

Abstract
Platelet-derived growth factor receptor (PDGFR) α and β have been suggested as potential targets for treatment of rhabdomyosarcoma, the most common soft tissue sarcoma in children. This study identifies biological activities linked to PDGF signaling in rhabdomyosarcoma models and human sample collections. Analysis of gene expression profiles of 101 primary human rhabdomyosarcomas revealed elevated PDGF-C and -D expression in all subtypes, with PDGF-D as the solely over-expressed PDGFRβ ligand. By immunohistochemistry, PDGF-CC, PDGF-DD and PDGFRα were found in tumor cells, whereas PDGFRβ was primarily detected in vascular stroma. These results are concordant with the biological processes and pathways identified by data mining. While PDGF-CC/PDGFRα signaling associated with genes involved in the reactivation of developmental programs, PDGF-DD/PDGFRβ signaling related to wound healing and leukocyte differentiation. Clinicopathological correlations further identified associations between PDGFRβ in vascular stroma and the alveolar subtype and with presence of metastases. Functional validation of our findings was carried out in molecularly distinct model systems, where therapeutic targeting reduced tumor burden in a PDGFR-dependent manner with effects on cell proliferation, vessel density and macrophage infiltration. The PDGFR-selective inhibitor CP-673,451 regulated cell proliferation through mechanisms involving reduced phosphorylation of GSK-3α and GSK-3β. Additional tissue culture studies demonstrated a PDGFR-dependent regulation of rhabdosphere formation/cancer cell stemness, differentiation, senescence and apoptosis. In summary, the study demonstrates a clinically relevant distinction in PDGF signaling in human rhabdomyosarcoma and also suggests continued exploration of the influence of stromal PDGFRs on sarcoma progression.
Keywords: PDGF, rhabdomyosarcoma, tyrosine kinase inhibition, tumor microenvironment, stroma
Introduction
Rhabdomyosarcomas (RMS) are the most common pediatric soft tissue sarcoma with two main histological subtypes, embryonal (ERMS) and alveolar (ARMS) (1). ERMS have the most favorable prognosis, but the estimated three-year overall survival is still only 47 % in children and adolescents with metastatic disease (2). The ARMS are typically associated with frequent expression of oncogenic PAX3-FOXO1 or PAX7-FOXO1 gene fusion products and a high propensity to metastasize (2). Both subtypes display features of developing skeletal muscle (3) and their genetic signatures and presence of PAX3-FOXO1 have been proposed to be useful for patient stratification into low- and high-risk groups (4-7). However, the oncogenic heterogeneity of RMS tumors makes the identification of suitable molecular targets for directed therapies challenging.
A potential therapeutic candidate for RMS is the tyrosine kinase platelet-derived growth factor receptor (PDGFR) α. PDGFRα is a direct target of the PAX3-FOXO1 fusion protein in p53-deficient cells (8, 9) and its expression has been reported to correlate with decreased failure-free survival in patients and increased tumorigenicity in mice (9-11). So far, there has been little evidence for other PDGF family members contributing to the biology of RMS. Classically, tumorigenic PDGF signaling follows as a consequence of activating point mutations, amplifications or translocations, often resulting in autocrine stimulatory loops (12, 13). PDGF ligand production is another mechanism of action to promote both autocrine signaling and paracrine crosstalk with infiltrating stromal cells in various tumors (14).
It has been demonstrated that the five homo– or heterodimeric ligands (PDGF-AA, -BB, -AB, -CC and -DD) have different receptor affinity in vivo; both PDGF-AA and PDGF-CC signal via PDGFRα, while PDGF-BB and PDGF-DD act preferentially via PDGFRβ (12). Moreover, PDGF-CC and PDGF-DD are secreted as latent growth factors, subsequently proteolytically activated by plasminogen activators before PDGFR binding (15-18). Accordingly, it is becoming increasingly evident that PDGF signaling is regulated at multiple levels in a context-dependent manner.
In this study, autocrine and paracrine PDGF signaling events are systematically analyzed with particular attention to the heterogeneity among RMS tumors. The data illustrates that PDGF activity is linked to microenvironmental changes with distinct cellular responses observed, not always predictable from PDGFR expression levels.
Materials and Methods
Gene expression profiles of RMS patients
The ITCC/CIT (Innovative Therapies for Children with Cancer/Carte d’Identité des Tumeurs) gene expression profile dataset has been previously described (6, 7). Additional information about the gene expression profiling and data handling can be found in Supplementary Materials and Methods.
Tissue microarrays and clinicopathological correlations
Three independent tissue microarrays were used for analysis of PDGF ligand and receptor expression in human RMS specimens. First, a commercially available array (Tissue Array Networks), representing RMS and leiomyosarcoma samples from 36 patients of various ages, was used to assess tumor compartment-specific protein localization. For clinicopathological correlations, only pediatric material was used. The second array has been previously described and contained material from 60 alveolar and 171 embryonal cases with a mean age of 6.3 years (19). The third array, also previously described, consisted of 25 alveolar and 54 embryonal cases with a mean age of 3.8 years (20). Immunostaining for PDGFRα and PDGFRβ was scored by a consultant pathologist (KT). Each tissue core was assigned a staining intensity score, where 0, 1, 2 and 3 indicated negative, weak, moderate and strong staining, respectively. The score for each patient was derived from the maximal score in all available cores for a patient where the percentage of cells stained was above 10 %. Kaplan-Meier survival analysis of PDGFR expression was performed separately for tumor cells and tumor stroma, using 1 % positively stained cells as cut-off for the latter. Survival characteristics were analyzed based on overall survival as clinical outcome using date of diagnosis as time zero.
Cell culture
Cells were kept at 37 °C in a humidified 5 % CO2 atmosphere. Dulbecco’s Modified Eagle Medium (DMEM) was used for all cell lines with the exception of RMS-YM, which was maintained in Roswell Park Memorial Institute (RPMI) 1640 medium. Supplements included 10 % fetal bovine serum (FBS), 2 mM glutamine, 100 U ml−1 penicillin and 100 μg ml−1 streptomycin unless otherwise stated.
Characterization of cell lines
Gene expression profiles for cell lines were available (21) and analyzed to evaluate PDGF-C and -D expression levels. PDGFRα expression was analyzed by immunoblotting of cell lysates using a rabbit antibody (Cell Signaling). Equal sample loading was confirmed with a calnexin-targeting goat antibody (Santa Cruz Biotechnology).
Modulation of PDGF activity in vitro
Tyrosine phosphorylation of PDGFα was investigated as previously described (16). Following PDGF treatment for 7 min at 37 °C, pAkt(Thr308) was detected with a rabbit antibody (Cell Signaling). Phosphorylated GSK-3α(Ser21) and GSK-3β(Ser9) were likewise detected with a rabbit antibody (Cell Signaling) after a one hour pre-treatment at 37 °C with 0.5 μM CP-673,451 (22) or vehicle (dimethyl sulfoxide). CP-673,451 was chosen for its reported ability to inhibit PDGFR phosphorylation with an otherwise limited substrate crossreactivity (23).
Cell proliferation/viability was analyzed using the CyQuant proliferation assay (Life Technologies). Pre-starved cells were treated every 24 hours with vehicle (dimethyl sulfoxide) or 0.5 μM CP-673,451 diluted in serum-reduced medium (1.5 % FBS) for 96 hours. The amount of nucleic acid present in lysed cells was normalized to the amount when treatment was initiated. Cell proliferation/viability in response to 300 ng/ml PDGF-CC (16) was likewise analyzed, but cells were then kept in serum-free medium and treated twice during a 48-hour period.
Apoptosis was studied after 96 hours treatment as above. Cells were then enzymatically detached and labeled using the Annexin-V-FLUOS staining Kit (Roche) according to the manufacturer’s instructions. Samples were analyzed with the BD FACSCalibur flow cytometer and the CellQuest software (BD Biosciences).
A cell cycle analysis was likewise performed after 96 hours treatment. Cell membranes were lysed with a hypotonic buffer (4 mM sodium citrate, 0.1 % Triton X-100) containing 0.1 mM propidium iodide and 50 μg/ml RNaseA (Life Technologies) for 20 min at 4 °C. Aggregates were removed by filtration trough a 40 μm cell strainer. The total nuclei fluorescence, FL2-A, was measured under exclusion of debris/aggregates via the FL2-W versus FL2-A plot. Data was analyzed with the ModFIT LT Cell Cycle analysis software (Verity Software House).
Rhabdosphere-forming capacity was analyzed in a limited dilution assay with or without pre-treatment with 0.5 μM CP-673,451 or vehicle for 72 hours. The cells were filtered through a 40 μm cell strainer to generate a single cell suspension and then seeded in a descending cell number/well (16, 8, 4, 2, 1) in ultra-low attachment 96-well plates (Corning) in 75 μl complete stem cell medium (Neurocult, LifeTechnologies) supplemented with 2 μg/ml heparin (STEMCELL Technologies), 2xB27 without vitamin A (Life Technologies), 10 ng/ml EGF and 10 ng/ml bFGF (both R&D Systems) containing either 0.5 μM CP-673,451 or vehicle. Fresh medium, 20 μl/well of 0.5 μM CP-673,451 or vehicle, was added every day. RD rhabdospheres larger than 100 μm were counted after ten days, RUCH2 rhabdospheres larger than 200 μm were counted after fourteen days. Results were analyzed using the online Extrem Limited Dilution Analysis software (24).
Xenograft studies
Experimental procedures were approved by the local committee for animal experiments. Two million RUCH2 cells in PBS were implanted subcutaneously into the flank of eight-week-old females and those with palpable tumors were then stratified into two groups based on tumor size (width2 × length × 0.52). The first cohort of SCID mice included thirteen tumor-bearing animals and the second, six animals. Freshly prepared CP-673,451, or poly ethylene glycol-400 as vehicle, was given once-daily by oral gavage for eighteen days and the animals were sacrificed four hours after the last treatment. After perfusion with PBS followed by 2 % paraformaldehyde (PFA), tumors were kept in 30 % sucrose at 4 °C overnight and then embedded for cryopreservation, or alternatively, postfixed in 2 % PFA at 4 °C overnight, dehydrated and paraffin embedded. For xenografts derived from the RMS cell line, five million cells in PBS/matrigel (BD Biosciences) were implanted into twenty SCID mice and CP-673,451 or vehicle was given for nine days. The Sorafenib treatment has been described elsewhere (25).
Immunostaining and immunoquantifications
Paraffin-embedded sections were deparaffinized and endogenous peroxidase activity was quenched by 3 % H2O2 for 10 min at room temperature. Immunostaining was then performed as previously described for PDGFRα and PDGFRβ (26) as well as PDGF-DD (18). For detection of PDGF-CC, the latter staining protocol was used with the exception of including an affinity-purified polyclonal rabbit anti-human PDGF-CC antibody. Staining specificities were confirmed in pelleted and paraffin-embedded COS-1 cells transiently transfected with human full-length PDGF-C or –D as previously described (16, 18) (Fig. S1). The PDGFR antibodies were chosen based on the results from a recent antibody screen, where PDGFR isoform-specific reactivities were investigated (27). Both unphosphorylated and phosphorylated receptors were recognized.
For immunofluorescence-based detection, cryosections were fixed in 4 % PFA (Ki67) or ice-cold acetone (F4/80, CD31/PECAM, phosphorylated (p) PDGFRs). Paraffin-embedded sections were deparaffinized and heat-induced antigen retrieval performed in citrate (Vector Laboratories) or DAKO target retrieval solution, high pH buffer (DAKO) prior staining (Podocalyxin and PDGFRβ respectively). Slides were incubated with blocking solution (DAKO) and primary antibodies in TNB buffer (PerkinElmer Life Sciences) overnight. Ki67 was detected with a rabbit antibody according to the manufacturer’s instructions (Novocastra Laboratories Ltd.). For recognition of macrophages, a rat F4/80 antibody (Serotec) was used. CD31/PECAM was detected with a rat antibody (BD Pharmingen) and pPDGFRs with a crossreacting mouse antibody against PDGFRβ(PY751) (Cell Signaling). Podocalyxin was detected with a goat antibody (R&D Systems) and PDGFRβ with the same antibody as above. Appropriate secondary antibodies (Life Technologies) were applied before mounting with Vectashield mounting medium with DAPI (Vector Laboratories). The AxioVision Rel. 4.6 Software (Carl Zeiss) was used for automated quantifications of immunostaining from typically six RUCH2 tumors/group. Vessel density in RMS tumors was analyzed in three tumors/group.
Results
PDGF ligands and receptors are expressed in human RMS
PDGF ligand and receptor expression was systematically assessed by microarray analysis of patient material from 101 RMS tumors and 36 skeletal muscle samples. For comparison, other small round blue cell tumors (SRBCT) and mesenchymal stem cells were also included. PDGF-C and PDGF-D were the only PDGF ligands consistently over-expressed compared to skeletal muscle, while PDGF-B was consistently under-expressed (Fig. 1A). Only PDGFRβ, among the receptors, showed significant over-expression in both ERMS and ARMS P3F-positive samples (Fig. 1B). Altogether, these data are indicative of a ligand-mediated PDGFR activation in RMS, which is also supported by the lack of identified PDGFR-activating mutations reported so far (28, 29).
Figure 1.

PDGF ligands and receptors are expressed in human RMS. Box and whisker plots representing the expression of A, PDGF-A, PDGF-B, PDGF-C, PDGF-D and B, their cognate receptors, PDGFRα and PDGFRβ, in 36 skeletal muscle control samples (Sk. Muscle), 32 mesenchymal stem cell samples (MSC), 50 neuroblastomas (NB), 18 Ewing sarcomas (ES), 20 Wilms tumors (WT), 39 medulloblastomas (MBL), 36 ERMS (ERMS), 34 ARMS PAX3-FOXO1 and 1 PAX3-NCOA1 fusion-positive (ARMS_P3F), 10 ARMS PAX7-FOXO1 fusion-positive (ARMS_P7F) and 20 ARMS fusion-negative (ARMS_NEG) samples based on gene expression profiling using the Affymetrix chip HG-133plus2 (*P<0.05, **P<0.01, ***P<0.001, Wilcoxon rank-sum test between Sk. Muscle and each RMS subtype).
PDGF ligands are confined to the RMS tumor cell compartment, while PDGFRs are expressed by both tumor cells and infiltrating stromal cells
Tumor compartment-specific expression of PDGF family members was analyzed by immunostaining of three different tissue microarrays. Based on the results from the RNA expression profiling data, PDGF-CC was selected as the only consistently over-expressed PDGFRα ligand and PDGF-DD as the only over-expressed PDGFRβ ligand. Production of PDGF-CC and PDGF-DD was shown to be almost exclusively located to tumor cells, while PDGFRα was frequently expressed in both the tumor and stromal cell compartment (Fig. 2A, 2C). PDGFRβ was occasionally detected in tumor cells, but mainly expressed in vascular stroma (Fig. 2A-2C).
Figure 2.
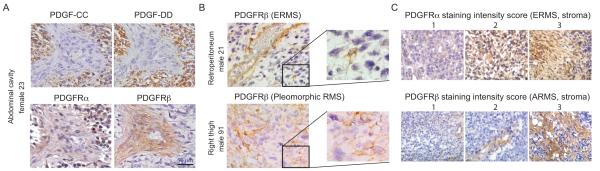
PDGF ligands are confined to the RMS tumor cell compartment, while PDGFRs are expressed by both tumor cells and infiltrating stromal cells. A, immunostaining of PDGF-CC, PDGF-DD, PDGFRα, and PDGFRβ in consecutive sections of human ERMS tissue cores. B, PDGFRβ expression in vascular stroma (top) and in tumor cells (bottom). C, stromal expression of PDGFRα and PDGFRβ in ERMS and ARMS, respectively, used for scoring and clinicopathological correlations. PDGFRα was notably also found in tumor cells.
Stromal PDGFRβ expression associates with the alveolar subtype
Protein levels of PDGFRs were assessed in two different tissue microarrays for clinicopathological correlations. PDGFRβ was again rarely detected in tumor cells, while its stromal staining was associated with the alveolar subtype (Table 1). PDGFRα expression was observed in tumor cells and stroma and was in both cases associated with the embryonal subtype (P<0.0033 and P<0.0329).
Table 1.
Associations of PDGFR expression in tumor stroma with metastatic status and subtype using a chi-squared test for trend. Metastatic status implies that the patient presented at Stage 4 with metastasis.
| PDGFRα in tumor stroma | PDGFRβ in tumor stroma | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Staining intensity score |
Metastasis | Subtype distribution | Staining intensity score |
Metastasis | Subtype distribution | ||||
| No | Yes | ERMS | ARMS | No | Yes | ERMS | ARMS | ||
| 0 | 88 | 30 | 125 | 62 | 0 | 76 | 9 | 121 | 17 |
| 1 | 14 | 3 | 25 | 8 | 1 | 23 | 8 | 36 | 26 |
| 2 | 16 | 0 | 22 | 3 | 2 | 29 | 16 | 44 | 27 |
| 3 | 4 | 0 | 3 | 1 | 3 | 12 | 4 | 12 | 10 |
| PDGFRα stromal staining is negatively associated with metastasis. |
PDGFRα stromal staining is positively associated with the ERMS subtype. |
PDGFRβ stromal staining is positively associated with metastasis. |
PDGFRβ stromal staining is positively associated with the ARMS subtype. |
||||||
| Chi squared test for trend statistic 6.4005; P=0.0114 |
Chi squared test for trend statistic 4.551; P=0.0329 |
Chi squared test for trend statistic 8.6819; P=0.0032 |
Chi squared test for trend statistic 19.02; P<0.0001 |
||||||
The material in the third and largest array (19) was separately analyzed for associations with proliferation and metastatic status. No significant association was seen between either PDGFR staining and the proliferation marker Ki67 (evaluated by the reference monoclonal antibody MIB-1, data not shown). Stromal expression of PDGFRα and PDGFRβ was respectively negatively and positively associated with the presence of metastases at diagnosis (Table 1).
PDGF-C expression is associated with genes involved in developmental programs, while PDGF-D is associated with those involved in wounding and immune system processes
To further investigate ligand-dependent PDGFR activation, the gene expression profile of the primary tumors was analyzed to identify sets of genes, which correlated to either PDGF-C or PDGF-D. Based on GO enrichment analysis, several genes positively correlating with PDGF-C expression were associated with various developmental processes (Table 2). This suggests that PDGF-C expression is linked with the reactivation of embryonic signaling pathways, which could facilitate tumor progression. PDGF-D expression correlated with genes associated with wounding, leukocyte differentiation and cell adhesion. When individual genes were investigated, PDGF-C was found to correlate with e.g. HOX genes, GLI3 and FZD7 (Table S1). PDGF-D, on the other hand, correlated with genes such as CXCL12, VWF and CD302. Our findings are consistent with some of the previously reported activities mediated by PDGFRα and PDGFRβ signaling (30).
Table 2.
PDGF-C expression is associated with genes involved in developmental programs, while PDGF-D is associated with those involved in wounding and immune system processes. A gene to GO BP conditional test for over-representation, where Exp Count is the expected number of genes to be found associated with each specific process (Term). This is obtained considering the number of genes (Size) associated with that process and how many of those being selected by the analysis (Count). The first six categories associated with each gene are shown.
| PDGF-C | ||||||
|---|---|---|---|---|---|---|
|
| ||||||
| GOBPID | P-value | Odds Ratio |
Exp Count |
Count | Size | Term |
| GO:0009954 | 0.000 | 32.961 | 0 | 6 | 18 | Proximal/distal pattern formation |
| GO:0009952 | 0.000 | 7.919 | 1 | 10 | 95 | Anterior/posterior pattern formation |
| GO:0045110 | 0.000 | 193.875 | 0 | 3 | 4 | Intermediate filament bundle assembly |
| GO:0048856 | 0.000 | 2.228 | 25 | 46 | 1695 | Anatomical structure development |
| GO:0007275 | 0.000 | 2.145 | 25 | 44 | 1705 | Multicellular organismal development |
| GO:0007389 | 0.000 | 4.427 | 3 | 11 | 178 | Pattern specification process |
|
| ||||||
| PDGF-D | ||||||
|
| ||||||
| GOBPID | P-value | Odds Ratio |
Exp Count |
Count | Size | Term |
|
| ||||||
| GO:0007155 | 0.000 | 6.203 | 3 | 12 | 477 | Cell adhesion |
| GO:0030318 | 0.000 | 75.597 | 0 | 3 | 11 | Melanocyte differentiation |
| GO:0009611 | 0.000 | 6.940 | 2 | 9 | 302 | Response to wounding |
| GO:0050878 | 0.000 | 15.424 | 0 | 5 | 73 | Regulation of body fluid levels |
| GO:0042246 | 0.000 | 60.464 | 0 | 3 | 13 | Tissue regeneration |
| GO:0002763 | 0.000 | 54.961 | 0 | 3 | 14 | Positive regulation of myeloid leukocyte differentiation |
To further analyze PDGF-DD/PDGFRβ signaling, RMS patients with high (top quartile) and low (bottom quartile) PDGF-D expressing tumors were compared using pre-defined gene sets (31). High expression of PDGF-D was associated with expression of genes involved in e.g. cell migration (Fig. S2A), leukocyte transendothelial migration and vascular smooth muscle contraction (Fig. S2B). These associations were not seen in the corresponding PDGF-C expression analysis (data not shown).
Therapeutic targeting of PDGFR signaling results in decreased cell proliferation, cell cycle arrest and apoptosis in a cell type-specific manner
To explore ligand-dependent PDGFR activity, cell lines were screened for PDGF-C and -D expression (Table S2) and a subset (Table S3) of high and low-expressing cell lines was analyzed for PDGFRα protein expression. RD and RUCH2 cells displayed high PDGFRα expression and were responsive to PDGF stimuli (Fig. 3A-B, S3A-B). By qRT-PCR analysis (Table S4), these two cell lines displayed the highest expression levels of PDGF-C and -D, compared to PDGF-A and –B (Fig. S3C). They also decreased their proliferation rate in response to the PDGFR inhibitor CP-673,451, whereas the PDGFR-negative cell line RMS did not (Fig. 3C). Furthermore, Akt and the downstream key regulators GSK-3α and GSK-3β were identified as targets after ligand-induced PDGFRα stimulation (Fig. 3D-E). This phosphorylation response was completely abolished when cells were pre-treated with CP-673,451. A striking heterogeneity was observed between the two PDGFR-positive cell lines; the RUCH2 cell line displayed an apoptotic response accompanied by a G2/M cell cycle arrest, whereas RD cells only responded with a modest S phase arrest (Fig. 3F-G).
Figure 3.

Therapeutic targeting of PDGFR signaling results in decreased cell proliferation, cell cycle arrest and apoptosis in a cell type-specific manner. A, immunoblotting of PDGFRα protein in RMS cell lysates. Calnexin was used as loading control. B, immunoblotting of pPDGFRα and total PDGFRα after ligand stimulation as indicated. C, cell proliferation response following PDGFR inhibition in two PDGFR-positive cell lines and one negative. D, immunoblotting of pAkt(Thr308) after PDGF ligand stimulation. PDGFRα was used as loading control. E, immunoblotting of pPDGFRα, total PDGFRα, pGSK-3α(Ser21) and pGSK-3β(Ser9) following treatments as indicated. Calnexin was used as loading control. F, apoptotic response analyzed by flow cytometry in PDGFR-positive cell lines following treatment with CP-673,451 or vehicle. G, cell cycle analysis by flow cytometry of PDGFR-positive cell lines following treatment with CP-673,451 or vehicle. Data are presented ± SD (*P<0.05, ***P<0.001, Student’s t test).
Impaired rhabdosphere-forming capacity and differentiation in cells devoid of PDGF activity
So far, the data indicated that PDGF-CC/PDGFRα signaling is involved in the reactivation of developmental pathways. Stemness potential in the presence of CP-673,451 or vehicle was therefore analyzed in an anchorage-independent growth assay. CP-673,451 impaired rhabdosphere-forming capacity in both RD and RUCH2 cultures (Fig. 4A) and this was likewise observed in a rhabdosphere-forming assay where adherent RUCH2 cells were pre-treated for 72 hours with CP-673,451 before seeding without treatments (Fig. 4B). In light of previous studies, RD cells were also used as a model system for RMS differentiation (32). In the presence of CP-673,451 under conditions facilitating cellular differentiation, hallmarks of senescence, i.e. enlarged cell size and presence of perinuclear SA-βgal activity, were observed (Fig. S4A). The absence of elongating cells initiating myogenesis suggested dysfunctional differentiation (Fig. S4B), further supported by an expression analysis of the myogenic markers MYL1 and MYOGENIN and the myogenic repressor HEY-1 (33) (Fig. S4C).
Figure 4.
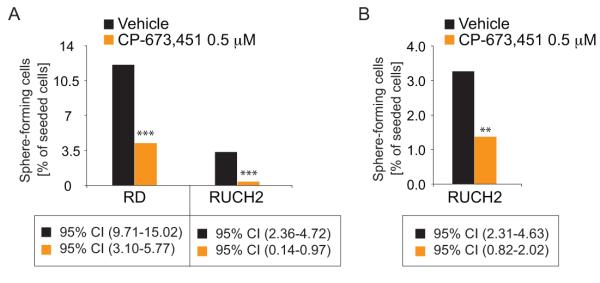
Impaired anchorage-independent rhabdosphere-forming capacity in cells devoid of PDGF activity. A, rhabdosphere formation in the presence of CP-673,451 or vehicle. B, rhabdosphere formation of cells pretreated 72 hours with CP-673,451 or vehicle in adherent cultures before seeding.
PDGFR inhibition reduces tumor growth and stromal cell infiltration
Based on the in vitro results, RUCH2 cells expressed PDGF-C and PDGF-D and demonstrated PDGFR dependency. Xenograft-bearing mice were treated with CP-673,451 or vehicle and growth inhibition due to the therapeutic regimen was detected after ten days of treatment (Fig. 5A). Tumor volume after dissection was analyzed in both cohorts as a separate endpoint. Control tumors had then reached an average size of 66 mm3 and tumors from mice treated with the active compound had reached a size of 32 mm3 (Student’s t test, P=0.017, n≥9 mice/group). No metastases were detected.
Figure 5.

PDGFR inhibition reduces tumor growth and stromal cell infiltration. A, growth of RUCH2 xenografts in response to CP-673,451 or vehicle (n≥6 mice/group). B, quantification of Ki67-immunopositive cells in RUCH2 tumor sections. C, quantification of pPDGFR-immunopositive cells in RUCH2 tumor sections. D, quantification of F4/80-immunopositive macrophages in RUCH2 tumor sections. E, quantification of vessel density in RUCH2 tumor sections immunostained for CD31/PECAM. F-G, quantification of vessel density in tumors derived from the RMS cell line and treated with CP-673,451, Sorafenib or vehicle. Sections were immunostained for the vessel marker Podocalyxin. A compensatory threshold was set to disregard the autofluorescence generated from massive cell death in tumors treated with Sorafenib. Data are presented ± SD (*P<0.05, **P<0.01, ***P<0.001, Student’s t test).
Immunostaining revealed a decreased number of cells in the proliferative phase in CP-673,451-treated animals (Fig. 5B). The effects on PDGFR phosphorylation was specifically visualized by immunostaining for phosphorylated PDGFRs. A reduced staining intensity was observed in sections from animals treated with the active compound (Fig. 5C).
Before evaluating the therapeutic effect on host-derived stroma, it was confirmed that human PDGF-DD (34) could activate mouse PDGFRβ (Fig. S5). Thereafter, immunostaining of RUCH2 xenografts revealed that CP-673,451 reduced the number of F4/80-positive macrophages (Fig. 5D) and CD31-positive vessels (Fig. 5E). Fibrotic streaks composed of collectively migrating fibroblasts were not found and consequently not quantified (data not shown).
Vessel density in tumors derived from the RMS cell line is reduced by the multi-targeting tyrosine kinase inhibitor Sorafenib, but not by CP-673,451
In primary tumors, PDGF-D expression correlated to sets of genes involved in blood vessel function. Vessel density was therefore separately analyzed in tumors derived from the RMS cell line after treatment with CP-673,451 or Sorafenib. PDGFRs were detected in infiltrating stroma (Fig. S6A), but not in tumor cells or vessels (data not shown). CP-673,451 neither altered tumor growth (Fig. S6B) nor vessel density (Fig. 5F), whereas Sorafenib treatment almost completely eliminated all blood vessels (Fig. 5G).
Discussion
Therapeutic inhibition of PDGF activity has proven beneficial in several types of sarcomas (35-37). PDGFRα was also recently associated with acquired resistance to IGF-1R antibody therapy in RMS (38). However, little is known about the pathobiology associated with PDGF signaling in RMS. We have therefore systematically analyzed PDGF ligand and receptor expression in human RMS samples and used animal and cell culture models for functional studies. The analyses suggest the existence of tumor compartment-specific effects of ligand-dependent PDGFRα and PDGFRβ signaling. An important aspect of the study is the indication of clinically relevant variations in the stromal compartment of RMS. Stromal PDGFR signaling has previously been extensively described in tumors of epithelial origin (27, 39, 40).
Supported by an analysis of the gene expression profile of 101 pediatric primary tumors and 36 normal skeletal muscle samples. PDGF-C and PDGF-D were identified as the only PDGF ligands with consistently elevated expression relative to skeletal muscle. Accordingly, we confirmed their presence in the hyperchromatic tumor cell compartment. PDGFRα seemed to be the predominating receptor isoform expressed by tumor cells, whereas PDGFRβ was mainly found in vascular stroma. Consistent with our findings, autocrine PDGFRα signaling has recently been associated with both RMS tumor cell survival and regulation of differentiation in pediatric malignancies (9, 41), whereas PDGFRβ is classically associated with blood vessel morphogenesis (42-44). Accumulating data hereby suggest that PDGF signaling in RMS includes both autocrine and paracrine signaling in different cell types.
The observed compartmentalization of PDGFRα and PDGFRβ expression urged us to elucidate clinical parameters associated with their expression in tumor stroma. PDGFRα was then found to associate with the embryonal subtype, whereas PDGFRβ associated with the alveolar subtype. This difference was not detected in our gene expression analysis of tumor lysates, but is likely explained by the relatively small proportion of infiltrating cells compared to the tumor cell mass. In light of previous findings about PDGF signaling in blood vessel morphogenesis (43) and metastatic spread of RMS cells (9, 10), associations with the presence of metastases were analyzed in a similar way. Stromal PDGFRα was then found to negatively associate with metastasis, whereas PDGFRβ positively associated with metastasis. These findings could reflect the difference in PDGFR expression observed between ERMS and ARMS, and the higher propensity of the latter to metastasize. Accordingly, multivariate analyses of data from even larger cohorts are needed in order to clarify whether the investigated correlations are independent of histology.
By analyzing gene expression profiling of primary tumor samples, tumorigenic PDGF activities were characterized; GO enrichments for biological processes suggested that PDGF-C may be involved in developmental processes, whereas PDGF-D associated with genes active in cell adhesion, wounding and immune system processes. These findings support a role of PDGF-DD/PDGFRβ signaling in stromal cell recruitment and are in line with previous studies on PDGFRβ as a regulator of interstitial fluid pressure, a well-established mechanism involving stromal cells and extracellular matrix constituents with direct implications for the delivery of therapeutic agents to tumor cells (45-47). To explore this further, we compared high PDGF-D expressing tumors with low PDGF-D expressing tumors on the basis of metagenes generated using GO and KEGG annotations. In this analysis, we observed enrichment for terms such as cell-adhesion molecules, leukocyte transendothelial migration and vascular smooth muscle contraction. This was detected for PDGF-D, and not PDGF-C, once again highlighting in vivo differences between PDGFRα and PDGFRβ signaling.
To mechanistically explore autocrine PDGFRα signaling, cell lines were screened for PDGF-C and -D expression. High PDGFRα protein expression was confirmed in RD and RUCH2 cells. These two cell lines were responsive to PDGF-CC stimulation and they displayed decreased cell proliferation/survival after PDGFR targeting by the PDGFR tyrosine kinase inhibitor CP-673,451. However, in line with our gene set enrichments based on human material, our in vitro data revealed a heterogeneity associated with PDGF signaling in RMS. Following PDGFR inhibition, the RUCH2 cell line morphologically displayed clear signs of cell death, mechanistically identified as an increase in apoptosis and a G2/M cell cycle arrest. RD cells, on the other hand, were largely unaffected under proliferative conditions. Under conditions facilitating myogenic differentiation, though, morphological changes, including signs of senescence, were observed in the presence of CP-673,451. An impaired differentiation capacity was also evident, which is in line with a recent study describing the need for active PDGFRα signaling in neuroblastoma differentiation (41). This is supportive of PDGF-CC/PDGFRα being involved in the intricate interplay between proliferation and differentiation as our findings from the data mining suggested. At the same time, in an anchorage-independent stem cell assay, both RD and RUCH2 cells displayed impaired ability to form rhabdospheres in the presence of CP-673,451, indicative of PDGFRα signaling being required for the maintenance of stemness characteristics of embryonal RMS cells. Together these results suggest that PDGFRα signaling regulates both cancer cell stemness and myogenic differentiation.
For further validation of the identified correlations in the RMS patient data set, the RUCH2 cell line, with comparably high PDGF-D expression, was selected for development of a mouse xenograft model. These cells displayed the highest response to PDGF-CC stimulation in vitro and in the corresponding in vivo model, therapeutic targeting of PDGFRs decreased tumor burden. Additional effects were noted on vessel density and the number of infiltrating macrophages. Whether our therapeutic regimen mechanistically targeted PDGFRβ-positive pericytes involved in the angiogenic process (22, 48), or alternatively, downregulated VEGF expression in tumorigenic cells (49) is not known. It is however likely that both these previously characterized PDGF/PDGFR-driven processes would explain how PDGF signaling contributes to angiogenesis in RUCH2 xenografts.
Angiogenesis was also explored in xenografts derived from the RMS cell line. These cells were not responsive to PDGFR inhibition in vitro or in vivo. Blood vessel density was also not altered in these tumors following treatment with CP-673,451. PDGFRβ expression was only detected in stroma, and consequently, these results indicate that targeting of PDGFR signaling in stroma is insufficient to affect RMS tumor growth. They further support that if PDGF signaling regulates blood vessel characteristics it is most likely via an intimate crosstalk between PDGFR-positive mural cells and endothelial cells, mechanistically distinct from e.g. VEGF-driven angiogenesis acting directly on the vascular endothelial cells. In a comparative analysis, tumors derived from the RMS cell line were in contrast highly responsive to Sorafenib, known for its ability to target VEGFRs.
Whether or not vascular PDGFRs are transiently expressed during RMS tumor formation and progression is still unclear, but this was at least not evident from a therapeutic perspective in our study. Overall, very little is known about stromagenesis, including blood vessel morphogenesis and e.g. immune regulatory processes, in RMS biology. Macrophage infiltration has been linked to PDGF-D expression in skeletal muscle (50) and our findings indicate that PDGF-D has similar immune regulatory functions in the corresponding tumor tissue. Immune modulation was also recently investigated in a conditional mouse model of RMS (9). PDGFRα expression was in these mice linked to disease progression, but did not regulate immune responses. This is in line with our results that rather suggest PDGFRβ to be involved in this regulation, not PDGFRα.
Taken together, through analysis of gene expression profiling of 101 RMS patient samples and subsequent in vitro and in vivo validation, we found that PDGF activity can support RMS growth and regulate fundamental cellular behavior. This includes tumor cell proliferation/survival, apoptosis/senescence, cancer cell stemness/differentiation, immune system processes and blood vessel characteristics -typical processes known to require extracellular matrix remodeling. We could also link these activities to PDGF-CC/PDGFRα signaling and PDGF-DD/PDGFRβ signaling in a tumor compartment-specific manner. Our findings suggest that stromal PDGFR signaling should be further studied in RMS subtypes and other sarcomas.
Supplementary Material
Acknowledgements
We thank Inger Bodin for technical assistance. Carina Hellberg and Carl-Henrik Heldin (Ludwig Institute for Cancer Research Ltd., Uppsala Branch) kindly provided PDGF-BB and antiserum against human PDGFRs. CP-673,451, owned by Arog Pharmaceuticals LLC, was a valuable gift from Pfizer. The Childrenś Cancer and Leukaemia Group assisted with tumor collection.
Financial support: This study was supported by the Ludwig Institute for Cancer Research Ltd. (M. Ehnman, E. Folestad, U. Eriksson), The Swedish Cancer Society (U. Eriksson, A. Östman, K. Pietras), The Childhood Cancer Foundation (M. Ehnman, K. Pietras), The Swedish Research Council Linnaeus grant to the STARGET consortium (U. Eriksson, A. Östman, K. Pietras), Karolinska Institutet (U. Eriksson), The Chris Lucas Trust (J. Selfe, E. Missiaglia), The Institute of Cancer Research (J. Shipley), The Royal Marsden Hospital Charity Panel (K. Thway), and Anders Otto Swärds stiftelse (E. Folestad). Cancer Research UK (J. Shipley C5066/A10399) funded tumor collection and Carte d'Identite Program of the Ligue Nationale Contre le Cancer supported the expression profiling. The authors thank NHS funding to the NIHR Biomedical Research Centre.
Footnotes
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Meza JL, Anderson J, Pappo AS, Meyer WH. Analysis of prognostic factors in patients with nonmetastatic rhabdomyosarcoma treated on intergroup rhabdomyosarcoma studies III and IV: the Children’s Oncology Group. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2006;24(24):3844–51. doi: 10.1200/JCO.2005.05.3801. [DOI] [PubMed] [Google Scholar]
- 2.Breneman JC, Lyden E, Pappo AS, Link MP, Anderson JR, Parham DM, et al. Prognostic factors and clinical outcomes in children and adolescents with metastatic rhabdomyosarcoma--a report from the Intergroup Rhabdomyosarcoma Study IV. J Clin Oncol. 2003;21(1):78–84. doi: 10.1200/JCO.2003.06.129. [DOI] [PubMed] [Google Scholar]
- 3.Morgenstern DA, Rees H, Sebire NJ, Shipley J, Anderson J. Rhabdomyosarcoma subtyping by immunohistochemical assessment of myogenin: tissue array study and review of the literature. Pathol Oncol Res. 2008;14(3):233–8. doi: 10.1007/s12253-008-9012-5. [DOI] [PubMed] [Google Scholar]
- 4.Davicioni E, Finckenstein FG, Shahbazian V, Buckley JD, Triche TJ, Anderson MJ. Identification of a PAX-FKHR gene expression signature that defines molecular classes and determines the prognosis of alveolar rhabdomyosarcomas. Cancer Res. 2006;66(14):6936–46. doi: 10.1158/0008-5472.CAN-05-4578. [DOI] [PubMed] [Google Scholar]
- 5.Wachtel M, Dettling M, Koscielniak E, Stegmaier S, Treuner J, Simon-Klingenstein K, et al. Gene expression signatures identify rhabdomyosarcoma subtypes and detect a novel t(2;2)(q35;p23) translocation fusing PAX3 to NCOA1. Cancer Res. 2004;64(16):5539–45. doi: 10.1158/0008-5472.CAN-04-0844. [DOI] [PubMed] [Google Scholar]
- 6.Williamson D, Missiaglia E, de Reynies A, Pierron G, Thuille B, Palenzuela G, et al. Fusion gene-negative alveolar rhabdomyosarcoma is clinically and molecularly indistinguishable from embryonal rhabdomyosarcoma. J Clin Oncol. 2010;28(13):2151–8. doi: 10.1200/JCO.2009.26.3814. [DOI] [PubMed] [Google Scholar]
- 7.Missiaglia E, Williamson D, Chisholm J, Wirapati P, Pierron G, Petel F, et al. PAX3/FOXO1 Fusion Gene Status Is the Key Prognostic Molecular Marker in Rhabdomyosarcoma and Significantly Improves Current Risk Stratification. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2012 doi: 10.1200/JCO.2011.38.5591. [DOI] [PubMed] [Google Scholar]
- 8.Epstein JA, Song B, Lakkis M, Wang C. Tumor-specific PAX3-FKHR transcription factor, but not PAX3, activates the platelet-derived growth factor alpha receptor. Mol Cell Biol. 1998;18(7):4118–30. doi: 10.1128/mcb.18.7.4118. PMCID: 108996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Taniguchi E, Nishijo K, McCleish AT, Michalek JE, Grayson MH, Infante AJ, et al. PDGFR-A is a therapeutic target in alveolar rhabdomyosarcoma. Oncogene. 2008;27(51):6550–60. doi: 10.1038/onc.2008.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Armistead PM, Salganick J, Roh JS, Steinert DM, Patel S, Munsell M, et al. Expression of receptor tyrosine kinases and apoptotic molecules in rhabdomyosarcoma: correlation with overall survival in 105 patients. Cancer. 2007;110(10):2293–303. doi: 10.1002/cncr.23038. [DOI] [PubMed] [Google Scholar]
- 11.Blandford MC, Barr FG, Lynch JC, Randall RL, Qualman SJ, Keller C. Rhabdomyosarcomas utilize developmental, myogenic growth factors for disease advantage: a report from the Children’s Oncology Group. Pediatr Blood Cancer. 2006;46(3):329–38. doi: 10.1002/pbc.20466. [DOI] [PubMed] [Google Scholar]
- 12.Andrae J, Gallini R, Betsholtz C. Role of platelet-derived growth factors in physiology and medicine. Genes & development. 2008;22(10):1276–312. doi: 10.1101/gad.1653708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pietras K, Sjoblom T, Rubin K, Heldin CH, Ostman A. PDGF receptors as cancer drug targets. Cancer Cell. 2003;3(5):439–43. doi: 10.1016/s1535-6108(03)00089-8. [DOI] [PubMed] [Google Scholar]
- 14.Pietras K, Ostman A. Hallmarks of cancer: interactions with the tumor stroma. Experimental cell research. 2010;316(8):1324–31. doi: 10.1016/j.yexcr.2010.02.045. [DOI] [PubMed] [Google Scholar]
- 15.Fredriksson L, Li H, Fieber C, Li X, Eriksson U. Tissue plasminogen activator is a potent activator of PDGF-CC. Embo J. 2004;23(19):3793–802. doi: 10.1038/sj.emboj.7600397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fredriksson L, Ehnman M, Fieber C, Eriksson U. Structural requirements for activation of latent platelet-derived growth factor CC by tissue plasminogen activator. J Biol Chem. 2005;280(29):26856–62. doi: 10.1074/jbc.M503388200. [DOI] [PubMed] [Google Scholar]
- 17.Ustach CV, Kim HR. Platelet-derived growth factor D is activated by urokinase plasminogen activator in prostate carcinoma cells. Mol Cell Biol. 2005;25(14):6279–88. doi: 10.1128/MCB.25.14.6279-6288.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ehnman M, Li H, Fredriksson L, Pietras K, Eriksson U. The uPA/uPAR system regulates the bioavailability of PDGF-DD: implications for tumour growth. Oncogene. 2009;28(4):534–44. doi: 10.1038/onc.2008.410. [DOI] [PubMed] [Google Scholar]
- 19.Wachtel M, Runge T, Leuschner I, Stegmaier S, Koscielniak E, Treuner J, et al. Subtype and prognostic classification of rhabdomyosarcoma by immunohistochemistry. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2006;24(5):816–22. doi: 10.1200/JCO.2005.03.4934. [DOI] [PubMed] [Google Scholar]
- 20.Tonelli R, McIntyre A, Camerin C, Walters ZS, Di Leo K, Selfe J, et al. Antitumor Activity of Sustained N-Myc Reduction in Rhabdomyosarcomas and Transcriptional Block by Antigene Therapy. Clinical cancer research : an official journal of the American Association for Cancer Research. 2012;18(3):796–807. doi: 10.1158/1078-0432.CCR-11-1981. [DOI] [PubMed] [Google Scholar]
- 21.Missiaglia E, Selfe J, Hamdi M, Williamson D, Schaaf G, Fang C, et al. Genomic imbalances in rhabdomyosarcoma cell lines affect expression of genes frequently altered in primary tumors: an approach to identify candidate genes involved in tumor development. Genes Chromosomes Cancer. 2009;48(6):455–67. doi: 10.1002/gcc.20655. [DOI] [PubMed] [Google Scholar]
- 22.Roberts WG, Whalen PM, Soderstrom E, Moraski G, Lyssikatos JP, Wang HF, et al. Antiangiogenic and antitumor activity of a selective PDGFR tyrosine kinase inhibitor, CP-673,451. Cancer Res. 2005;65(3):957–66. [PubMed] [Google Scholar]
- 23.Homsi J, Daud AI. Spectrum of activity and mechanism of action of VEGF/PDGF inhibitors. Cancer Control. 2007;14(3):285–94. doi: 10.1177/107327480701400312. [DOI] [PubMed] [Google Scholar]
- 24.Hu Y, Smyth GK. ELDA: extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J Immunol Methods. 2009;347(1-2):70–8. doi: 10.1016/j.jim.2009.06.008. [DOI] [PubMed] [Google Scholar]
- 25.Maruwge W, D’Arcy P, Folin A, Brnjic S, Wejde J, Davis A, et al. Sorafenib inhibits tumor growth and vascularization of rhabdomyosarcoma cells by blocking IGF-1R-mediated signaling. Onco Targets Ther. 2008;1:67–78. doi: 10.2147/ott.s3833. PMCID: 2994208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nupponen NN, Paulsson J, Jeibmann A, Wrede B, Tanner M, Wolff JE, et al. Platelet-derived growth factor receptor expression and amplification in choroid plexus carcinomas. Mod Pathol. 2008;21(3):265–70. doi: 10.1038/modpathol.3800989. [DOI] [PubMed] [Google Scholar]
- 27.Paulsson J, Sjöblom T, Micke P, Pontén F, Landberg G, Heldin CH, et al. Prognostic significance of stromal platelet-derived growth factor beta-receptor expression in human breast cancer. Am J Pathol. 2009;175(1):334–41. doi: 10.2353/ajpath.2009.081030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shukla N, Ameur N, Yilmaz I, Nafa K, Lau CY, Marchetti A, et al. Oncogene mutation profiling of pediatric solid tumors reveals significant subsets of embryonal rhabdomyosarcoma and neuroblastoma with mutated genes in growth signaling pathways. Clinical cancer research : an official journal of the American Association for Cancer Research. 2012;18(3):748–57. doi: 10.1158/1078-0432.CCR-11-2056. PMCID: 3271129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bai Y, Li J, Fang B, Edwards A, Zhang G, Bui M, et al. Phosphoproteomics identifies driver tyrosine kinases in sarcoma cell lines and tumors. Cancer research. 2012 doi: 10.1158/0008-5472.CAN-11-3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ostman A. PDGF receptors-mediators of autocrine tumor growth and regulators of tumor vasculature and stroma. Cytokine & growth factor reviews. 2004;15(4):275–86. doi: 10.1016/j.cytogfr.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 31.Kim SY, Volsky DJ. PAGE: parametric analysis of gene set enrichment. BMC Bioinformatics. 2005;6:144. doi: 10.1186/1471-2105-6-144. PMCID: 1183189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bouche M, Canipari R, Melchionna R, Willems D, Senni MI, Molinaro M. TGF-beta autocrine loop regulates cell growth and myogenic differentiation in human rhabdomyosarcoma cells. Faseb J. 2000;14(9):1147–58. doi: 10.1096/fasebj.14.9.1147. [DOI] [PubMed] [Google Scholar]
- 33.Buas MF, Kabak S, Kadesch T. The Notch effector Hey1 associates with myogenic target genes to repress myogenesis. J Biol Chem. 2010;285(2):1249–58. doi: 10.1074/jbc.M109.046441. PMCID: 2801253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bergsten E, Uutela M, Li X, Pietras K, Ostman A, Heldin CH, et al. PDGF-D is a specific, protease-activated ligand for the PDGF beta-receptor. Nat Cell Biol. 2001;3(5):512–6. doi: 10.1038/35074588. [DOI] [PubMed] [Google Scholar]
- 35.Wang YX, Mandal D, Wang S, Hughes D, Pollock RE, Lev D, et al. Inhibiting platelet-derived growth factor beta reduces Ewing’s sarcoma growth and metastasis in a novel orthotopic human xenograft model. In Vivo. 2009;23(6):903–9. [PubMed] [Google Scholar]
- 36.McDermott U, Ames RY, Iafrate AJ, Maheswaran S, Stubbs H, Greninger P, et al. Ligand-dependent platelet-derived growth factor receptor (PDGFR)-alpha activation sensitizes rare lung cancer and sarcoma cells to PDGFR kinase inhibitors. Cancer Res. 2009;69(9):3937–46. doi: 10.1158/0008-5472.CAN-08-4327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hartmann JT. Systemic treatment options for patients with refractory adult-type sarcoma beyond anthracyclines. Anticancer Drugs. 2007;18(3):245–54. doi: 10.1097/CAD.0b013e3280124e41. [DOI] [PubMed] [Google Scholar]
- 38.Huang F, Hurlburt W, Greer A, Reeves KA, Hillerman S, Chang H, et al. Differential mechanisms of acquired resistance to insulin-like growth factor-i receptor antibody therapy or to a small-molecule inhibitor, BMS-754807, in a human rhabdomyosarcoma model. Cancer research. 2010;70(18):7221–31. doi: 10.1158/0008-5472.CAN-10-0391. [DOI] [PubMed] [Google Scholar]
- 39.Östman A, Augsten M. Cancer-associated fibroblasts and tumor growth--bystanders turning into key players. Curr Opin Genet Dev. 2009;19(1):67–73. doi: 10.1016/j.gde.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 40.Pietras K, Pahler J, Bergers G, Hanahan D. Functions of paracrine PDGF signaling in the proangiogenic tumor stroma revealed by pharmacological targeting. PLoS Med. 2008;5(1):e19. doi: 10.1371/journal.pmed.0050019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mei Y, Wang Z, Zhang L, Zhang Y, Li X, Liu H, et al. Regulation of neuroblastoma differentiation by forkhead transcription factors FOXO1/3/4 through the receptor tyrosine kinase PDGFRA. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(13):4898–903. doi: 10.1073/pnas.1119535109. PMCID: 3323967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hellström M, Kalén M, Lindahl P, Abramsson A, Betsholtz C. Role of PDGF-B and PDGFR-beta in recruitment of vascular smooth muscle cells and pericytes during embryonic blood vessel formation in the mouse. Development. 1999;126(14):3047–55. doi: 10.1242/dev.126.14.3047. [DOI] [PubMed] [Google Scholar]
- 43.Abramsson A, Lindblom P, Betsholtz C. Endothelial and nonendothelial sources of PDGF-B regulate pericyte recruitment and influence vascular pattern formation in tumors. J Clin Invest. 2003;112(8):1142–51. doi: 10.1172/JCI18549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Magnusson PU, Looman C, Ahgren A, Wu Y, Claesson-Welsh L, Heuchel RL. Platelet-derived growth factor receptor-beta constitutive activity promotes angiogenesis in vivo and in vitro. Arterioscler Thromb Vasc Biol. 2007;27(10):2142–9. doi: 10.1161/01.ATV.0000282198.60701.94. [DOI] [PubMed] [Google Scholar]
- 45.Klosowska-Wardega A, Hasumi Y, Burmakin M, Ahgren A, Stuhr L, Moen I, et al. Combined anti-angiogenic therapy targeting PDGF and VEGF receptors lowers the interstitial fluid pressure in a murine experimental carcinoma. PLoS One. 2009;4(12):e8149. doi: 10.1371/journal.pone.0008149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pietras K, Rubin K, Sjöblom T, Buchdunger E, Sjöquist M, Heldin CH, et al. Inhibition of PDGF receptor signaling in tumor stroma enhances antitumor effect of chemotherapy. Cancer Res. 2002;62(19):5476–84. [PubMed] [Google Scholar]
- 47.Heldin CH, Rubin K, Pietras K, Ostman A. High interstitial fluid pressure - an obstacle in cancer therapy. Nature reviews. 2004;4(10):806–13. doi: 10.1038/nrc1456. [DOI] [PubMed] [Google Scholar]
- 48.Pietras K, Hanahan D. A multitargeted, metronomic, and maximum-tolerated dose “chemo-switch” regimen is antiangiogenic, producing objective responses and survival benefit in a mouse model of cancer. J Clin Oncol. 2005;23(5):939–52. doi: 10.1200/JCO.2005.07.093. [DOI] [PubMed] [Google Scholar]
- 49.Li H, Fredriksson L, Li X, Eriksson U. PDGF-D is a potent transforming and angiogenic growth factor. Oncogene. 2003;22(10):1501–10. doi: 10.1038/sj.onc.1206223. [DOI] [PubMed] [Google Scholar]
- 50.Uutela M, Wirzenius M, Paavonen K, Rajantie I, He Y, Karpanen T, et al. PDGF-D induces macrophage recruitment, increased interstitial pressure, and blood vessel maturation during angiogenesis. Blood. 2004;104(10):3198–204. doi: 10.1182/blood-2004-04-1485. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.