, washed, and stimulated by F(ab’)2 fragment-mediated cross-linking for the indicated amounts of time. Time 0, no cross-linking. Lysates were prepared and protein subjected to immunoblotting. PKB and p38 phosphorylation was analysed using phospho-specific antibodies; β-COP served as loading control. A representative experiment from four independent experiments is shown.
To test whether ARAP3 affected FcγR signalling directly, we stimulated cells in solution by antibody-mediated FcγR cross-linking and measured ROS production. We observed very similar ROS production by primed or unprimed Arap3PH*/PH* neutrophils compared to controls on cross-linking FcγRII/III (Fig. 5C; we were unable to obtain meaningful results on cross-linking FcγRIV in a similar manner). We also measured PKB and p38 phosphorylation on FcγRII/III cross-linking in solution. Again we observed very similar activations in control and Arap3PH*/PH* neutrophils (Fig 5D). This work suggested, that the presence of the PH domain mutation in ARAP3 did not affect signalling through FcγR per se, but rather, that we had observed an indirect effect when assaying Arap3PH*/PH* neutrophils that had been plated onto immobilised immune complexes.
Adhesion-dependent RhoA activation is increased in Arap3PH*/PH* neutrophils
ARAP3 is a functional GAP protein for RhoA and Arf6 that is activated by PI3K and Rap. To analyse the mechanism underlying our observations, we carried out activity assays. Since Rap is known to regulate integrins, we assayed Rap1-GTP in Arap3PH*/PH* neutrophils that were kept in suspension or plated onto polyRGD. Rap1-GTP was significantly increased on plating neutrophils of either genotype onto polyRGD, but we detected no significant differences between genotypes in suspension cells or those plated onto polyRGD (Fig. 6A). We next analysed Arf6-GTP in neutrophils that were kept in suspension or plated onto polyRGD. Arf6 was strongly activated by plating control and Arap3PH*/PH* neutrophils onto polyRGD. No difference was observed between the two genotypes (Fig. 6B). RhoA was also significantly activated on plating control and Arap3PH*/PH* neutrophils onto polyRGD. Our measurements indicated that RhoA fold activation was significantly higher in Arap3PH*/PH* than in control neutrophils (Fig. 6C), suggesting that the incorporation of the PH* mutation into ARAP3 affected RhoA signalling in neutrophils. Since RhoA and Rac are known to cross-talk, we also analysed Rac. In agreement with previously published data from human neutrophils (20), neither Rac1 nor Rac2 were activated in neutrophils by plating onto polyRGD. We did not observe any differences in Rac1 and Rac2 activities between wild-type control and Arap3PH*/PH* neutrophils (not shown). To summarise, RhoA signalling was affected in Arap3PH*/PH* neutrophils plated onto integrin ligands, which is suggestive of an involvement of RhoA signalling in the adhesion-dependent Arap3PH*/PH* phenotype we observed.
Figure 6. RhoA activation is affected in Arap3PH*/PH* neutrophils.

(A-C) Wild-type control (WT) and Arap3PH*/PH* (PH*) neutrophils were kept in suspension or plated onto polyRGD-coated tissue culture plastic. Cells were lysed with ice-cold lysis buffer. (A, B) Clarified lysates were subjected to ‘pull-down’ assays using GST-Ral GDS as bait to determine GTP-loaded fractions of Rap (A) and using GST-MT2 bait to determine GTP-loaded Arf6 (B). Results obtained from a minimum of five independent experiments were pooled and plotted (mean ± SEM, left); representative examples are shown (right). L, lysates, PD, pull-downs. Blots were probed with an antibody specific for Rap1 (A) and Arf6 (B). (C) Clarified lysates were employed in RhoA G-LISA assays to determine GTP-loaded RhoA. Results from five pooled, independent experiments are presented (mean ± SEM). (A-C) Raw data were analysed by paired T-tests. * p<0.05; ** p<0.001. (D) Indirect assessment of localisation of RhoA activation. Neutrophils were or were not stimulated for the indicated time with 1μM fMLF in solution, allowed to settle on a glass coverslip for 3 minutes, fixed and stained using phalloidin, to visualise filamentous actin and anti phospho myosin light chain. Representative cells from three independent experiments are shown.
We monitored phospho myosin light chain (pMLC), a RhoA effector, as an indirect read-out for the localisation of RhoA activation in the neutrophils. In wild-type control cells F-actin and pMLC staining were mutually exclusive. In contrast, pMLC localization was perturbed in Arap3PH*/PH* neutrophils that had or had not been stimulated with fMLF (Fig 6D and not shown), suggestive of mislocalized RhoA-GTP in these cells.
Arap3PH*/PH* neutrophils have a chemotaxis defect in vitro
To test whether uncoupling ARAP3 from PI3K affects the ability of neutrophils to chemotax, we performed Dunn chamber chemotaxis assays. We assayed cells migrating towards fMLF, a commonly used chemoattractant in this context, which has been shown to support PI3K and p38 MAPK mediated chemotaxis. We also analyzed cells migrating towards MIP2, the murine equivalent of human IL-8, that has been shown to promote chemotaxis independently of p38, in a PI3K-dependent fashion (21, 22). Tracking of cell trajectories (Fig. 7A) followed by computational analysis of the tracks showed that Arap3PH*/PH* neutrophils travelled longer total, but shorter euclidian distances exhibiting reduced directionality compared to controls (Fig. 7B,C) when migrating towards fMLF. When the cells moved towards MIP2 (Fig 7D-F), the defect observed was more severe, with Arap3PH*/PH* neutrophils migrating shorter accumulated and euclidian distances than their control counterparts whilst the directionality defect remained. These measurements confirmed that uncoupling ARAP3 from PI3K caused an in vitro neutrophil chemotaxis defect, the nature of which is dependent on the chemoattractant used and the signalling intermediates it relies on.
Figure 7. Arap3PH*/PH* neutrophils have a chemotaxis defect.

(A-F) In vitro chemotaxis assays. Bone marrow derived Arap3PH*/PH* and control neutrophils were allowed to chemotax towards 300nM fMLF (A-C) or towards 10nM MIP2 (D-F) in Dunn chambers. Movements were recorded by time-lapse imaging. (A,D) Pooled tracks of individual cells from experiments carried out with three separate cell preparations were plotted using the Ibidi chemotaxis tool plug-in into ImageJ. The source of chemoattractant is at the below. The tracks were analysed using the Ibidi chemotaxis tool’s statistics features. Accumulated and Euclidian distances and directionality are plotted (mean ± SEM; B,C; E,F). (G) Neutrophil recruitment to the peritoneum in a model for sterile peritonitis. Bone marrow chimeras (generated with four bone marrow donors per genotype) were intraperitoneally injected with 0.25ml thioglycollate containing broth. Mice were sacrificed 4.5 hours after injection, their peritonea were flushed and Mac1high GR1-positive neutrophils were counted. Pooled results obtained from two separate experiments are plotted. (B-G) Data were analysed by T-tests (Mann Whitney). * p<0.05; *** p<0.001. (H-I) Serum transfer arthritis. Twelve wild-type and 13 Arap3PH*/PH* bone marrow chimeras were injected with 150μl arthritic serum and six wild-type and six Arap3PH*/PH* bone marrow chimeras were injected with 150μl control serum in two separate experiments. Joints were scored daily for two weeks. Ankle thickness and clinical score are plotted. Circles, and dotted lines, wild-type bone marrow chimeras; triangles and full lines, Arap3PH*/PH* bone marrow chimeras. Blue symbols, control serum; red symbols, arthritogenic serum. The area under the graph was compared by T-test (Mann Whitney); ankle thickness, p=0.053; clinical score, p=0.023. (I) Wax sections of decalcified joints from chimeras reconstituted with control (WT) or Arap3PH*/PH* (PH*) bone marrows induced as indicated were H&E stained to visualise leukocyte infiltration on day 4 after serum injection. Representative examples from sections obtained with 6 arthritogenic and 2 control serum-injected mice mice in two independent experiments are shown.
Arap3PH*/PH* cells have a recruitment defect in vivo
To analyse in vivo chemotaxis, we assessed neutrophil recruitment to the peritoneum of control and Arap3PH*/PH* bone marrow chimeras in which sterile peritonitis had been induced by thioglycollate. In this assay, significantly reduced numbers of Arap3PH*/PH* neutrophils were recruited to the peritoneum (Fig. 7G). In the lights of the elevated responses of Arap3PH*/PH* neutrophils we had observed on plating neutrophils onto immobilised immune complexes in vitro, we next tested the effect of uncoupling ARAP3 from PI3K in the context of a model of an autoimmune disease. For this we made use of the well-characterised K/BxN serum transfer model for the effector phase of rheumatoid arthritis (23, 24). In this model, arthritogenic K/BxN serum is transferred to healthy mice, resulting in neutrophil-dependent inflammation of their joints. Sera were injected into lethally irradiated recipient mice reconstituted with Arap3PH*/PH* or matched control bone marrows, and ankle thickness and clinical score were measured over 14 days (Fig. 7H). Unexpectedly, arthritogenic serum-injected Arap3PH*/PH* bone marrow chimeras were less affected than control chimeras. Hence, Arap3PH*/PH* neutrophils conferred mild protection from immunoarthritis. The inflammatory response in the K/BxN model depends on efficient neutrophil recruitment to the joints. We analysed this in ankle sections from chimeras that had been injected with control or arthritogenic serum, and observed reduced tissue infiltration in joints from Arap3PH*/PH* chimeras (Fig 7I). This suggests, the protection from arthritis in these animals was due to a leukocyte recruitment defect conferred by uncoupling ARAP3 from activation by PI3K.
Discussion
We recently showed that ARAP3 is an important regulator of neutrophils, involved in the modulation of integrin inside-out signalling, and regulating responses downstream of integrin ligation. ARAP3 is regulated by the PI3K lipid product PtdIns(3,4,5)P3 and by Rap-GTP. To analyse the regulatory contribution of PI3K on ARAP3 in the neutrophil, we analysed neutrophils in which ARAP3 harboured a PH domain point mutation that uncoupled it from activation by PI3K. The present work argues that ARAP3 is an important PI3K effector in neutrophils.
We demonstrated before that ARAP3 is regulated by PI3K catalytically, and it is also recruited to the plasma membrane in a PI3K and PH domain-dependent fashion. Therefore, the regulatory role of PI3K upstream of ARAP3 could contain direct and/or indirect components. PI3K has been shown to regulate integrins in several cell types (25-27) and our present work argues that it does this in the neutrophil at least in part by signalling through ARAP3. However, lack of PtdIns(3,4,5)P3 mediated recruitment of the ARAP3 PH domain point mutant to the plasma membrane could also have impinged on ARAP3’s ability to be regulated by Rap, an established regulator of integrin inside-out signalling (28-30). We are currently devising an ARAP3 Ras binding domain point mutation knock-in to address this possibility.
Interestingly, we noted that PKB phosphorylation, a commonly used read-out of PI3K activity, was increased in Arap3PH*/PH* neutrophils that had been plated onto polyRGD or immobilised immune complexes (Figs 2 and 4). In contrast, Rap-GTP was not affected by genotypes (Fig 6), suggesting that Rap is not involved in a feed-back loop involving ARAP3 and integrin signalling, whilst PI3K might be.
We carried out activity assays to elucidate the mechanism downstream of ARAP3 in this complex phenotype. Plating neutrophils onto polyRGD caused activation of RhoA and Arf6, but only RhoA fold-activation was increased in the knock-in cells, suggesting a regulatory role for RhoA downstream of ARAP3. Indeed, RhoA has been shown in a variety of cell types to regulate integrins (26, 31-34). Although RhoA and Rac have been shown to have an antagonistic relationship in many experimental systems (35), cross-talk between Rho and Rac does not appear to play a major role in this context. We noticed that the distribution of pMLC was disturbed in Arap3PH*/PH* neutrophils, suggesting indirectly that their RhoA-GTP localization may be affected. This will be investigated further in future work. Potentially, perturbed distribution of RhoA-GTP could have contributed to the chemotaxis defect we observed.
In our in vitro assays, immune complex induced responses were found to be increased in Arap3PH*/PH* neutrophils in addition to integrin-dependent ones. Since activating cells by cross-linking their FcγRII/III did not cause similarly increased responses, uncoupling ARAP3 from activation through PI3K does not directly affect signalling through FcγRII/III. We cannot exclude a potential direct effect on the activating FcγRIV (7), which acts together with FcγRIII to regulate immobilised immune complex-dependent activation of neutrophils (15), since we were unable to test this receptor experimentally in similar cross-linking experiments. An alternative explanation for our data could be, that cross-talk between β2 integrins and FcγRs takes place when FcγR-dependent signalling is induced by immobilised immune complexes. The pre-activated β2 integrins of Arap3PH*/PH* neutrophils might be able to modulate FcγR-dependent signalling. Cross-talk between integrin and FcγR signalling has previously been described both in vitro (36-38) and with in vivo models (39-41). Whilst the molecular mechanisms of such cross-talk has yet to be elucidated, it has been clearly demonstrated that both classes of receptors share common downstream signalling adaptors and further components of their signalling cascades (42).
We observed a chemotaxis defect with Arap3PH*/PH* neutrophils in vitro, which was more pronounced when MIP2 rather than fMLF was used as a chemoattractant, in-line with the distinct dependency on PI3K by these two chemoattractants (21, 22). We also noted evidence for recruitment defects to the peritoneum and joints, respectively, on inducing sterile inflammation in these locations. Neutrophil recruitment to the periphery is known to depend on β2 integrins (43), and our data suggests that PI3K-dependent, ARAP3-mediated fine tuning of β2 integrin inside-out signalling is required for such recruitment to occur in an efficient manner. Interestingly, our result are in-line with findings from several other laboratories who reported that constitutively activating β2 integrins (Mac1 or LFA1) either genetically by introducing activating mutations or pharmacologically by employing specific small molecule agonists results in reduced leukocyte extravasation, reduced neutrophil recruitment in sterile peritonitis and reduced inflammation in a number of animal model systems (44, 45).
Acknowledgments
We thank Simon Walker for help with image analysis and Anne Segond-Pichon for help with statistical analysis, Falk Nimmerjahn (University of Erlangen, Germany) and Nick Ktistakis for antibodies, Anthony Green (Cambridge Institute for Medical Research) for use of a blood cell counter, Su Kulkarni, Tamara Chessa, George Damoulakis and Alison Condliffe (Cambridge University Medical School) for helpful discussions.
This work was supported by the Biotechnology and Biological Sciences Research Council; SVe was supported by a Biotechnology and Biological Sciences Research Council David Phillips Fellowship (BB/C520712). SVo was supported by a Leonardo da Vinci Programme studentship. AM and ZJ were supported by the Wellcome Trust and the European Research Council.
References
- 1.Nathan C. Neutrophils and immunity: challenges and opportunities. Nat Rev Immunol. 2006;6:173–182. doi: 10.1038/nri1785. [DOI] [PubMed] [Google Scholar]
- 2.Hawkins PT, Stephens LR, Suire S, Wilson M. PI3K signaling in neutrophils. Curr Top Microbiol Immunol. 2010;346:183–202. doi: 10.1007/82_2010_40. [DOI] [PubMed] [Google Scholar]
- 3.Krugmann S, Anderson KE, Ridley SH, Risso N, McGregor A, Coadwell J, Davidson K, Eguinoa A, Ellson CD, Lipp P, Manifava M, Ktistakis N, Painter G, Thuring JW, Cooper MA, Lim ZY, Holmes AB, Dove SK, Michell RH, Grewal A, Nazarian A, Erdjument-Bromage H, Tempst P, Stephens LR, Hawkins PT. Identification of ARAP3, a novel PI3K effector regulating both Arf and Rho GTPases, by selective capture on phosphoinositide affinity matrices. Mol Cell. 2002;9:95–108. doi: 10.1016/s1097-2765(02)00434-3. [DOI] [PubMed] [Google Scholar]
- 4.Krugmann S, Williams R, Stephens L, Hawkins PT. ARAP3 is a PI3K- and rap-regulated GAP for RhoA. Curr Biol. 2004;14:1380–1384. doi: 10.1016/j.cub.2004.07.058. [DOI] [PubMed] [Google Scholar]
- 5.Gambardella L, Anderson KE, Nussbaum C, Segonds-Pichon A, Margarido T, Norton L, Ludwig T, Sperandio M, Hawkins PT, Stephens L, Vermeren S. The GTPase-activating protein ARAP3 regulates chemotaxis and adhesion-dependent processes in neutrophils. Blood. 2011;118:1087–1098. doi: 10.1182/blood-2010-10-312959. [DOI] [PubMed] [Google Scholar]
- 6.Craig HE, Coadwell J, Guillou H, Vermeren S. ARAP3 binding to phosphatidylinositol-(3,4,5)-trisphosphate depends on N-terminal tandem PH domains and adjacent sequences. Cell Signal. 2010;22:257–264. doi: 10.1016/j.cellsig.2009.09.025. [DOI] [PubMed] [Google Scholar]
- 7.Nimmerjahn F, Bruhns P, Horiuchi K, Ravetch JV. FcgammaRIV: a novel FcR with distinct IgG subclass specificity. Immunity. 2005;23:41–51. doi: 10.1016/j.immuni.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 8.Gambardella L, Hemberger M, Hughes B, Zudaire E, Andrews S, Vermeren S. PI3K signaling through the dual GTPase-activating protein ARAP3 is essential for developmental angiogenesis. Sci Signal. 2010;3:ra76. doi: 10.1126/scisignal.2001026. [DOI] [PubMed] [Google Scholar]
- 9.Condliffe AM, Davidson K, Anderson KE, Ellson CD, Crabbe T, Okkenhaug K, Vanhaesebroeck B, Turner M, Webb L, Wymann MP, Hirsch E, Ruckle T, Camps M, Rommel C, Jackson SP, Chilvers ER, Stephens LR, Hawkins PT. Sequential activation of class IB and class IA PI3K is important for the primed respiratory burst of human but not murine neutrophils. Blood. 2005;106:1432–1440. doi: 10.1182/blood-2005-03-0944. [DOI] [PubMed] [Google Scholar]
- 10.Ferguson GJ, Milne L, Kulkarni S, Sasaki T, Walker S, Andrews S, Crabbe T, Finan P, Jones G, Jackson S, Camps M, Rommel C, Wymann M, Hirsch E, Hawkins P, Stephens L. PI(3)Kgamma has an important context-dependent role in neutrophil chemokinesis. Nat Cell Biol. 2007;9:86–91. doi: 10.1038/ncb1517. [DOI] [PubMed] [Google Scholar]
- 11.Utomo A, Cullere X, Glogauer M, Swat W, Mayadas TN. Vav proteins in neutrophils are required for FcgammaR-mediated signaling to Rac GTPases and nicotinamide adenine dinucleotide phosphate oxidase component p40(phox) J Immunol. 2006;177:6388–6397. doi: 10.4049/jimmunol.177.9.6388. [DOI] [PubMed] [Google Scholar]
- 12.Anderson KE, Boyle KB, Davidson K, Chessa TA, Kulkarni S, Jarvis GE, Sindrilaru A, Scharffetter-Kochanek K, Rausch O, Stephens LR, Hawkins PT. CD18-dependent activation of the neutrophil NADPH oxidase during phagocytosis of Escherichia coli or Staphylococcus aureus is regulated by class III but not class I or II PI3Ks. Blood. 2008;112:5202–5211. doi: 10.1182/blood-2008-04-149450. [DOI] [PubMed] [Google Scholar]
- 13.Welch HC, Condliffe AM, Milne LJ, Ferguson GJ, Hill K, Webb LM, Okkenhaug K, Coadwell WJ, Andrews SR, Thelen M, Jones GE, Hawkins PT, Stephens LR. P-Rex1 regulates neutrophil function. Curr Biol. 2005;15:1867–1873. doi: 10.1016/j.cub.2005.09.050. [DOI] [PubMed] [Google Scholar]
- 14.Mocsai A, Zhou M, Meng F, Tybulewicz VL, Lowell CA. Syk is required for integrin signaling in neutrophils. Immunity. 2002;16:547–558. doi: 10.1016/s1074-7613(02)00303-5. [DOI] [PubMed] [Google Scholar]
- 15.Jakus Z, Nemeth T, Verbeek JS, Mocsai A. Critical but overlapping role of FcgammaRIII and FcgammaRIV in activation of murine neutrophils by immobilized immune complexes. J Immunol. 2008;180:618–629. doi: 10.4049/jimmunol.180.1.618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jakus Z, Simon E, Frommhold D, Sperandio M, Mocsai A. Critical role of phospholipase Cgamma2 in integrin and Fc receptor-mediated neutrophil functions and the effector phase of autoimmune arthritis. J Exp Med. 2009;206:577–593. doi: 10.1084/jem.20081859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nigrovic PA, Binstadt BA, Monach PA, Johnsen A, Gurish M, Iwakura Y, Benoist C, Mathis D, Lee DM. Mast cells contribute to initiation of autoantibody-mediated arthritis via IL-1. Proc Natl Acad Sci U S A. 2007;104:2325–2330. doi: 10.1073/pnas.0610852103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nathan CF. Neutrophil activation on biological surfaces. Massive secretion of hydrogen peroxide in response to products of macrophages and lymphocytes. J Clin Invest. 1987;80:1550–1560. doi: 10.1172/JCI113241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Abram CL, Lowell CA. The ins and outs of leukocyte integrin signaling. Annu Rev Immunol. 2009;27:339–362. doi: 10.1146/annurev.immunol.021908.132554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dib K, Melander F, Axelsson L, Dagher MC, Aspenstrom P, Andersson T. Down-regulation of Rac activity during beta 2 integrin-mediated adhesion of human neutrophils. J Biol Chem. 2003;278:24181–24188. doi: 10.1074/jbc.M302300200. [DOI] [PubMed] [Google Scholar]
- 21.Heit B, Tavener S, Raharjo E, Kubes P. An intracellular signaling hierarchy determines direction of migration in opposing chemotactic gradients. J Cell Biol. 2002;159:91–102. doi: 10.1083/jcb.200202114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Heit B, Liu L, Colarusso P, Puri KD, Kubes P. PI3K accelerates, but is not required for, neutrophil chemotaxis to fMLP. J Cell Sci. 2008;121:205–214. doi: 10.1242/jcs.020412. [DOI] [PubMed] [Google Scholar]
- 23.Kouskoff V, Korganow AS, Duchatelle V, Degott C, Benoist C, Mathis D. Organ-specific disease provoked by systemic autoimmunity. Cell. 1996;87:811–822. doi: 10.1016/s0092-8674(00)81989-3. [DOI] [PubMed] [Google Scholar]
- 24.Korganow AS, Ji H, Mangialaio S, Duchatelle V, Pelanda R, Martin T, Degott C, Kikutani H, Rajewsky K, Pasquali JL, Benoist C, Mathis D. From systemic T cell self-reactivity to organ-specific autoimmune disease via immunoglobulins. Immunity. 1999;10:451–461. doi: 10.1016/s1074-7613(00)80045-x. [DOI] [PubMed] [Google Scholar]
- 25.Katagiri K, Hattori M, Minato N, Irie S, Takatsu K, Kinashi T. Rap1 is a potent activation signal for leukocyte function-associated antigen 1 distinct from protein kinase C and phosphatidylinositol-3-OH kinase. Mol Cell Biol. 2000;20:1956–1969. doi: 10.1128/mcb.20.6.1956-1969.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Constantin G, Majeed M, Giagulli C, Piccio L, Kim JY, Butcher EC, Laudanna C. Chemokines trigger immediate beta2 integrin affinity and mobility changes: differential regulation and roles in lymphocyte arrest under flow. Immunity. 2000;13:759–769. doi: 10.1016/s1074-7613(00)00074-1. [DOI] [PubMed] [Google Scholar]
- 27.Schoenwaelder SM, Ono A, Nesbitt WS, Lim J, Jarman K, Jackson SP. Phosphoinositide 3-kinase p110 beta regulates integrin alpha IIb beta 3 avidity and the cellular transmission of contractile forces. J Biol Chem. 2010;285:2886–2896. doi: 10.1074/jbc.M109.029132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li Y, Yan J, De P, Chang HC, Yamauchi A, Christopherson KW, 2nd, Paranavitana NC, Peng X, Kim C, Munugalavadla V, Kapur R, Chen H, Shou W, Stone JC, Kaplan MH, Dinauer MC, Durden DL, Quilliam LA. Rap1a null mice have altered myeloid cell functions suggesting distinct roles for the closely related Rap1a and 1b proteins. J Immunol. 2007;179:8322–8331. doi: 10.4049/jimmunol.179.12.8322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chrzanowska-Wodnicka M, Smyth SS, Schoenwaelder SM, Fischer TH, White GC., 2nd Rap1b is required for normal platelet function and hemostasis in mice. J Clin Invest. 2005;115:680–687. doi: 10.1172/JCI22973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sebzda E, Bracke M, Tugal T, Hogg N, Cantrell DA. Rap1A positively regulates T cells via integrin activation rather than inhibiting lymphocyte signaling. Nat Immunol. 2002;3:251–258. doi: 10.1038/ni765. [DOI] [PubMed] [Google Scholar]
- 31.Laudanna C, Campbell JJ, Butcher EC. Role of Rho in chemoattractant-activated leukocyte adhesion through integrins. Science. 1996;271:981–983. doi: 10.1126/science.271.5251.981. [DOI] [PubMed] [Google Scholar]
- 32.Giagulli C, Scarpini E, Ottoboni L, Narumiya S, Butcher EC, Constantin G, Laudanna C. RhoA and zeta PKC control distinct modalities of LFA-1 activation by chemokines: critical role of LFA-1 affinity triggering in lymphocyte in vivo homing. Immunity. 2004;20:25–35. doi: 10.1016/s1074-7613(03)00350-9. [DOI] [PubMed] [Google Scholar]
- 33.Xu K, Sacharidou A, Fu S, Chong DC, Skaug B, Chen ZJ, Davis GE, Cleaver O. Blood vessel tubulogenesis requires Rasip1 regulation of GTPase signaling. Dev Cell. 2011;20:526–539. doi: 10.1016/j.devcel.2011.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vielkind S, Gallagher-Gambarelli M, Gomez M, Hinton HJ, Cantrell DA. Integrin regulation by RhoA in thymocytes. J Immunol. 2005;175:350–357. doi: 10.4049/jimmunol.175.1.350. [DOI] [PubMed] [Google Scholar]
- 35.Guilluy C, Garcia-Mata R, Burridge K. Rho protein crosstalk: another social network? Trends Cell Biol. 2011;21:718–726. doi: 10.1016/j.tcb.2011.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhou M, Todd RF, 3rd, van de Winkel JG, Petty HR. Cocapping of the leukoadhesin molecules complement receptor type 3 and lymphocyte function-associated antigen-1 with Fc gamma receptor III on human neutrophils. Possible role of lectin-like interactions. J Immunol. 1993;150:3030–3041. [PubMed] [Google Scholar]
- 37.Zhou MJ, Brown EJ. CR3 (Mac-1, alpha M beta 2, CD11b/CD18) and Fc gamma RIII cooperate in generation of a neutrophil respiratory burst: requirement for Fc gamma RIII and tyrosine phosphorylation. J Cell Biol. 1994;125:1407–1416. doi: 10.1083/jcb.125.6.1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jakus Z, Berton G, Ligeti E, Lowell CA, Mocsai A. Responses of neutrophils to anti-integrin antibodies depends on costimulation through low affinity Fc gamma Rs: full activation requires both integrin and nonintegrin signals. J Immunol. 2004;173:2068–2077. doi: 10.4049/jimmunol.173.3.2068. [DOI] [PubMed] [Google Scholar]
- 39.Chen H, Mocsai A, Zhang H, Ding RX, Morisaki JH, White M, Rothfork JM, Heiser P, Colucci-Guyon E, Lowell CA, Gresham HD, Allen PM, Brown EJ. Role for plastin in host defense distinguishes integrin signaling from cell adhesion and spreading. Immunity. 2003;19:95–104. doi: 10.1016/s1074-7613(03)00172-9. [DOI] [PubMed] [Google Scholar]
- 40.Watts GM, Beurskens FJ, Martin-Padura I, Ballantyne CM, Klickstein LB, Brenner MB, Lee DM. Manifestations of inflammatory arthritis are critically dependent on LFA-1. J Immunol. 2005;174:3668–3675. doi: 10.4049/jimmunol.174.6.3668. [DOI] [PubMed] [Google Scholar]
- 41.Tsuboi N, Ernandez T, Li X, Nishi H, Cullere X, Mekala D, Hazen M, Kohl J, Lee DM, Mayadas TN. Regulation of human neutrophil Fcgamma receptor IIa by C5a receptor promotes inflammatory arthritis in mice. Arthritis Rheum. 2011;63:467–478. doi: 10.1002/art.30141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mocsai A, Abram CL, Jakus Z, Hu Y, Lanier LL, Lowell CA. Integrin signaling in neutrophils and macrophages uses adaptors containing immunoreceptor tyrosine-based activation motifs. Nat Immunol. 2006;7:1326–1333. doi: 10.1038/ni1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Andrew DP, Spellberg JP, Takimoto H, Schmits R, Mak TW, Zukowski MM. Transendothelial migration and trafficking of leukocytes in LFA-1-deficient mice. Eur J Immunol. 1998;28:1959–1969. doi: 10.1002/(SICI)1521-4141(199806)28:06<1959::AID-IMMU1959>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 44.Semmrich M, Smith A, Feterowski C, Beer S, Engelhardt B, Busch DH, Bartsch B, Laschinger M, Hogg N, Pfeffer K, Holzmann B. Importance of integrin LFA-1 deactivation for the generation of immune responses. J Exp Med. 2005;201:1987–1998. doi: 10.1084/jem.20041850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Maiguel D, Faridi MH, Wei C, Kuwano Y, Balla KM, Hernandez D, Barth CJ, Lugo G, Donnelly M, Nayer A, Moita LF, Schurer S, Traver D, Ruiz P, Vazquez-Padron RI, Ley K, Reiser J, Gupta V. Small molecule-mediated activation of the integrin CD11b/CD18 reduces inflammatory disease. Sci Signal. 2011;4:ra57. doi: 10.1126/scisignal.2001811. [DOI] [PMC free article] [PubMed] [Google Scholar]