

Abstract
Productive engagement of MHC Class I by inhibitory NK cell receptors depends on the peptide bound by the MHC class I molecule. Peptide:MHC complexes that bind weakly to killer cell immunoglobulin-like receptors (KIR) can antagonize the inhibition mediated by high affinity peptide:MHC complexes and cause NK cell activation. We show that low affinity peptide:MHC complexes stall inhibitory signalling at the step of SHP-1 recruitment and do not go on to form the KIR microclusters induced by high affinity peptide:MHC, which are associated with Vav dephosphorylation and downstream signalling. Furthermore the low affinity peptide:MHC complexes prevented the formation of KIR microclusters by high affinity peptide:MHC. Thus peptide antagonism of NK cells is an active phenomenon of inhibitory synapse disruption.
INTRODUCTION
Natural killer (NK) cells are an important component of the innate immune system that provide a rapid immune response through cytokine secretion and direct lysis of stressed, infected, or transformed cells (1). Their functions are controlled by a balance of signals transduced by activating and inhibitory receptors. The inhibitory receptors include Killer Cell Ig–like Receptors (KIR), CD94:NKG2A, the Leukocyte Immunoglobulin-like Receptors (LILR) and NKR-P1. The KIR and CD94:NKG2A receptors have MHC class I ligands. During infection or tumorigenesis MHC class I may be down regulated leading to loss of inhibitory signals (2). KIR specificity for MHC class I is determined by oligomorphic motifs on MHC class I, such as the Bw4 motif for KIR3DL1, or residue 80 for the HLA-C specific inhibitory KIR (3). Additionally these receptors are sensitive to the peptide bound by MHC class I, and so inhibition of NK cells expressing specific KIR may be mediated by only a subset of expressed peptide:MHC complexes (4-9). In particular the inhibitory KIR, KIR2DL2 and KIR2DL3 recognise a subset of HLA-C allotypes with an asparagine at position 80 and binding of these receptors to HLA-C is modulated by residues 7 and 8 of the bound peptide. In general, large hydrophobic residues are permissive at P7 and small residues permissive at P8 (7, 10). We have recently shown that a peptide variant, which by itself does not inhibit KIR2DL2/3-positive NK cells can antagonise the inhibition due to a peptide that strongly inhibits NK cells, as opposed to being functionally neutral (10). This suggests that NK cells could be sensitive to small changes in peptide repertoire, in addition to MHC class I down-regulation.
Following engagement of cognate MHC class I on a target cell the KIR form microclusters at the inhibitory immune synapse (11). Inhibitory signalling by KIR is subsequently determined by the presence of Immunoreceptor Tyrosine-based Inhibitory Motifs (ITIMs; V/I/LxYxxL/V) in their cytoplasmic tails. Phosphorylation of these ITIMs leads to recruitment of Src homology protein tyrosine phosphatase (SHP) 1 or 2 (12-15). SHP-1/2 dephosphorylates Vav-1 and leads to a block in membrane-proximal NK cell activation signals (16). This block is thought to precede actin cytoskeletal rearrangement (17, 18). Recent work has shown that the activating receptors 2B4 and CD2 can colocalise at inhibitory synapses with inhibitory KIR indicating that inhibitory signals do not prevent recruitment of at least some activating receptors to the immune synapse (19), although there is evidence that they may alter the membrane organisation of some receptors such as NKG2D (17). Our previous work has shown that an antagonist peptide bound to MHC class I can recruit inhibitory KIR to the contact area between effector and target cell but does not induce inhibitory signalling (10). Thus inhibitory signalling can be fine-tuned by the peptide:MHC complexes presented to NK cells. Here we set out to investigate the mechanism by which KIR engaged by antagonist peptide:MHC complexes interferes with inhibitory signalling.
Materials and Methods
Cell Lines and Culture
We used, as target cells, a TAP deficient cell line 721.174 (20), which was pulsed exogenously at 26°C with VAPWNSFAL (FA), VAPWNSDAL (DA) or an equal mix of both peptides (Peptide Protein Research, Hampshire, UK). NKL cells, which lack KIR expression (with the exception of KIR2DL4), have been transfected with a functional KIR2DL3 (NKL-2DL3) or an ITIM mutated KIR2DL3 (NKL-2DL3.2YF), both conjugated to eGFP. In the ITIM mutated KIR2DL3, tyrosines in position 282 and 312 (Y282 & Y312) have been replaced by a phenylalanine. The KIR2DL3-GFP fusion constructs were generated by subcloning RT-PCR amplified cDNAs encoding KIR2DL3 into plasmid pcDNA3.1 (Invitrogen, Life technologies Ltd, Paisley, UK) already containing the eGFP sequence. Substitution of KIR2DL3 residues Tyr282 and Tyr312 with phenylalanine was achieved by sequential site-directed mutagenesis by polymerase chain reaction (PCR) using the Expand High Fidelity PCR System and dNTPack (Roche Diagnostics Ltd, Burgess Hill, UK). For expression in NKL, KIR2DL3-GFP was cloned into retroviral vector pIB2 and the Phoenix packaging cell line was used to produce retroviral particles.
721.174 was cultured in R10 medium (RPMI 1640 medium supplemented with 1% penicillin/streptomycin (Invitrogen) and 10% FBS (Globepharm, Guildford, UK)) while the NK cell lines, NKL, NKL-2DL3 and NKL-2DL3.2YF were cultured in R10 medium supplemented with 100IU recombinant IL-2 (gift from National Cancer Institute Biometric Research Branch).
CD107a assay
Peripheral blood mononuclear cells (PBMCs), isolated using Hypaque-Ficoll density centrifugation, were stimulated overnight with 1ng/mL rHuIL-15 (R&D Systems, Abingdon UK). 721.174 cells were loaded with peptide at 26°C overnight and re-suspended with the PBMCs at an effector-to-target (E:T) ratio of 5:1 in fresh R10 medium containing peptides and CD107a-Alexa Fluor 647 mAb (ebioscience Ltd, Hatfield UK). Cells were incubated at 26 °C for 4hrs with 6μg/mL Golgi-Stop (BD Biosciences, Oxford UK) added after 1hr-incubation. Cells were stained with CD3-PerCP (Biolegend UK Ltd, Cambridge UK), CD56-PE and CD158b-FITC (anti-KIR2DL2/3 & 2DS2, BD Biosciences) then analyzed on a BD Accuri C6 flow cytometer with BD CFlow® Software (BD Biosciences).
Immunoprecipitation and Western Blotting
721.174 cells were cultured with 20μM peptide overnight at 26°C. NKL-2DL3 and 721.174 cells were co-incubated at a 1:1 Effector:Target ratio for 5 min at 37 °C then resuspended in cell lysis buffer (20 mM Tris-HCl, pH 7.6, 150 mM NaCl, 1 mM EDTA, 1 mM sodium orthovanadate, and 0.5% Triton X-100). Total cellular proteins or proteins immunoprecipitated with CD158b (clone GL183, AbD Serotec, Oxford, UK) were subjected to SDS-PAGE and analyzed by Western blotting. Antibodies recognizing phospho-Vav1 (Y174, clone EP510Y, Abcam, Cambridge, UK), Vav1 (Cell Signalling Technology, Hitchin, UK), phospho-p42/p44 MAPK (T202/Y204, clone 107G2, CST), SHP-1 (clone C14H6, CST), CD158b (clone GL183, AbD Serotec) and β-actin (Cambridge Bioscience, Cambridge, UK) were used with HRP-conjugated secondary Abs (Millipore, Watford, UK). Membranes were stripped using the Western Blot Recycling Kit (Alpha Diagnostics, San Antonio, Tx). Protein bands were detected by chemiluminescence (Supersignal Westpico Chemiluminescent Substrate; Perbio Science, Cramlingotn, UK) using the ChemiDoc-It Imaging System with Vision Works software (UVP) and quantified with ImageJ software (National Institutes of Health).
Contact time
721.174 cells were cultured with 10μM peptide overnight at 26°C. NKL cells were stained with 1μM DiO (Vybrant DiO; Invitrogen) in RPMI medium for 30 min at 37°C then washed and re-suspended in R10 medium. 721.174 was mixed at a 2:1 (E:T) with either DiO-stained NKL, NKL-2DL3 or NKL-2DL3.2YF in one well of a chambered coverslide. Live cells were imaged at 37°C, 5% CO2, with R10 as the imaging medium by resonance scanning confocal microscopy with laser lines of 488 nm and a 63× 1.2 numerical aperture (NA) water immersion objective (TCS SP5 RS, Leica Microsystems, Milton Keynes, UK). Images were acquired (every 5 sec) with Leica Application Suite Advanced Fluorescence software and analyzed with Leica confocal software (both from Leica).
KIR2DL3 accumulation
721.174 cells were cultured with 10μM peptide overnight at 26°C, then co-incubated at an 2:1 (E:T) ratio with either NKL-2DL3 or NKL-2DL3.2YF for 10 min at 37 °C. Conjugates were fixed in 2% paraformaldehyde for 30 min at 37 °C and imaged by resonance scanning confocal microscopy with laser lines of 488 nm and a 63× 1.2 numerical aperture (NA) oil immersion objective (TCS SP2 RS, Leica). Images were acquired with Leica Application Suite Advanced Fluorescence (Leica) and analyzed with ImageJ (NIH) software. The increase in fluorescence intensity at the immune synapse was calculated as a ratio of the average fluorescence intensity along the NKL-721.174 interface compared with the average fluorescence intensity along the NKL plasma membrane not in contact with another cell, with both values corrected for background fluorescence as measured within an empty region of the image.
Statistical Analysis
All statistical analyses were performed using GraphPad Prism, version 5 (GraphPad Software, La Jolla, CA).
Results
An antagonistic peptide disrupts inhibitory signalling
We have previously shown that a variant of the naturally processed HLA-Cw*0102-restricted noname peptide VAPWNSFAL (FA) is a strong inhibitor of KIR2DL3+ primary NK cells and that a second variant VAPWNSDAL (DA) acts as a peptide antagonist for this subset (10). On KIR2DL3+ primary NK cells, the increase of membrane CD107a expression (a marker of degranulation) due to HLA-Cw*0102 positive, TAP-deficient 721.174 B cells (.174) is lost in the presence of FA (FA 174, Figure 1A, 1B). In contrast, DA doesn’t modify membrane CD107a expression (DA 174) and further prevents the FA-induced decrease (DA:FA 174). To understand how these peptides affect NK cell signal transduction we analysed the responses of NKL cells transfected with KIR2DL3 (NKL-2DL3) to .174 cells, presenting FA and DA peptides on HLA-Cw*0102 (20). Because NKL constitutively express membrane CD107a, we could not use surface expression of this molecule as a readout of NKL cell activity (Figure S1). We therefore measured the duration of intercellular contacts as a marker of productive engagement between NKL and .174 target cells by live cell imaging, as previous work has established that contact times are shorter if there is a productive inhibitory interaction (21). There was no significant difference in the contact times between untransfected NKL cells and .174 cells in the presence or absence of peptide (Figure 1C). In contrast, for NKL-2DL3 the strong KIR binding peptide FA dramatically reduced the contact time between NKL-2DL3 and 721.174 cells compared to no peptide or the antagonist peptide DA (from 279 ±29sec to 169 ±13sec, Figure 1D). This decrease was abrogated by using an equimolar mix of DA and FA, consistent with our previous data showing that DA can antagonize the inhibition due to FA . Moreover these differences in contact time were dependent on inhibitory signalling as they were abrogated in the NKL cell line transfected with a KIR2DL3 construct in which the tyrosine residues within the two ITIMs had been mutated to phenylalanine (NKL-2DL3.2YF) (Figure 1E).
Figure 1. The DA peptide anatagonises the inhibition of NK cells due to the FA peptide.

(A, B) CD107a assay gated on CD3-CD56+ NK cells using 721.174 either unloaded (No Pep) or loaded with the indicated peptides. A dot plot from one representative experiment is shown in (A) and the mean value ±SEM of three experiments in (B). (C-E) Unpulsed 721.174 cells (No Pep) or cells loaded with FA, DA, or both peptides at an equimolar ratio (DA/FA) were used as target cells for either NKL (C), NKL-2DL3 (D) or NKL-2DL3.2YF (E) cells in live-cell imaging experiments. Movies were recorded for 40min in a 37°C-chamber by confocal microscopy. The contact times of between 36 to 63 conjugates were determined and plotted for each peptide condition. The mean contact time in seconds ± SEM is shown (gray bars). Data were analysed using a one-way ANOVA and a Newman-Keul’s post-test for multiple comparisons. For all panels significant p values are indicated as follows: *p <0.05; **p < 0.01; *** p < 0.001.
As Vav1 is the primary substrate of SHP-1 we investigated the effects of the DA/FA mix on inhibitory signalling. Consistent with our observations on contact time, FA alone induced dephosphorylation of the SHP-1 substrate Vav1 while the addition of DA to FA restored Vav1 phosphorylation (Figure 2A, 2B). Similarly, downstream signalling through Erk1 and Erk2, required for actin reorganisation and NK cell degranulation (22, 23), is reduced in response to FA alone, but restored in the agonist:antagonist FA:DA peptide mix (Figure 2C, 2D). Furthermore titration of the peptide concentrations within the agonist:antagonist mix resulted in significantly greater Vav1 phosphorylation in the presence of DA, than in its absence (Figure 3A, 3B). Overall we found that in the presence of small amounts of FA peptide there was a substantial decrease in Vav1 phosphorylation which was signifcantly decreased in the presence of the DA peptide (Figure 3C). These data are similar to those observed for titrations of the same peptides in CD107a assays of primary NK cells (10). In those experiments in the in the presence of the antagonist peptide, there is a linear decrease in Vav1 phosphorylation which was not observed for the single peptide. Similarly in these experiments linear regression analysis of the data in Figure 3C demonstrated that there was a linear decrease in Vav1 phosphorylation for the peptide mix experiments (r2=0.94), but not for the single FA peptide experiments (r2=0.67), for which a one phase decay curve provided a superior fit (r2=0.97) (Figure S2).
Figure 2. A peptide antagonist abrogates inhibitory signalling.

Analysis of cell lysates for Vav1 and Erk signalling by western blotting. 721.174 incubated without exogenous peptide (No Pep, lane 2) or 721.174 loaded with DA (DA, lane 3), FA (FA, lane 4) or both peptides (DA/FA, lane 5) were incubated with NKL-2DL3 for 5min at 37°C. Cell lysates were analysed by western blot for pVav1 (A, B) and pErk1/2 (C, D) phosphorylation and also for Vav1 (A, bottom blot) and β-actin (C, bottom blot). Lysates of NKL-2DL3 (lane 1) and 721.174 (lane 6) alone were used to determined the basal level of phosphorylation in the two cell lines. Blots from one representative experiment are shown for Vav1 (A) and Erk1/2 (C). Data from 3 independent experiments (Vav1, B) or 8 independent experiments (Erk1/2, D) were used to calculate the ratio of phosphorylation (p-Vav1/ Vav1, p-Erk1/ β-actin or p-Erk2/ β-actin) using ImageJ software and plotted as mean value ±SEM. The ratio of NKL-L3 (lane 1) and 721.174 (lane 6) are shown to demonstrate the basal ratios of phosphorylation in the cell lines (B & D). For all panels significant p values are indicated by *p <0.05. Data were analysed using a one-way ANOVA and a Newman-Keul’s post-test for multiple comparisons. For all panels significant p values are indicated as follows: *p <0.05.
Figure 3. Comparison of inhibition of Vav1 pohosphorylation in the presence or absence of antagonist peptide.
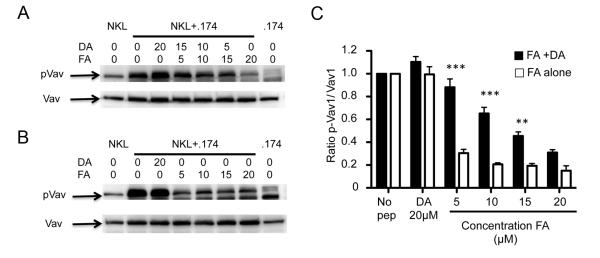
Analysis of cell lysates for Vav1 by western blotting. 721.174 cells were incubated without exogenous peptide, or in the presence of various concentrations of the inhibitory peptide (FA) in the presence (A) or absence (B) of the antagonist peptide DA and the lysates assayed for Vav1 phosphorylation. The peptide concentrations for each condition in μM are indicated. The mean and SEMs of three independent experiments, normalised to Vav1 phosphorylation in the absence of peptide, are shown in (C). For the FA+DA columns, the DA peptide was added to make the total the total peptide concentration 20μM as indicated in panel (A) lanes 2-7, and for the FA alone columns peptide concentrations are as in panel (B) lanes 2-7. Data at each concentration were compared using a one-way ANOVA and a Newman-Keul’s post-test for multiple comparisons. Significantly different levels of Vav1 phosphorylation between FA alone and FA+DA at the same concentrations of the FA peptides are indicated with *** (p<0.001) and ** (p<0.01). Other comparisons were non-significant.
An antagonistic peptide disrupts tight clustering of KIR at the inhibitory synapse
To investigate how DA might affect the initiation of inhibitory signalling, we compared the inhibitory synapses formed in the presence of DA, FA or the FA:DA peptide mix. We used NKL cells expressing KIR2DL3 C-terminally tagged with eGFP to determine the distribution of KIR2DL3 at the immune synapse between target and effector cells. The increase in intensity of eGFP fluorescence at the contact area as compared to a non-contact area was then determined. The addition of FA, DA, or the DA:FA mix induced similar levels of KIR2DL3 accumulation at the contact area between NK cells and .174 targets (Figure 4A: 1.58 ±0.04, 1.54 ±0.04 and 1.55 ±0.03 fold respectively). A cut-off of 1.5 times background (mean of control peptide±3 SEMs) was defined as KIR accumulation at the immune synapse that was significantly above random fluctuations of fluorescence within the membrane. Differences in the percentage of conjugates demonstrating a ≥1.5 fold increase in KIR at the immune synapse were not significantly different between FA, DA, and the DA:FA mix (Figure 4B). Therefore the amount of KIR recruited to the immune synapse was similar under inhibitory or antagonistic conditions.
Figure 4. An antagonistic peptide disrupts tight clustering of KIR2DL3 at the inhibitory synapse.

(A-E) Unpulsed 721.221 cells (No Pep) or cells loaded with control peptide (GILG), FA, DA, or both DA and FA (DA/FA) peptides were used as target cells for NKL-2DL3. After a 10 minute co-incubation at 37°C, the conjugates were fixed and imaged by confocal microscopy. (A) The GFP intensity of 86 to 116 conjugates from 3 independent experiments was quantified for each peptide condition. KIR2DL3 accumulation was determined by comparing the intensity at the interface between the effector and target cells, with that of the NKL-2DL3 plasma membrane at a noncontact area and the data plotted as a fold increase in intensity. The mean fold increase in intensity ±SEM is shown in red bars. Data were analysed using one-way ANOVA and Newman-Keul’s post test to compare individual groups. No significant differences were noted between the FA, DA, and DA/FA conditions. (B) Using the same experiments as for (A) the percentage of conjugates showing a KIR2DL3 accumulation with a fold increase ≥ at 1.5 was determined. Data were analysed by Student’s T-test between GILG and other conditions. (C) Confocal microscopy image of 721.174 and NKL-2DL3 conjugates. 721.174 cells were loaded with FA, DA or both peptides at an equimolar ratio (DA/FA). The 3D projection of the interface between NKL-2DL3 and 721.174 is labelled with a pseudo-colour scale and the arrows show the plane used for the plot profile. On the plot profile, the grey area shows the area with a KIR2DL3 accumulation greater than 1.5 times background. (D, E) Analysis of microclusters at the inhibitory synapse. The experiments from (A) were reanalyzed to determine the density of KIR2DL3 accumulation within the area of more than 1.5 times KIR2DL3 accumulation (gray area in (C)). The intensity of KIR2DL3 accumulation area within this region was compared with that of NKL-2DL3 plasma membrane at a noncontact area (D). The means ±SEM are shown in red, and statistical analysis performed as for (A). Significant values for FA versus other conditions are shown. No significant difference was observed between the DA and DA/FA conditions. (E) The percentage of conjugates showing a density of KIR2DL3 accumulation with a fold increase ≥ at 2.5 plotted as mean value ± SEM. FA was compared with the other conditions using a Student’s T-test. For all panels significant p values are indicated as follows: *p <0.05; **p < 0.01; *** p < 0.001.
However, 3D-reconstruction of the contact areas showed that FA and DA induced the formation of differently structured synapses. FA induced the formation of very tight and bright clusters whilst DA induced a more diffuse accumulation lacking large areas of intense clustering (Figure 4C). Moreover, synapses formed when the FA:DA peptide mix was presented demonstrated a diffuse clustering of KIR, similar to the synaptic patterning induced by DA alone. To determine the density of KIR2DL3 accumulation at the inhibitory synapse we measured the fluorescence intensity within the area of detectable KIR2DL3 clustering (gray area in the profile plots), as opposed to the total contact area between NKL and .174 cells (Figure 4C). The density of KIR2DL3 was higher in the presence of FA (Figure 4D) and showed that only FA induced tight clustering of KIR as defined by a fold increase ≥2.5 between the fluorescence intensity within the synapse compared to the non-contact area (Figure 4E). These tight clusters are similar in dimension to the KIR-based inhibitory signalling microclusters described by Treanor et al (11). Microcluster formation was present in 35 ±7% of FA conjugates as compared to 7 ±4% of DA conjugates and 7 ±6% in the FA:DA mix. Thus DA prevents the formation of microclusters containing tightly clustered KIR2DL3 which are associated with productive inhibitory signalling. Peptide antagonism therefore influences inhibitory synapse organization.
Peptide antagonism requires recruitment of SHP-1
To determine the mechanism which leads to the formation of tight microclusters of KIR2DL3 we investigated if the KIR2DL3-associated inhibitory signal transduction machinery was involved with this process. Images of conjugates formed between NKL-2DL3.2YF and .174 demonstrated that in the absence of functional ITIMs, KIR2DL3 accumulated diffusely at the interface between the two cells and did not form tight clusters, indicating that KIR association with ITIM-mediated signalling components is critical for tight cluster formation. However, immunoprecipitation of KIR2DL3 demonstrated that each of the stimulating conditions (DA, FA or the FA:DA mix) lead to similar levels of SHP-1 recruitment to KIR2DL3 (Figure 5A). Thus, although the association of SHP-1 with KIR2DL3 is necessary for the formation of tight KIR clusters, simply recruiting this phosphatase to KIR2DL3 does not define the critical step within the process of tight cluster formation.
Figure 5. Tight clustering of KIR2DL3 requires inhibitory signalling.

(A) 721.174 cells either unpulsed or loaded with the indicated peptides were incubated with NKL-2DL3 cells. Cell lysates were immunoprecipitated using an anti-KIR2DL3 mAb then analysed by western blot for SHP-1 (top blot) and KIR2DL3 (bottom blot). One representative experiment out of 2 is shown. (B-E) 721.174 cells either unpulsed or loaded with the indicated peptides were co-incubated with NKL-2DL3.2YF cells. After 10min co-culture at 37°C, the conjugates were fixed and imaged by confocal microscopy (B). The 3D projection of the interface between NKL-2DL3.2YF and 721.174 is labelled with a pseudo-colour scale. (C) The GFP intensity of 48 to 71 conjugates from 3 independent experiments was quantified for each peptide condition. The KIR2DL3 accumulation was determined by comparing the intensity of GFP at the interface between 721.174 and NKL-2DL3.2YF and that of the NKL-2DL3.2YF plasma membrane at a noncontact area and plotted as a fold increase in intensity. The mean of fold increase in intensity ±SEM are shown in red. Data were analysed using one-way ANOVA and Newman-Keul’s post test to compare individual groups. Only significant p values are indicated and comparisons between GILG, FA DA and DA/FA were not significant (D, E) The percentage of conjugates showing KIR2DL3.2YF accumulation with a fold increase at ≥1.5 (D) and at ≥2.5 (E) was determined plotted as mean value ±SEM. Data were analysed by Student’s T-test to show significant differences between FA and other conditions. For all panels significant p values are indicated as follows: *p <0.05; **p < 0.01; *** p < 0.001.
In the absence of functional ITIMs most conjugates showed a diffuse accumulation of KIR2DL3 (Figure 5B, 5C). However the levels of KIR2DL3 accumulation at the contact area between NK cells and .174 targets and the frequency of clustering (number of conjugates showing fold increase >1.5) observed with both FA (1.7 ±0.06 fold and 69.5 ±0.5% of conjugates) and also the FA:DA mix (1.7 ±0.05 fold and 62.0 ±6.5% of conjugates), was higher than with DA alone (1.4 ±0.05 fold and 41.7 ±6.9% of conjugates) (Figure 5D). Furthermore although the number of conjugates with tight clustering of KIR2DL3 at the synapse (fold increase >2.5) was low for FA (12.0 ±3.0% of conjugates), and also for FA:DA (6.8 ±2.4% of conjugates), these were both higher than for DA alone (0.0 ±0.0% of conjugates)(Figure 5E). Thus in the absence of ITIMs the FA:DA peptide mix behaves more like FA alone, whereas in their presence the mix behaves more like the DA peptide alone. Thus peptide:MHC complexes with low affinity for KIR do not disrupt the passive association of high affinity complexes with KIR, indicating that peptide antagonism is an active process of signal disruption.
Discussion
Peptide antagonism is a mechanism by which NK cells can be released from inhibition in the absence of MHC class I down-regulation. The peptides VAPWNSFAL (FA) and VAPWNSDAL (DA) both bind HLA-Cw*0102 with similar affinities, but whilst FA is strongly inhibitory, DA does not inhibit NK cells expressing KIR2DL2 or KIR2DL3. In binding assays using KIR-Fc fusion constructs, DA induces no discernible binding of KIR-Fc fusion constructs to HLA-Cw*0102 (10). Despite this apparent lack of function the DA peptide can induce clustering of KIR2DL3 to the inhibitory synapse. This is most likely because the avidity of the KIR:peptide:MHC interaction is greater at the synapse between two cells as compared to that measured in solution. This has recently been demonstrated for TCR:peptide:MHC class I interaction in which the Kd of the interaction between the TCR and the peptide:MHC class I complex was up to 8 times lower as measured by TIRF microscopy at an in vitro “synapse” as compared to a solution measurement (24). Nevertheless, although the DA peptide can recruit KIR and SHP-1 to the immune synapse, it was only the high affinity peptide FA that induced inhibition of signalling as determined by a reduction in NK:target cell contact time and also in terms of Vav1 dephosphorylation. In this system inhibitory signalling correlated directly with the ability to form tight inhibitory micro-clusters (11), and only the FA peptide induced the formation of these structures. The .174 cells express low levels of Cw*0102 on the cell surface in the absence of exogenous peptide (20). In the absence of TAP it most likely binds low affinity peptides as has been found for the other alleles expressed by this cell line, HLA-A*0201 and HLA-B*5101 (25). These HLA-C:peptide complexes do not recruit KIR to the immune synapse, nor induce SHP-1 recruitment to KIR, presumably as they are unstable. However HLA-C:DA complexes, which have a low avidity for KIR are sufficient to perform both of these functions, and thus this peptide uncouples SHP-1 recruitment from inhibitory signalling.
The use of the two peptides FA and DA separate early synaptic events (recruitment of KIR to the interface and SHP-1 recruitment) from later events (microcluster formation and inhibitory signalling). The DA peptide induced only early synaptic events and prevented the formation of microclusters related to FA. Thus peptide antagonism operates at the early stage of synapse formation. In the absence of ITIMs, and hence SHP-1 recruitment, the peptide mix behaved more like the FA peptide. However, in the presence of intact ITIMs the mix behaves more like the DA peptide. In the absence of ITIMs the patterning of KIR at the immune synapse is most likely due to the passive recruitment by peptide:MHC complexes and hence is determined by the affinity of the peptide:MHC complex for KIR. Thus the DA peptide:MHC complexes did not alter recruitment of KIR-2DL3.2YF by the FA complexes to the synapse.
KIR were originally shown to be recruited to the interface between effector and target cells in the absence of actin polarization, and adhesion molecules, implying that formation of the inhibitory synapse is a passive process (17, 26). However, recent work has demonstrated that formation of inhibitory synapses is more complex. In addition to recruiting SHP-1 and SHP-2, KIR can also bind to β-arrestin 2 and the F-actin binding protein supervillin (27, 28). These molecules are reported to facilitate SHP-1 recruitment to KIR, and may assist in the recruitment of KIR to signalling scaffolds. Additionally, KIR ligation leads to the phosphorylation of Crk indicating that KIR can have a positive signalling effect. Moreover, the activating receptor 2B4 has been found to co-localise with KIR at the immune synapse. Taken together these data show that inhibitory signalling is a multi-step process. Recruitment of SHP-1 to KIR may stabilize the low affinity interaction between KIR2DL3 and DA:MHC, but not to the extent that it permits triggering of tight cluster formation. In our experiments mutation of the KIR ITIMs, prevented microcluster formation, implying that there is a requirement for SHP association and that microcluster formation is an active process. It may be related to a co-association of KIR with activating signalling complexes, which has been suggested to be one explanation for the association of KIR with phosphotyrosine moieties in micro-clusters and is consistent with the co-localisation of KIR and 2B4 at the immune synapse (19, 29).
Our data help to dissect out this multi-step process in that: passive recruitment of KIR to the immune synapse can occur without SHP-1 recruitment; DA can recruit SHP-1 but not induce inhibitory micro-cluster formation; and productive inhibitory signalling requires both high affinity peptide:MHC:KIR interactions and ITIM signalling. This is consistent with a threshold for inhibition as described by Almeida et al, and hence also for micro-cluster formation (30). In this model a threshold level of high-affinity peptide:MHC complexes are needed to engage KIR and bind SHP-1 and this threshold may not be reached if SHP-1 is also sequestered by low affinity peptide:MHC class I complexes.
Peptide antagonism is a novel mechanism for releasing NK cells from dominant inhibitory signals, and is a feature of peptide:MHC class I complexes with a low affinity for KIR. This effect, and its in vivo significance, will require testing in more physiological systems of viral infection and tumours. Our observation of altered KIR synapse patterning in the presence of an agonist:anatagonist peptide pairing differs from observation of T cell receptor antagonists. In this system there is a decrease in MHC class I recruitment to the central supramolecular activation complex (cSMAC), but formation of the cSMAC is not abrogated (31). Thus the feature of synaptic patterning is not shared between these two agonist:antagonist systems, consistent with there being distinct mechanisms of peptide antagonism in immunocytes. Our data suggest that KIR provide NK cells with the ability to scan the peptide repertoire of a target cell and if the critical threshold of high affinity peptide:MHC complexes is not reached then the cell will be released from inhibition. Peptide antagonists can thus fine tune the inhibition of NK cells.
Supplementary Material
Acknowledgements
We would like to thank Dr Hans Schuppe for his assistance in confocal microscopy experiments.
This work was supported by The Wellcome Trust and The Medical Research Council, UK
Abbreviations
- KIR
Killer Cell Immunoglobulin-like Receptors
- SHP-1
Src Homology Protein Tyrosine Phosphatase
- ITIM
Immunoreceptor Tyrosine-based Inhibitory Motifs
- TIMP
Tissue Inhibitor of Metalloproteinase
References
- 1.Lanier LL. NK Cell Recognition. Annu. Rev. Immunol. 2005;23:225–274. doi: 10.1146/annurev.immunol.23.021704.115526. [DOI] [PubMed] [Google Scholar]
- 2.Raulet DH, Vance RE. Self-tolerance of natural killer cells. Nat Rev Immunol. 2006;6:520–531. doi: 10.1038/nri1863. [DOI] [PubMed] [Google Scholar]
- 3.Vilches C, Parham P. KIR: diverse, rapidly evolving receptors of innate and adaptive immunity. Annu. Rev. Immunol. 2002;20:217–251. doi: 10.1146/annurev.immunol.20.092501.134942. [DOI] [PubMed] [Google Scholar]
- 4.Malnati MS, Peruzzi M, Parker KC, Biddison WE, Ciccone E, Moretta A, Long EO. Peptide specificity in the recognition of MHC class I by natural killer cell clones. Science. 1995;267:1016–1018. doi: 10.1126/science.7863326. [DOI] [PubMed] [Google Scholar]
- 5.Rajagopalan S, Long EO. The direct binding of a p58 killer cell inhibitory receptor to human histocompatibility leukocyte antigen (HLA)-Cw4 exhibits peptide selectivity. J. Exp. Med. 1997;185:1523–1528. doi: 10.1084/jem.185.8.1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mandelboim O, Wilson SB, Vales-Gomez M, Reyburn HT, Strominger JL. Self and viral peptides can initiate lysis by autologous natural killer cells. Proc. Natl. Acad. Sci. U. S. A. 1997;94:4604–4609. doi: 10.1073/pnas.94.9.4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boyington JC, Motyka SA, Schuck P, Brooks AG, Sun PD. Crystal structure of an NK cell immunoglobulin-like receptor in complex with its class I MHC ligand. Nature. 2000;405:537–543. doi: 10.1038/35014520. [DOI] [PubMed] [Google Scholar]
- 8.Hansasuta P, Dong T, Thananchai H, Weekes M, Willberg C, Aldemir H, Rowland-Jones S, Braud VM. Recognition of HLA-A3 and HLA-A11 by KIR3DL2 is peptide-specific. Eur. J. Immunol. 2004;34:1673–1679. doi: 10.1002/eji.200425089. [DOI] [PubMed] [Google Scholar]
- 9.Vivian JP, Duncan RC, Berry R, O’Connor GM, Reid HH, Beddoe T, Gras S, Saunders PM, Olshina MA, Widjaja JM, Harpur CM, Lin J, Maloveste SM, Price DA, Lafont BA, McVicar DW, Clements CS, Brooks AG, Rossjohn J. Killer cell immunoglobulin-like receptor 3DL1-mediated recognition of human leukocyte antigen B. Nature. 2011;479:401–405. doi: 10.1038/nature10517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fadda L, Borhis G, Ahmed P, Cheent K, Pageon SV, Cazaly A, Stathopoulos S, Middleton D, Mulder A, Claas FH, Elliott T, Davis DM, Purbhoo MA, Khakoo SI. Peptide antagonism as a mechanism for NK cell activation. Proc. Natl. Acad. Sci. U. S. A. 2010;107:10160–10165. doi: 10.1073/pnas.0913745107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Treanor B, Lanigan PM, Kumar S, Dunsby C, Munro I, Auksorius E, Culley FJ, Purbhoo MA, Phillips D, Neil MA, Burshtyn DN, French PM, Davis DM. Microclusters of inhibitory killer immunoglobulin-like receptor signaling at natural killer cell immunological synapses. J Cell Biol. 2006;174:153–161. doi: 10.1083/jcb.200601108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Burshtyn DN, Scharenberg AM, Wagtmann N, Rajagopalan S, Berrada K, Yi T, Kinet JP, Long EO. Recruitment of tyrosine phosphatase HCP by the killer cell inhibitor receptor. Immunity. 1996;4:77–85. doi: 10.1016/s1074-7613(00)80300-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Olcese L, Lang P, Vely F, Cambiaggi A, Marguet D, Blery M, Hippen KL, Biassoni R, Moretta A, Moretta L, Cambier JC, Vivier E. Human and mouse killer-cell inhibitory receptors recruit PTP1C and PTP1D protein tyrosine phosphatases. J. Immunol. 1996;156:4531–4534. [PubMed] [Google Scholar]
- 14.Burshtyn DN, Lam AS, Weston M, Gupta N, Warmerdam PA, Long EO. Conserved residues amino-terminal of cytoplasmic tyrosines contribute to the SHP-1-mediated inhibitory function of killer cell Ig-like receptors. J. Immunol. 1999;162:897–902. [PubMed] [Google Scholar]
- 15.Fry AM, Lanier LL, Weiss A. Phosphotyrosines in the killer cell inhibitory receptor motif of NKB1 are required for negative signaling and for association with protein tyrosine phosphatase 1C. J. Exp. Med. 1996;184:295–300. doi: 10.1084/jem.184.1.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stebbins CC, Watzl C, Billadeau DD, Leibson PJ, Burshtyn DN, Long EO. Vav1 dephosphorylation by the tyrosine phosphatase SHP-1 as a mechanism for inhibition of cellular cytotoxicity. Mol Cell Biol. 2003;23:6291–6299. doi: 10.1128/MCB.23.17.6291-6299.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Davis DM, Chiu I, Fassett M, Cohen GB, Mandelboim O, Strominger JL. The human natural killer cell immune synapse. Proc. Natl. Acad. Sci. U. S. A. 1999;96:15062–15067. doi: 10.1073/pnas.96.26.15062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Long EO. Negative signaling by inhibitory receptors: the NK cell paradigm. Immunol Rev. 2008;224:70–84. doi: 10.1111/j.1600-065X.2008.00660.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schleinitz N, March ME, Long EO. Recruitment of activation receptors at inhibitory NK cell immune synapses. PLoS ONE. 2008;3:e3278. doi: 10.1371/journal.pone.0003278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Young NT, Mulder A, Cerundolo V, Claas FH, Welsh KI. Expression of HLA class I antigens in transporter associated with antigen processing (TAP)-deficient mutant cell lines. Tissue Antigens. 1998;52:368–373. doi: 10.1111/j.1399-0039.1998.tb03057.x. [DOI] [PubMed] [Google Scholar]
- 21.Culley FJ, Johnson M, Evans JH, Kumar S, Crilly R, Casasbuenas J, Schnyder T, Mehrabi M, Deonarain MP, Ushakov DS, Braud V, Roth G, Brock R, Kohler K, Davis DM. Natural killer cell signal integration balances synapse symmetry and migration. PLoS Biol. 2009;7:e1000159. doi: 10.1371/journal.pbio.1000159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Djeu JY, Jiang K, Wei S. A view to a kill: signals triggering cytotoxicity. Clin Cancer Res. 2002;8:636–640. [PubMed] [Google Scholar]
- 23.Chen X, Allan DS, Krzewski K, Ge B, Kopcow H, Strominger JL. CD28-stimulated ERK2 phosphorylation is required for polarization of the microtubule organizing center and granules in YTS NK cells. Proc. Natl. Acad. Sci. U. S. A. 2006;103:10346–10351. doi: 10.1073/pnas.0604236103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huppa JB, Axmann M, Mortelmaier MA, Lillemeier BF, Newell EW, Brameshuber M, Klein LO, Schutz GJ, Davis MM. TCR-peptide-MHC interactions in situ show accelerated kinetics and increased affinity. Nature. 2010;463:963–967. doi: 10.1038/nature08746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Weinzierl AO, Rudolf D, Hillen N, Tenzer S, van Endert P, Schild H, Rammensee HG, Stevanovic S. Features of TAP-independent MHC class I ligands revealed by quantitative mass spectrometry. Eur. J. Immunol. 2008;38:1503–1510. doi: 10.1002/eji.200838136. [DOI] [PubMed] [Google Scholar]
- 26.Faure M, Barber DF, Takahashi SM, Jin T, Long EO. Spontaneous clustering and tyrosine phosphorylation of NK cell inhibitory receptor induced by ligand binding. J. Immunol. 2003;170:6107–6114. doi: 10.4049/jimmunol.170.12.6107. [DOI] [PubMed] [Google Scholar]
- 27.Yu MC, Su LL, Zou L, Liu Y, Wu N, Kong L, Zhuang ZH, Sun L, Liu HP, Hu JH, Li D, Strominger JL, Zang JW, Pei G, Ge BX. An essential function for beta-arrestin 2 in the inhibitory signaling of natural killer cells. Nat. Immunol. 2008;9:898–907. doi: 10.1038/ni.1635. [DOI] [PubMed] [Google Scholar]
- 28.Liu HP, Yu MC, Jiang MH, Chen JX, Yan DP, Liu F, Ge BX. Association of supervillin with KIR2DL1 regulates the inhibitory signaling of natural killer cells. Cell Signal. 2011;23:487–496. doi: 10.1016/j.cellsig.2010.11.001. [DOI] [PubMed] [Google Scholar]
- 29.Peterson ME, Long EO. Inhibitory receptor signaling via tyrosine phosphorylation of the adaptor Crk. Immunity. 2008;29:578–588. doi: 10.1016/j.immuni.2008.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Almeida CR, Davis DM. Segregation of HLA-C from ICAM-1 at NK cell immune synapses is controlled by its cell surface density. J. Immunol. 2006;177:6904–6910. doi: 10.4049/jimmunol.177.10.6904. [DOI] [PubMed] [Google Scholar]
- 31.Sumen C, Dustin ML, Davis MM. T cell receptor antagonism interferes with MHC clustering and integrin patterning during immunological synapse formation. J Cell Biol. 2004;166:579–590. doi: 10.1083/jcb.200404059. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.