

Abstract
Background & Aims
Early reports suggested androgen/androgen receptor (AR) signals promote hepatocarcinogenesis. However, all antiandrogen clinical trials failed in advanced hepatocellular carcinoma (HCC) without reasonable explanations. We have examined AR functions in HCC cancer metastasis in this study.
Methods
We examined hepatic AR roles in HCC metastasis by comparing liver hepatocyte AR knockout and wildtype in carcinogen-induced HCC mouse model. We examined tumor histology, cancer metastatic risks, and cancer survival in vivo, as well as cell anoikis and migration using primary hepatic tumor culture in vitro. We also examined therapeutic potentials of AR expression combined with molecular targeting agent, Sorafenib, in HCC metastasis mouse model.
Results
We found a novel cancer phenotype in which mice lacking hepatic AR developed more undifferentiated tumors and larger tumor size at later metastatic stage. These mice also died earlier with increased lung metastasis, suggesting hepatic-AR may play dual yet opposite roles to promote HCC initiation but suppress HCC metastasis. Mechanistic dissection found that hepatic AR could enhance anoikis and suppress migration of HCC cells via suppression of p38 phosphorylation/activation and the NFκB-MMP9 pathway, respectively. In addition, the in vivo pre-clinical trials concluded that a combination therapy of increased AR expression and reduced multiple-kinase inhibitor (Sorafenib) exhibited better therapeutic efficacy.
Conclusions
Our study demonstrated that AR could orchestrate intrahepatic signalling hierarchies and cellular behavior, consequently affect HCC progression. Results from combination therapy shed a light on developing new therapeutic paradigm for battling HCC at later metastatic stage.
Keywords: Androgen receptor (AR), Hepatocellular carcinoma (HCC), Cancer Metastasis
INTRODUCTORY STATMENT
Hepatocellular carcinoma (HCC) was ranked the 7th cause of cancer death in the U.S and 5th worldwide (10). Androgen and androgen receptor (AR) signals have been suspected to regulate malignant transformation and progression of HCC (11, 12). However, the amount of AR expression during HCC remains inconclusive with reports showing AR is either up- or down-regulated (3, 4, 6, 7, 13-15). Furthermore, clinical studies using anti-androgens had disappointing results with little beneficial effects on patients (1, 16), or even worse survival (16).
Tumor cell capacity to survive in detached environment (circulation) or the ability to invade out of primary liver tumor either homing to distant organs or micrometastasis to neighboring tissue can be critical to the cancer metastasis. The recurrence of HCC, even after hepatic transplantation surgery, could be due to re-homing of circulating HCC cells (17) residing in the vascular system(18). Since AR roles in HCC at later metastatic stage remain unclear, using conditional knockout AR strategy, we examined hepatic AR functions in HCC metastasis.
EXPERIMENTAL PROCEDURES
Patient enrollment
From 2005 to 2010, primary HCC tumors of diameter less than 3 centimeter and metastatic tumors were collected. Detailed patient information is described in the supplementary data. A written informed consent was obtained from these patients. These studies were approved by the Institutional Review Board of Chang Gung Memorial Hospital, and China Medical University Hospital in Taiwan.
Maintenance of animals, generation of L-AR−/y mice, and HCC metastasis
All of the animal experiments followed the Guidance of the Care and Use of Laboratory Animals of the US National Institutes of Health and with approval from the Department of Laboratory Animal Medicine at the University of Rochester Medical Center. The strategy to generate flox-AR gene-targeting mice has been described earlier (4). Briefly, we mated male Alb-Cre (19) (Cre recombinase under control of Albumin promoter; Jackson Lab., B6.Cg-Tg(Alb-cre)21Mgn/J) mice with flox-AR/AR heterozygous (ARflox/X; B6) female mice to produce L-AR−/y males. Each type of transgenic mice expresses flox-AR and Cre alleles in tail genomic DNA. We genotyped 21-day-old pups from tail snips by PCR, as previously described (20). To induce HCC in the mice liver, we injected 12-days old pups with HCC initiator, Nʼ-Nʼ-Diethylnitrosamine (DEN; 20 mg/kg/mice; Sigma-Aldrich) (21). The male DEN-injected mice were sacrificed at 30-, 40-, 50-, and 60-wks of age. The nude mice used for tail vein injection experiments were 6-wks old 20-25 gm male nude mice (Charles River; Crl: CD1-Foxn1nu Origin).
Spontaneous HCC development and Tail vein injection of HCC cells for in vivo metastasis assay and Sorafenib treatments
Carcinogen-induced mice HCC procedure was described in Suppl. text, and in Ma et al. (4). SKAR− and SKAR+ cells, parental and AR stable clone of SKhep1 cells, respectively, were cultured in 150-mm flask, maintained in DMEM with 10% FCS, 1% P/S and 1% NEAA. When the cells reached ~70-80% confluence, they were detached by detaching buffer (0.1 mg/ml trypsin, and 5 mM EDTA), and 2×106 cells/100 μl were injected into the tail veins of 8-wks old athymic nude mice. One month after injection, the mice were treated with/wo Sorafenib (Bayer; 30 mg/kg/mice; daily) for another month. The Sorafenib stock solutions were prepared weekly at 4X by dissolving 0.1g in 4 ml solvent (Cremophor CL:Ethanol=1:1), and stored at -20°C. For injection, we diluted the 4X Sorafenib with distilled H2O.
The experiments were composed of 24 nude mice, which were randomly assigned to four experimental groups, including placebo and Sorafenib treatments in SKAR− cells xenografted mice; placebo and Sorafenib treatments in SKAR+ cells xenografted mice. The dosage of Sorafenib is based on the minimal dosages used in murine models of allograft transplantation. The mice randomized to Sorafenib treatment group were given 30 mg/kg/mice in 100 μL by gavage (orally, daily) starting on day 30 after tumor cells injection. All control mice received an equal volume of carrier solution by gavage. The mice were sacrificed 5 wks after treatment. At necropsy, we observed the visceral organs, and calculated the tumor foci. Both primary tumors and metastatic site tumors were stained for AR and p-p38.
Other materials and methods (including Maintenance of animals, generation of L-AR−/y mice, and HCC metastasis, in vitro cell culture/maintenance, lentiviral-based gene delivery, Reagents, histology, trichrome staining, and immunohistochemistry, Transfection and reporter gene assay, Cell migration, anoikis assays; Statistical analysis) all described in online supplemental materials.
RESULTS
Mice lacking hepatic AR developed an earlier onset of metastatic HCC tumors and low cancer survival rates
An early study suggested that hepatic AR promotes hepatocarcinogenesis during normal hepatocytes transformation and in mice treated with carcinogen-DEN (4). This conflicted with the concepts of clinical trials using anti-androgens to treat HCC patients (16, 22-25). We therefore decided to further dissect hepatic AR roles beyond the HCC initiation stage; especially at HCC later metastatic stage using mouse models similar to those we established earlier (4).
As expected, we found that male mice lacking liver hepatocyte AR (L-AR−/y, LARKO) developed HCC later as compared to wild type littermates (AR+/y, WT), which was consistent with previous studies (4). Yet surprisingly, we found those L-AR−/y mice died earlier compared to AR+/y mice (Fig. 1A). Similar results with lower survival rate also occurred in female LARKO mice (L-AR−/−) as compared to their WT littermates (Fig. 1A, right panel). Measurements of the tumor growth (liver weight/body weight) in these mice found the HCC tumor growth in the WT mice is initially faster as compared to LARKO mice before 36-wks. However, the tumor size was not distinguishable between these two groups at 40-wks, and the trend was even reversed at 50-, and 60-wks (Fig. 1B, left panel).
Figure 1. Loss of AR promotes metastasis with poorer prognosis in DEN-HCC mice.
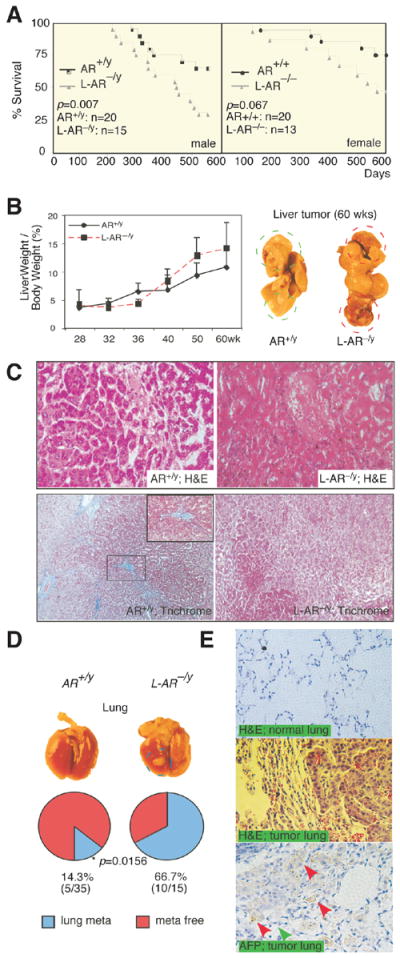
(A) Cancer survival of the male and female ARKO and L-ARKO mice. Mantel-Haenszel log-rank test of ARKO versus L-ARKO HCC mice survival in male (AR+/y and L-AR−/y, left panel) and female (AR+/+ and L-AR−/−, right panel). Comparing ARKO and L-ARKO P= 0.007 while it is 0.067 in female. (B) Liver weight/body weight (LW/BW) of HCC tumors in 28-, 32-, 36-, 40-, 50-, and 60-wks old mice. 28-wks old tumors from each genotype were set as basal level for comparisons (left panel). Gross observation on 60-wks old HCC mice (right panel). AR+/y (WildType mice); L-AR−/y (Liver specific AR knockout mice). (C) More malignant Histological pattern of 60-wks HCC tumors in L-AR−/y than in AR+/y mice. H&E staining (upper panel; 400X), and Trichrome stain (lower panel; 200X). Blue color indicates ECM deposition. (D) Gross observation of the lungs from AR+/y and L-AR−/y mice at 60-wks (upper panel). Percent of lung metastasis vs. normal lung occurring in the AR+/y and L-AR−/y mice at 60-wks old HCC tumors (lower panel). * P value indicated a statistical significance by Fisher test. (E) Histological study of the lungs from mice with 60-wks old metastatic HCC tumors. Upper panel: H&E staining of normal lung in L-AR−/y mice. Middle panel: H&E staining of metastatic tumor in L-AR−/y mouse lung. Lower panel: α-fetoprotein (AFP) staining of metastatic tumor of L-AR−/y mice lung. Red arrows indicate positive AFP staining of metastatic liver tumor cells. Green arrow indicates negative AFP staining of normal lung aveoli epithelia.
The malignancy of HCC in 60-wks old mice also showed more severe tumor appearance (red, vascular rich, soft) in the L-AR−/y livers as compared to livers with less malignant appearance (pale, collagen containing, hard) in AR+/y mice (Fig. 1B, right panel). Histological analysis of 60-week-old mice tumors found enlarged caniculi/sinusoid structure, malignant cytological pattern, and some necrotic, inflammatory lesions with undifferentiated histological pattern, which is in sharp contrast to the well cytologically differentiated HCC in AR+/y (Fig. 1C, upper panel). Trichrome staining (ECM/collagen deposition) also revealed more ECM deposition in the WT tumor liver, suggesting better liver healing in the WT mice as compared to L-AR−/y (Note-I changed this because fig. used this L-AR−/y) mice (Fig. 1C, lower panel).
In addition to the more malignant features observed in primary HCC tumors of L-AR−/y mice, we found higher lung metastasis risks in 60-wks old L-AR−/y mice as compared to WT mice (66.67 % vs. 14.29 %) (Fig.1D). H&E staining data in the normal portions of the lungs metastatic foci demonstrated normal healthy aveoli epithelia and intact cavity structure (Fig. 1E, upper panel). In contrast, lung tumor lesions showed HCC-like cells in terms of size and stronger Eosin staining (Fig. 1E, middle panel). Staining of α-fetoprotein (AFP, a HCC marker; positive staining showed in Suppl. Fig 1A) in these lungs with metastatic HCC tumors further confirmed the liver origin of these tumor cells (Fig. 1E, lower panel).
Together, results from Fig. 1A-E clearly demonstrated that mice lacking hepatic AR developed more malignant HCC with higher lung metastatic risk and died earlier.
AR suppresses p38 phosphorylation/activation during HCC progression
It is of great clinical relevance to seek for the potential mechanisms by which the hepatic AR switches its function from promoting HCC initiation to suppressing HCC metastasis. We first examined the AR expression in the human HCC patients (Suppl. Table 1) and found highly expressed AR in the HCC primary tumors with sizes smaller than 3cm diameter, which is in contrast to HCC metastatic tumors with much less AR expression (Suppl. Table 2, and Fig. 2A, upper panels). Interestingly, we found p38 phosphorylation, an important HCC therapeutic target, was reversely expressed compared to AR (Suppl. Table 2, Fig. 2A lower right panel).
Figure 2. Loss of AR leads to increased p38 phosphorylation, and reduced anoikis.
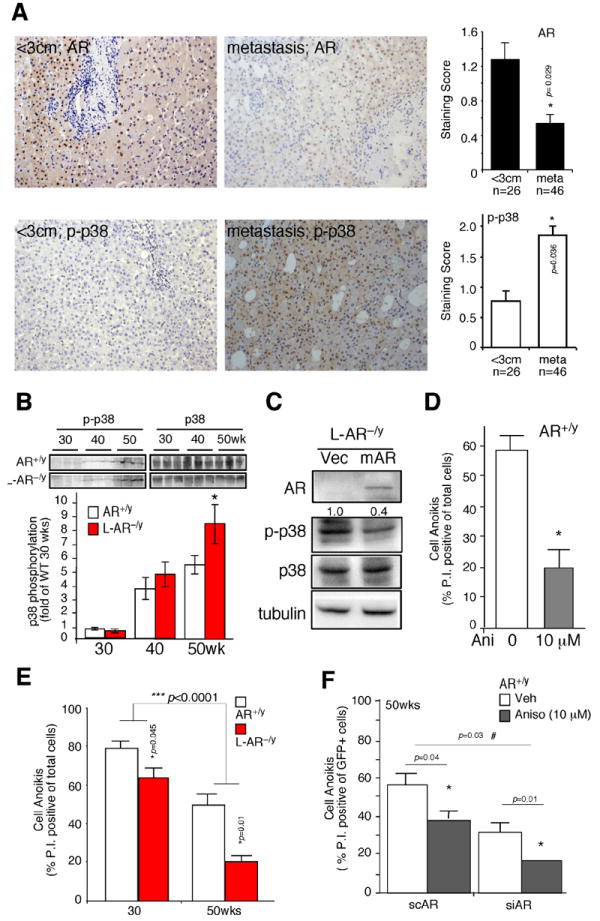
(A) AR and phospho-p38 (p-p38) expression in human HCC. The Immunohistochemistry staining of AR and p-p38 expression in tumors less than 3cm, and metastatic tumors. The quantitative results of AR and p-p38 based on staining scoring (ranking 0~3) in slides reviewed by two independent board certified pathologists. * indicates p-value significantly different (<0.05) comparing two groups. (B) Phospho-p38 (p-p38) and total p38 expression in HCC tumors of 30-, 40-, and 50-wks old mice (upper panel). The lower panel is the band densitometry quantitation of p-p38/p38 of AR+/y (n=6) and L-AR−/y mice (n=7) at each time point. Mean value of HCC in 30-wks old AR+/y mice was set as basal and compared. (C) Lentiviral transduction of vector (Vec) and mouse AR cDNA (mAR) in the hepatic cells from L-AR−/y HCC. Twenty μg total protein was loaded for SDS-PAGE, and Immunoblots were used to detect the protein levels of AR, p-p38, p38, and tubulin. (D) Anoikis was observed in the hepatic cultured cells from AR+/y mice treated with the p38 agonist, anisomycin (10 μM). (E) Cell anoikis was observed using hepatic culture cells from liver tumors of 30- and 50-wks old AR+/y and L-AR−/y mice. (F) Cell anoikis was measured in the primary hepatic cells from AR+/y, with introduced lentiviral-based AR scramble RNA (scAR) or small interference (siRNA), and treated with/without Anisomycin 10 μM (left panel). All the animal experiments were confirmed in at least three pairs of WT and LARKO littermates. Anoikis data were collected from three individual sets of experiments, and standard deviation (SD) was applied to represent error bars. * Represents significant difference with p-value <0.05.
An earlier study suggested that p38 phosphorylation/activation (p-p38) was enhanced during HCC progression (26). We also found the p-p38 increased from 30-wks to 40-wks and 50-wks in our mice treated with carcinogen-DEN (Fig. 2B), and p38 was highly phosphorylated/activated in more malignant HCC (Suppl. Fig. 1B). More importantly, we found loss of hepatic AR in L-AR−/y mice resulted in further increased p-p38 as compared to that in AR+/y mice at age of 50-week-old (Fig. 2B). To confirm that loss of hepatic AR results in enhanced p-p38, primary HCC cells isolated from L-AR−/y mice were cultured and infected with lentiviral-AR cDNA. Fig. 2C showed addition of AR cDNA resulted in the suppression of p-p38.
Together, results from Fig. 2A-C suggested that p38 is more active in the advanced state of HCC progression and loss of hepatic AR might lead to enhanced p-p38.
AR reverses p38-mediated cell anoikis resistance
The pathophysiological consequences of AR suppressed p-p38 in HCC were then examined by determining cell anoikis (27). We first demonstrated that p38 could modulate cell anoikis in primary cultured HCC cells isolated from AR+/y mice. As shown in Fig. 2D, addition of anisomycin, a p38 agonist (28), could reduce cell anoikis significantly. We then examined AR effects on cell anoikis using primary HCC cells from 30-wks (early stage), and 50-wks (pre- metastasis stage) old mice. We found loss of hepatic AR resulted in differential suppression effects on cell anoikis in 50-wks old tumor cells (WT: L-AR−/y = 55%±6%: 20%±4%; p=0.01) as compared to those at 30-wks (WT: L-AR−/y = 78%±4%: 66%±6%; p=0.045) (Fig. 2E). The AR differential suppression on two stages of cell anoikis reached statistical significance (p<0.0001). Next, using (siRNA) knockdown of AR (Suppl. Fig. 1C) in 50-wk old primary WT tumor cells treated with anisomycin, we tested AR and p38 effects on the cell anoikis. As shown in Fig. 2F, we found anisomycin reduces anoikis in the 50-wks old WT mice scramble treated hepatic cells (57%±8% to 39%±4%; p=0.04). However, when comparing cells in the AR siRNA treated groups, we found anisomycin has more dramatic impact on cell anoikis (36%±4% to 18%±0.4%; p=0.01) (Fig. 2F). The AR-related anisomycin suppression on cell anoikis could reach to statistical significance (p=0.03).
Our data consistently show that Anisomycin reduces anoikis, and AR enhances anoikis; furthermore, Anisomycin treatment of sc/siAR infected WT primary cells (Fig. 2F) showed the anoikis between sc vs. siAR RNA p-value is 0.003, which is consistent with our hypothesis. When comparing Anisomycin effect on scAR (Lane 1 vs. 2; p=0.04) and siAR (Lane 3 vs. 4; p=0.01) cells, the anisomycin was differentially impacted in sc vs. siAR cells. Together, the results from Fig. 2E-F suggested that the hepatic AR enhanced cell anoikis, at least in part, by modulating p38 phosphorylation.
AR enhances cell anoikis through suppression of p-p38 in human HCC cells
To further confirm that AR could enhance cell anoikis in the HCC cells, we repeated those experiments using mouse HCC cells with human HCC cells using previously established SKAR+ cells (4) (SKhep1 cell with AR stable expression) and HepG2-AR cells (8). We demonstrated that addition of AR in the human HCC SKAR+ and HepG2-AR cells resulted in increased cell anoikis (Fig. 3A, D). We also demonstrated that addition of AR led to suppression of p-p38 (Fig. 3B, E), and addition of p38 agonist Anisomycin reduced cell anoikis while expression of AR reversed that effect (Fig. 3C, F). A previous report indicated that FasL expression was associated with cell anoikis (29), which was also observed in our system (Suppl. Fig. 2A). Furthermore, Anisomycin could reduce, yet, addition of AR could enhance, FasL expression while the cells were detached (Suppl. Fig. 2A).
Figure 3. AR suppresses p38 phosphorylation in human HCC to enhance cell anoikis.

(A, D) AR enhances cell anoikis in SKhep1 and HepG2 cells. The cells were stably transfected with human AR cDNA. Less cell anoikis was observed in AR negative hepatoma cells, SKhep1 (AR−) and HepG2 cells (AR−) as compared to AR positive transfectant (AR+ in SKhep1, and AR+ in HepG2). The cells were treated with DHT 10 nM for 24 hrs, then detached, and further incubated in poly-HEMA coated plates for 48 hrs. Cells were stained with P.I. (5 μM) and measured by Flowcytometer to distinguish dead cells. The Y-axis indicates the fold of anoikis when comparing with AR- cells. (B, E) AR suppresses p38 phosphorylation in SKhep1 and HepG2 cells. AR, p-p38, and p38 were measured in AR- and AR+ cells using immunoblot assay. Tubulin served as loading control. (C, F) AR reverses p38-mediated anoikis resistance in hepatoma cells. The methods were described in Supplemental materials. The data was from average of four independent sets of experiments. * Represents significant difference between two groups using student T-test with p-value <0.01.
Together, the results from Figs. 2 and 3 strongly suggested that AR might increase cell anoikis via suppression of p-p38. As early studies suggested that cells with anoikis resistance ability is positively correlated with increased tumor metastasis (27, 30), it is possible that higher AR expression could negatively modulate p38-mediated cell anoikis resistance in HCC progression, which might be one reason why hepatic AR could switch from promotion of HCC initiation to suppression of HCC metastasis at the metastatic stage.
AR suppresses cell migration through suppression of NFκ B-MMP9 pathway
In addition to cell anoikis, cell invasiveness (from one foci to multiple foci within liver) is another important step contributing to the liver tumor metastasis (30). We noticed that the expression of MMP9, an important liver cancer migration marker (17, 31), was higher in the HCC tumors of L-AR−/y mice as compared to those in AR+/y mice (Fig. 4A). We then examined the AR effect on cell migration in the AR+/y primary cultured HCC cells. As shown in Fig. 4B, knocking down AR with siRNA in the AR+/y primary cultured HCC cells enhanced cell migration, and addition of AR in L-AR−/y primary cultured HCC cells reduced cell migration.
Figure 4. AR suppresses cell migration through suppression of NFκB-MMP9 pathway.
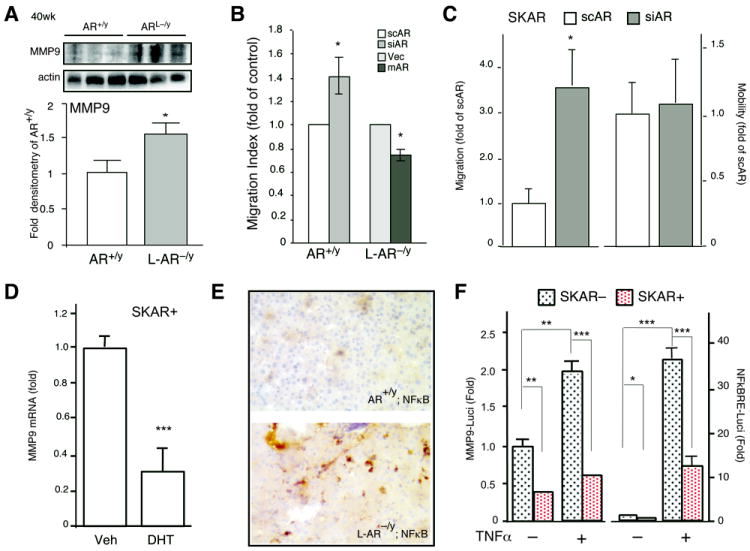
(A) Protein expression of MMP9 in HCC from 50-wks old AR+/y and L-AR−/y mice. Mean value of AR+/y HCC was set as basal level for comparisons. Actin level served as the loading control. (B) Cell migration was examined by matrigel-coated millipore chamber in AR+/y and L-AR−/y primary hepatic cultured cells. The scAR or siRNA in AR+/y cells, and Vec or mAR in L-AR−/y cells were transduced by lentiviral-based delivery system. (C) SKAR+ cell migration and mobility were determined by matrigel-coated and non-coated chamber assays, respectively. Human AR positive HCC cells, SKAR+, were stably transfected with scAR or siAR to examine migration and mobility. (D) MMP9 mRNA expression in SKAR+. (E) NFκB stains in 60-wks old AR+/y and L-AR−/y HCC. Less positive signal can be detected in AR+/y HCC; in contrast, there is significantly increased positive nuclear staining in L-AR−/y HCC. (F) MMP9-promoter driven luciferase (MMP9-Luci) was measured and parental (SKAR−), SKAR+ cells treated with/without TNFα (1 ng/ml) for 18 hrs (left side). NFκB response element driven luciferase activity (NFκBRE-Luci) was measured in SKAR− and SKAR+ cells treated with/without TNFα (1 ng/ml) for 18 hrs (right side). Thymidine kinase promoter driven renilla plasmid was cotransfected as control to normalize transfection efficiency. The results were averaged from 3 independent set of experiments containing triplicated results, * represents significant difference with p-value <0.05.
As knockdown of AR also increased migration, but not cell mobility, in SKAR+ human HCC cells (Fig. 4C; Suppl. Fig. 2C), we further examined MMP9 mRNA/protein expression in the SKAR+ cells, and found addition of DHT reduced MMP9 mRNA and protein expression (Fig. 4D). As an early study suggested that prostate epithelial AR could suppress MMP9 expression via modulation of NFκB activity (9), we examined NFκB expression in the mice HCC. Indeed, our data showed loss of hepatic AR led to higher expression/activation of NFκB in 60-wks old livers (Fig. 4E). Mechanistic dissection revealed that AR could suppress the TNFα-induced NFκBRE-Luciferase activity (9, 32) and MMP9 promoter-luciferase activity in SKAR+ human HCC cells (Fig. 4F). Together, results from Fig. 4, Suppl. Fig. 2B, C suggest that hepatic AR could also function through the NFκB-MMP9 pathway to modulate cell migration ability to suppress HCC metastasis.
Better therapeutic strategy via combination therapy of targeting AR and p38 with moderate dose of Sorafenib
With the contradictory AR functions (tumor initiation vs. migration/anoikis) taken into consideration in HCC therapy, we hypothesized that applying current molecular targeting agents (suppressing cell growth and migration) combined with the addition of AR (suppressing cell migration and anoikis) might benefit the current therapeutic paradigm. As the above results demonstrated that AR could play negative roles on HCC metastasis, we were encouraged to determine if we could enhance the therapeutic efficacy of HCC survival by targeting hepatic AR. Sorafenib (Bayer), a molecular target agent that has passed phase-III clinical trials via targeting multiple kinases, yet has higher I.C.50 on p38 (33), has been applied to treat advanced HCC patients with some benefits and less complications (34). We first titrated the Sorafenib dose on human SKhep1 HCC cells and found 5 μM of Sorafenib had a moderate cytotoxicity effect during 2 days treatment (Fig. 5A). Using this dose, we found Sorafenib reduced ERK phosphorylation (pERK) significantly, yet had little influence on p-p38 (Fig. 5B, lane 3 vs. 1). However, adding AR with 5 μM of Sorafenib resulted in abolished p-p38 (Fig. 5B, lane 4 vs. 2). Furthermore, we found that Sorafenib treatment alone could enhance cell anoikis and reduce cell migration in the SKhep1 cells (Fig. 5C, D; lane 2 vs. 1), and addition of AR alone could also enhance SKhep1 cell anoikis and suppress SKhep1 cell migration (Fig. 5C, D, lane 3 vs. 1). As expected, the combination treatment of adding AR plus Sorafenib resulted in additive enhancement of cell anoikis and suppression of migration (Fig. 5C, D, lanes 4 vs. 3). Similar additive results also occurred when we replaced SKhep1 with HepG2 cells (Suppl. Fig. 3, and Fig. 5E, F).
Figure 5. Combining Sorafenib and expression of AR effects on HCC cell growth, anoikis, and migration.
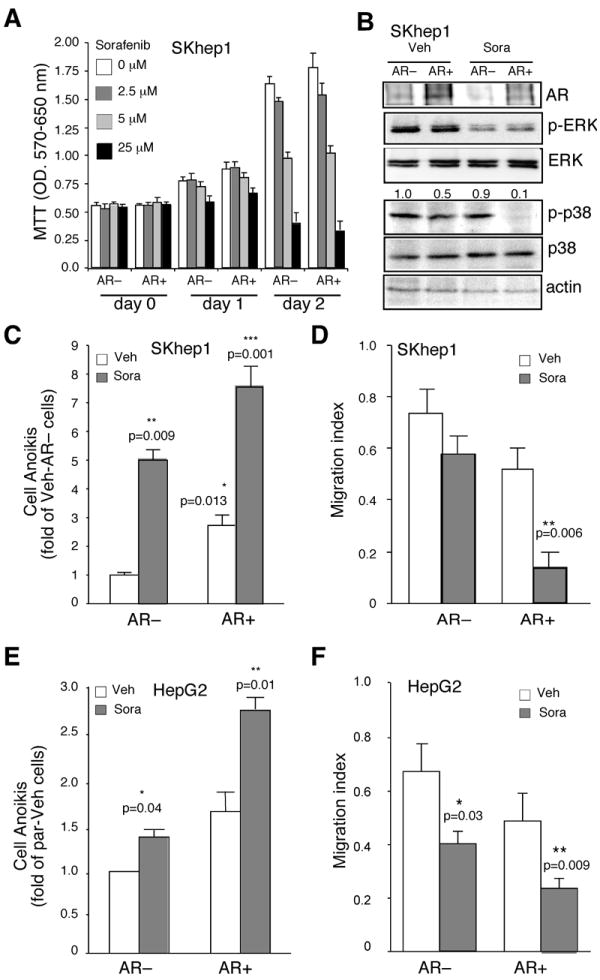
(A) Dose dependent effect of Sorafenib (Sora; 0, 2.5, 5, and 25 μM) on SKhep1 cell growth and viability. MTT assays were performed in AR negative (AR-), and AR positive (AR+) SKhep1 cells harvested at different times (day 0, 1, and 2). (B) p38 phosphorylation was robustly suppressed by combination of AR expression and moderate dose Sorafenib treatment. Cell extracts from AR- and AR+ cells treated with 5 μM Sorafenib for 16~18 hrs. ERK, p-ERK, p38, and p-p38 were detected. (C, E) Cell anoikis rate under subminimal dose of Sorafenib treatments on AR− or AR+ cells (SKhep1, 5 μM; HepG2, 2.5 μM). 106 cells were maintained in suspension culture for 48 hrs, harvested, stained with P.I., and subjected to Flowcytometric assay to determine cell death. The Y-axis represents fold changes compared to vehicle treatments of AR negative (par or AR-) cells in each experiment. (D, F) Subminimal dose of Sorafenib effect on cells migration with/without AR expression (SKhep1, 5 μM; HepG2, 2.5 μM). 5×105 cells were subjected to Boydenʼs chamber coating with/without Matrigel, and further incubated for 16-hrs (non-Matrigel coating), or 24 hrs (Matrigel coating). The cells migration assay was described in Methods. The Y-axis represents migration index. The migration index indicates the value of cell counting of Matrigel coating/non-coating of each treatment. * indicates <0.05, ** <0.01, and *** <0.005 p-value calculated by paired T-test.
To further prove the above in vitro findings from cell lines in Fig. 5, we injected SKhep1 cells with or without AR expression (AR− or AR+ cells) into nude mice via tail-veins to establish in vivo metastatic tumors. One month after cell injection, we treated the mice with Sorafenib or placebo orally (gavage feeding) at 30 mg/kg/mouse/day for another month and then observed HCC cancer survival rate and tumor metastasis. We found that addition of Sorafenib did improve the cancer survival in AR− mice (p=0.0158), while most of AR+ mice remained alive (Fig. 6A; p<0.0001). We then examined the mice for metastatic tumors in pleural cavity, peritoneal cavity, lymph nodes, visceral organs etc. at the time of death or sacrifice (Fig. 6B). The result showed that tumors were mainly located in the lungs (Fig. 6C) and several visceral organs. After calculating the metastatic risk, we found tumors could be observed in all AR−/placebo treatment mice. Injection of Sorafenib could improve metastasis free rate in the AR− group (28.6% metastasis free in Sorafenib vs. 0% placebo injection; Fig. 6B). On the other hand, addition of AR without Sorafenib injection (AR+/placebo) led to 25% of mice being metastasis free (compared to 0% in the AR−/placebo mice), indicating AR alone is already able to suppress tumor metastasis. As expected, the combination of AR expression with Sorafenib injection led to better therapeutic efficacy with significant increase of metastasis free mice (66.7% vs. 0%; p=0.0109).
Figure 6. Therapeutic evaluation of combining Sorafenib and AR expression in HCC metastasis mouse model.
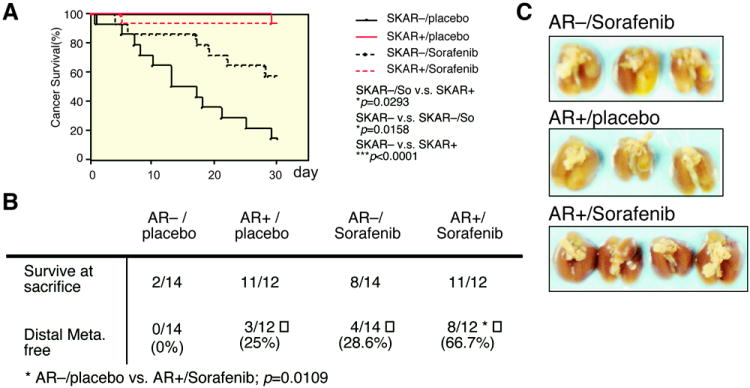
(A) Cancer survival of HCC metastasis model to observe combining low-dose Sorafenib (30 μg/ml/kg) and AR expression. Red line (SKAR− cells injected) and black line (SKAR+ cells injected) indicate the cancer survival of mice. Solid line (placebo), and dashed line (Sorafenib) indicate differential therapeutic effects of Sorafenib. The cancer survival and p-value were calculated and drawn using Kaplan–Meier estimator. * indicates the p-value <0.05, while *** indicates p-value <0.001. (B) Metastatic tumor in HCC metastasis mouse model. The mice from (A) were sacrificed or autopsied after one-month treatment and observed for metastasis tumors. The p-values were calculated by two-tailed Chi-square test where * indicates p=0.0109 while comparing metastasis free lungs in SKAR+/Sorafenib group to other three treatments. (C) Metastatic lung tumor from tail-vein injected HCC metastasis mouse model. Three groups of mice lung shown from top to bottom are: AR−/Sorafenib, AR+/placebo, and AR+/Sorafenib. The AR−/placebo group was absent due to early death during treatment.
Together, both in vitro and in vivo results from Fig. 5 and 6 demonstrated the beneficial and additive effect of combining AR expression and Sorafenib treatment in the HCC therapy.
DISCUSSION
Bimodal and opposite roles of hepatic AR in HCC initiation vs. metastasis
Using either DEN-induced HCC mouse model (4) or low-DEN with HBV-induced HCC mouse model (8, 35), we demonstrated that hepatic AR could promote hepatocarcinogenesis. These findings were opposite to the current findings showing hepatic AR could suppress HCC metastasis. This opposite roles of AR does not just occur in HCC. Indeed, AR in prostate cancer was also found to play dual yet opposite roles (36, 37). Interestingly, the potential mechanisms for prostate AR dual roles could be due to the differential AR signals in different prostate cells: being a proliferator in prostate stroma cells, an survivor in prostate luminal epithelial cells, and an suppressor in prostate basal intermediate epithelial cells (36, 37). In contrast, we believe the reasons for hepatic AR dual roles in HCC initiation vs. metastasis may be due to different intracellular signals within hepatocytes at different stages, as we demonstrated that hepatic AR-modulated p38 signals becomes more significant in the HCC metastasis. However, we do not exclude the potential contributors originating from other liver cells. For example, Kupffer-macrophage cells with various cytokines expression have been reported to play important roles for HCC progression (38). Our earlier report (39) also found macrophage AR might influence wound healing via modulating TNFα expression. Importantly, TNFα could also trigger p38 activation (40) and plays an important role in HCC metastasis (41). It will be interesting in the future to see if AR in Kupffer cells may also contribute to the HCC progression.
Differential expression of AR in initiation stage vs. metastasis stage
We noticed that AR expression was significantly reduced in metastastic tumors as compared to those in primary tumors in HCC patients (Fig. 2A). The reduced AR expression in metastatic tumors is echoed by early reports that AR expression in prostate metastatic tumors was lower than that found in primary prostate tumors (36). Similar observations also occurred in bladder tumors showing 75% of early superficial tumors expressed AR as compared to 21% found in invasive tumors (42). Interestingly, while all three types of tumors showed a similar conclusion that AR expression in metastatic tumors is less than that found in low staging primary tumors, the positive correlation of AR expression with tumor grades in the primary tumors during progression has never been established.
Inconsistent clinical trials using anti-androgens in HCC
Several clinical trials using various anti-androgens to treat HCC resulted in failed attempts without clear reasons (16, 22, 24). Three hypotheses might be able to explain these controversies. First, early (4) and current studies pointed out that AR expression, but not the classical androgens concentration, plays key role to influence HCC. Yet most of the anti- androgens used in clinical trials were developed to reduce/antagonize androgens binding to AR. Second, conclusions drawn in this report (AR dual roes in HCC) implied that targeting AR should be stage dependent. Third, the heterogeneity of cancer grading might result in differential cellular responses where the clonal selection process rapidly occurs within tumors. In this study, we have demonstrated the second possibility might be, at least in part, the potential answer.
Potential new therapy for HCC
Therapy with Sorafenib to treat HCC showed better efficacy with less systemic toxicity (34). However, complications with bleeding or even life threatening consequences (43) remain concerns. Here we found that a combination of increased AR expression with a moderate dose (5 μM) of Sorafenib resulted in better efficacy to treat HCC. Early Sorafenib phase I clinical trials indicated 6.0~7.7 μM (equal to serum concentration of 3.75~4.91 mg/L) of Sorafenib resulted in effective treatment with tolerable complications (44). Our finding that AR can be sensitized with a lower dose of Sorafenib (5 μM) that results in robust therapeutic effects may provide an individual approach to treat HCC patients. Any potential compound(s) to increase/stabilize AR expression or technology to increase AR gene delivery into liver might provide a potential method to achieve this purpose.
To sum up, there are two major concepts offered in this study and worthy of future investigations: one is to evaluate therapeutic efficiency by distinguishing AR+ patients before Sorafenib intervention. We expect that patients with more AR+ in HCC metastatic tumors may acquire better response and their dose of Sorafenib can be lower to obtain the maximal therapeutic effect with fewer side effects. The second concept is to develop technologies that include increasing/stabilization AR expression or AR gene delivery in advanced HCC patients, combined with other molecular targeting agents to evaluate therapeutic effects.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We appreciate Dean Dr. Fu-Jen Tsai for technical and resources support in Medical Research Core Facility, Office of Research & Development, China Medical University, Taiwan. We especially thank Karen Wolf for manuscript proofreading.
Financial Support:
This work was supported by George Whipple Professorship Endowment, NIH grant (CA122295), National Science Council of Taiwan (NSC100-2314-B039001-MY2), Taiwan Department of Health Clinical Trial and Research Center of Excellence to China Medical University (DOH100-TD-B-111-004), and CMU/H (CMU99-N2-06; CMR-100-159).
Ma and CL. Hsu designed and executed the experiments. CC. Yeh, MH. Wu, CK. Huang, assisted in some experimental techniques. YC. Hung, LB. Jeng, TY Lin, and S. Yeh helped with patient tissue histological diagnosis, scientific discussion and manuscript editing. C. Chang coordinated and supported.
Abbreviations
- AR
Androgen receptor
- HCC
Hepatocellular carcinoma
- NFκB
Nuclear Factor kappa B
- MMP9
Matrix metallopeptidase 9
- DEN
Diethylnitrosamine
Footnotes
All authors claim no conflict of interest in this work.
References
- 1.Di Maio M, Daniele B, Pignata S, Gallo C, De Maio E, Morabito A, Piccirillo MC, et al. Is human hepatocellular carcinoma a hormone-responsive tumor? World J Gastroenterol. 2008;14:1682–1689. doi: 10.3748/wjg.14.1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ishizu H, Une Y, Gondo H, Kameda H, Uchino J. Expression of sex-hormone receptors and clinicopathological findings in hepatocellular-carcinoma. Int J Oncol. 1994;4:1349–1352. doi: 10.3892/ijo.4.6.1349. [DOI] [PubMed] [Google Scholar]
- 3.Tavian D, De Petro G, Pitozzi A, Portolani N, Giulini SM, Barlati S. Androgen receptor mRNA under-expression in poorly differentiated human hepatocellular carcinoma. Histol Histopathol. 2002;17:1113–1119. doi: 10.14670/HH-17.1113. [DOI] [PubMed] [Google Scholar]
- 4.Ma WL, Hsu CL, Wu MH, Wu CT, Wu CC, Lai JJ, Jou YS, et al. Androgen receptor is a new potential therapeutic target for the treatment of hepatocellular carcinoma. Gastroenterology. 2008;135:947–955. 955, e941–945. doi: 10.1053/j.gastro.2008.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ohnishi S, Murakami T, Moriyama T, Mitamura K, Imawari M. Androgen and estrogen receptors in hepatocellular carcinoma and in the surrounding noncancerous liver tissue. Hepatology. 1986;6:440–443. doi: 10.1002/hep.1840060320. [DOI] [PubMed] [Google Scholar]
- 6.Barone M, Margiotta M, Scavo MP, Gentile A, Francioso D, Papagni S, Castellaneta A, et al. Possible involvement of androgen receptor alterations in hepatocarcinogenesis. Dig Liver Dis. 2009;41:665–670. doi: 10.1016/j.dld.2008.12.099. [DOI] [PubMed] [Google Scholar]
- 7.Nagasue N, Yu L, Yukaya H, Kohno H, Nakamura T. Androgen and oestrogen receptors in hepatocellular carcinoma and surrounding liver parenchyma: impact on intrahepatic recurrence after hepatic resection. Br J Surg. 1995;82:542–547. doi: 10.1002/bjs.1800820435. [DOI] [PubMed] [Google Scholar]
- 8.Wu MH, Ma WL, Hsu CL, Chen YL, Ou JH, Ryan CK, Hung YC, et al. Androgen receptor promotes hepatitis B virus-induced hepatocarcinogenesis through modulation of hepatitis B virus RNA transcription. Sci Transl Med. 2010;2:32ra35. doi: 10.1126/scitranslmed.3001143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Altuwaijri S, Lin HK, Chuang KH, Lin WJ, Yeh S, Hanchett LA, Rahman MM, et al. Interruption of nuclear factor kappaB signaling by the androgen receptor facilitates 12-O-tetradecanoylphorbolacetate-induced apoptosis in androgen-sensitive prostate cancer LNCaP cells. Cancer Res. 2003;63:7106–7112. [PubMed] [Google Scholar]
- 10.Jemal A, Siegel R, Xu J, Ward E. Cancer Statistics. CA: A Cancer Journal for Clinicians. 2010;60:277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 11.Ostrowski JL, Ingleton PM, Underwood JC, Parsons MA. Increased hepatic androgen receptor expression in female rats during diethylnitrosamine liver carcinogenesis. A possible correlation with liver tumor development. Gastroenterology. 1988;94:1193–1200. doi: 10.1016/0016-5085(88)90012-1. [DOI] [PubMed] [Google Scholar]
- 12.Yoon G, Kim JY, Choi YK, Won YS, Lim IK. Direct activation of TGF-beta1 transcription by androgen and androgen receptor complex in Huh7 human hepatoma cells and its tumor in nude mice. J Cell Biochem. 2006;97:393–411. doi: 10.1002/jcb.20638. [DOI] [PubMed] [Google Scholar]
- 13.Liang L, Lu M, Huang J. Antiandrogen treatment for nude mice model with ectopic transplanted human HCC. Zhonghua Yi Xue Za Zhi. 1998;78:299–300. [PubMed] [Google Scholar]
- 14.Wang AG, Lee KY, Kim SY, Choi JY, Lee KH, Kim WH, Wang HJ, et al. The expression of estrogen receptors in hepatocellular carcinoma in Korean patients. Yonsei Med J. 2006;47:811–816. doi: 10.3349/ymj.2006.47.6.811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cho H, Lim IK. Expression of androgen receptor and its implication in hepatoma cells. Cancer Lett. 1997;115:135–140. doi: 10.1016/s0304-3835(97)04732-0. [DOI] [PubMed] [Google Scholar]
- 16.Hépatocellulaire GdEedTdC. Randomized trial of leuprorelin and flutamide in male patients with hepatocellular carcinoma treated with tamoxifen. Hepatology. 2004;40:1361–1369. doi: 10.1002/hep.20474. [DOI] [PubMed] [Google Scholar]
- 17.Qin LX, Tang ZY. Recent progress in predictive biomarkers for metastatic recurrence of human hepatocellular carcinoma: a review of the literature. J Cancer Res Clin Oncol. 2004;130:497–513. doi: 10.1007/s00432-004-0572-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jeng KS, Sheen IS, Tsai YC. Circulating messenger RNA of alpha-fetoprotein: a possible risk factor of recurrence after resection of hepatocellular carcinoma. Arch Surg. 2004;139:1055–1060. doi: 10.1001/archsurg.139.10.1055. [DOI] [PubMed] [Google Scholar]
- 19.Postic C, Shiota M, Niswender KD, Jetton TL, Chen Y, Moates JM, Shelton KD, et al. Dual roles for glucokinase in glucose homeostasis as determined by liver and pancreatic beta cell-specific gene knock-outs using Cre recombinase. J Biol Chem. 1999;274:305–315. doi: 10.1074/jbc.274.1.305. [DOI] [PubMed] [Google Scholar]
- 20.Yeh S, Tsai MY, Xu Q, Mu XM, Lardy H, Huang KE, Lin H, et al. Generation and characterization of androgen receptor knockout (ARKO) mice: an in vivo model for the study of androgen functions in selective tissues. Proc Natl Acad Sci U S A. 2002;99:13498–13503. doi: 10.1073/pnas.212474399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tejura S, Rodgers GR, Dunion MH, Parsons MA, Underwood JC, Ingleton PM. Sex-steroid receptors in the diethylnitrosamine model of hepatocarcinogenesis: modifications by gonadal ablation and steroid replacement therapy. J Mol Endocrinol. 1989;3:229–237. doi: 10.1677/jme.0.0030229. [DOI] [PubMed] [Google Scholar]
- 22.Forbes A, Wilkinson ML, Iqbal MJ, Johnson PJ, Williams R. Response to cyproterone acetate treatment in primary hepatocellular carcinoma is related to fall in free 5 alpha-dihydrotestosterone. Eur J Cancer Clin Oncol. 1987;23:1659–1664. doi: 10.1016/0277-5379(87)90446-9. [DOI] [PubMed] [Google Scholar]
- 23.Gupta S, Korula J. Failure of ketoconazole as anti-androgen therapy in nonresectable primary hepatocellular carcinoma. J Clin Gastroenterol. 1988;10:651–654. doi: 10.1097/00004836-198812000-00016. [DOI] [PubMed] [Google Scholar]
- 24.Matsuura B, Taniguchi Y, Ohta Y. Effect of antiandrogen treatment on chemical hepatocarcinogenesis in rats. J Hepatol. 1994;21:187–193. doi: 10.1016/s0168-8278(05)80393-9. [DOI] [PubMed] [Google Scholar]
- 25.Chao Y, Chan WK, Huang YS, Teng HC, Wang SS, Lui WY, Whang-Peng J, et al. Phase II study of flutamide in the treatment of hepatocellular carcinoma. Cancer. 1996;77:635–639. [PubMed] [Google Scholar]
- 26.Guo K, Liu Y, Zhou H, Dai Z, Zhang J, Sun R, Chen J, et al. Involvement of protein kinase C beta-extracellular signal-regulating kinase 1/2/p38 mitogen-activated protein kinase-heat shock protein 27 activation in hepatocellular carcinoma cell motility and invasion. Cancer Sci. 2008;99:486–496. doi: 10.1111/j.1349-7006.2007.00702.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Frisch SM, Francis H. Disruption of epithelial cell-matrix interactions induces apoptosis. J Cell Biol. 1994;124:619–626. doi: 10.1083/jcb.124.4.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cano E, Doza YN, Ben-Levy R, Cohen P, Mahadevan LC. Identification of anisomycin-activated kinases p45 and p55 in murine cells as MAPKAP kinase-2. Oncogene. 1996;12:805–812. [PubMed] [Google Scholar]
- 29.Laguinge LM, Samara RN, Wang W, El-Deiry WS, Corner G, Augenlicht L, Mishra L, et al. DR5 receptor mediates anoikis in human colorectal carcinoma cell lines. Cancer Res. 2008;68:909–917. doi: 10.1158/0008-5472.CAN-06-1806. [DOI] [PubMed] [Google Scholar]
- 30.Sahai E. Illuminating the metastatic process. Nat Rev Cancer. 2007;7:737–749. doi: 10.1038/nrc2229. [DOI] [PubMed] [Google Scholar]
- 31.Yuan RH, Jeng YM, Chen HL, Lai PL, Pan HW, Hsieh FJ, Lin CY, et al. Stathmin overexpression cooperates with p53 mutation and osteopontin overexpression, and is associated with tumour progression, early recurrence, and poor prognosis in hepatocellular carcinoma. J Pathol. 2006;209:549–558. doi: 10.1002/path.2011. [DOI] [PubMed] [Google Scholar]
- 32.Yoshizaki T, Sato H, Furukawa M, Pagano JS. The expression of matrix metalloproteinase 9 is enhanced by Epstein-Barr virus latent membrane protein 1. Proc Natl Acad Sci U S A. 1998;95:3621–3626. doi: 10.1073/pnas.95.7.3621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wilhelm SM, Carter C, Tang L, Wilkie D, McNabola A, Rong H, Chen C, et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004;64:7099–7109. doi: 10.1158/0008-5472.CAN-04-1443. [DOI] [PubMed] [Google Scholar]
- 34.Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, de Oliveira AC, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359:378–390. doi: 10.1056/NEJMoa0708857. [DOI] [PubMed] [Google Scholar]
- 35.DeLoia JA, Burk RD, Gearhart JD. Developmental regulation of hepatitis B surface antigen expression in two lines of hepatitis B virus transgenic mice. J Virol. 1989;63:4069–4073. doi: 10.1128/jvi.63.9.4069-4073.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Niu Y, Altuwaijri S, Lai KP, Wu CT, Ricke WA, Messing EM, Yao J, et al. Androgen receptor is a tumor suppressor and proliferator in prostate cancer. Proc Natl Acad Sci U S A. 2008;105:12182–12187. doi: 10.1073/pnas.0804700105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Niu Y, Chang TM, Yeh S, Ma WL, Wang YZ, Chang C. Differential androgen receptor signals in different cells explain why androgen-deprivation therapy of prostate cancer fails. Oncogene. 29:3593–3604. doi: 10.1038/onc.2010.121. [DOI] [PubMed] [Google Scholar]
- 38.Sander LE, Trautwein C, Liedtke C. Is interleukin-6 a gender-specific risk factor for liver cancer? Hepatology. 2007;46:1304–1305. doi: 10.1002/hep.21982. [DOI] [PubMed] [Google Scholar]
- 39.Lai JJ, Lai KP, Chuang KH, Chang P, Yu IC, Lin WJ, Chang C. Monocyte/macrophage androgen receptor suppresses cutaneous wound healing in mice by enhancing local TNF-alpha expression. J Clin Invest. 2009 doi: 10.1172/JCI39335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schindler JF, Monahan JB, Smith WG. p38 pathway kinases as anti-inflammatory drug targets. J Dent Res. 2007;86:800–811. doi: 10.1177/154405910708600902. [DOI] [PubMed] [Google Scholar]
- 41.Luedde T, Trautwein C. Intracellular survival pathways in the liver. Liver Int. 2006;26:1163–1174. doi: 10.1111/j.1478-3231.2006.01366.x. [DOI] [PubMed] [Google Scholar]
- 42.Boorjian S, Ugras S, Mongan NP, Gudas LJ, You X, Tickoo SK, Scherr DS. Androgen receptor expression is inversely correlated with pathologic tumor stage in bladder cancer. Urology. 2004;64:383–388. doi: 10.1016/j.urology.2004.03.025. [DOI] [PubMed] [Google Scholar]
- 43.Je Y, Schutz FA, Choueiri TK. Risk of bleeding with vascular endothelial growth factor receptor tyrosine-kinase inhibitors sunitinib and sorafenib: a systematic review and meta-analysis of clinical trials. Lancet Oncol. 2009;10:967–974. doi: 10.1016/S1470-2045(09)70222-0. [DOI] [PubMed] [Google Scholar]
- 44.Chen HX, Cleck JN. Adverse effects of anticancer agents that target the VEGF pathway. Nat Rev Clin Oncol. 2009;6:465–477. doi: 10.1038/nrclinonc.2009.94. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.