

Abstract
Glutathione (GSH) plays an important role in a multitude of cellular processes, including cell differentiation, proliferation, and apoptosis, and disturbances in GSH homeostasis are involved in the etiology and progression of many human diseases including cancer. While GSH deficiency, or a decrease in the GSH/glutathione disulphide (GSSG) ratio, leads to an increased susceptibility to oxidative stress implicated in the progression of cancer, elevated GSH levels increase the antioxidant capacity and the resistance to oxidative stress as observed in many cancer cells. The present review highlights the role of GSH and related cytoprotective effects in the susceptibility to carcinogenesis and in the sensitivity of tumors to the cytotoxic effects of anticancer agents.
1. Introduction
Reactive oxygen species (ROS) are physiologically produced by aerobic cells [1] and their production increases under conditions of cell injury [2]. Physiological levels of ROS mediate crucial intracellular signaling pathways and are essential for cell survival. However, an excess of ROS formation generates cell damage and death. To prevent the irreversible cell damage, the increase of ROS induces an adaptive response, consisting in a compensatory upregulation of antioxidant systems, aimed to restore the redox homeostasis [3].
Oxidative stress has long been implicated in cancer development and progression [4], suggesting that antioxidant treatment may provide protection from cancer [5]. On other hand, prooxidant therapies, including ionizing radiation and chemotherapeutic agents, are widely used in clinics, based on the rationale that a further oxidative stimulus added to the constitutive oxidative stress in tumor cells should, in fact, cause the collapse of the antioxidant systems, leading to cell death [6]. However, this latter approach has provided unsatisfactory results in that many primary tumors overexpress antioxidant enzymes at very high levels, leading to a resistance of cancer cells to drug doses [7].
Among the enzymatic systems involved in the maintenance of the intracellular redox balance, a main role is played by GSH [8] that participates, not only in antioxidant defense systems, but also in many metabolic processes [9].
Elevated GSH levels are observed in various types of tumors, and this makes the neoplastic tissues more resistant to chemotherapy [10, 11]. Moreover, the content of GSH in some tumor cells is typically associated with higher levels of GSH-related enzymes, such as γ-glutamylcysteine ligase (GCL) and γ-glutamyl-transpeptidase (GGT) activities, as well as a higher expression of GSH-transporting export pumps [11, 12]. Therefore, it is not surprising that the GSH system has attracted the attention of pharmacologists as a possible target for medical intervention against cancer progression and chemoresistance.
The main research in this field has been aimed at depleting GSH by a specific inhibition of GCL, a key enzyme of GSH biosynthesis. In this context, buthionine sulfoximine (BSO) is the most popular GSH-depleting agent studied in both preclinical and early clinical trials, but limitation on its availability has led to a search for alternatives [13, 14]. Recently, GSH analogues have been employed in order to sensitize tumors to cytotoxic effects of anticancer agents, by depleting GSH-related cytoprotective effects [15].
However, during the last decade, a new approach for the regulation of GSH-utilizing enzymes has emerged. It is also evident that many of the antioxidant enzymes are induced by GSH depletion at the transcriptional level which involves the binding of the nuclear factor (erythroid-derived 2)-like 2 (Nrf2) transcription factor to the antioxidant response element (ARE) in the promoter region of the genes encoding GCL and glutathione S-transferases [16].
2. GSH Biosynthesis
Glutathione (GSH) is a tripeptide formed by glutamic acid, cysteine, and glycine. The glutamic acid forms a particular gamma-peptic bond with cysteine by its gamma glutamyl group. Two forms of GSH are possible: the reduced form (GSH) which represents the majority of GSH, reaching millimolar concentration in the intracellular compartment, and the oxidized form (GSSG) that is estimated to be less than 1% of the total GSH. Intracellularly, the majority of GSH is found in the cytosol (about 90%), while mitochondria contain nearly 10% and the endoplasmic reticulum contains a very small percentage [17].
The synthesis of GSH from its constituent amino acids involves two ATP-requiring enzymatic steps: (1) the first step is rate-limiting and catalyzed by GCL which is composed of two subunits: one catalytic (GCLC) and one modifier (GCLM):
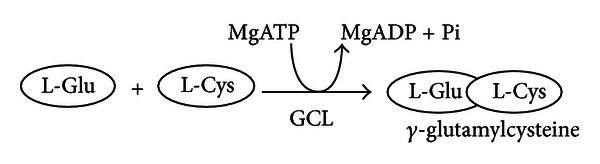
(2) The second step is catalyzed by GSH synthetase (GS) [19]:
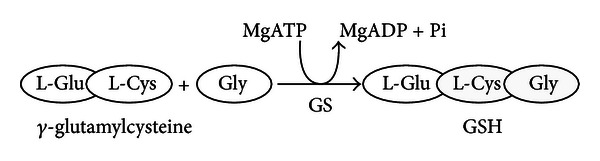
Although GS is generally thought not to be important in the regulation of GSH synthesis, accumulating evidence suggests that GS may play an important role, at least in certain tissues and/or under stressful conditions [20].
However, under normal physiological conditions, the rate of GSH synthesis is largely determined by two factors, that is, cysteine availability and GCL activity. Cysteine is normally derived from the diet, protein breakdown and in the liver, from methionine via transsulfuration (conversion of homocysteine to cysteine). Cysteine differs from other amino acids because its sulfhydryl form, cysteine, is predominant inside the cell whereas its disulfide form, cystine, is predominant outside the cell [21].
3. Glutathione Functions
The chemical structure of GSH determines its potential functions, and its broad distribution among all living organisms reflects its important biological role. A major function of GSH is the detoxification of xenobiotics and some endogenous compounds. These substances are electrophiles and form conjugates with GSH, either spontaneously or enzymatically, in reactions catalyzed by GSH-S-transferases (GST) [22]. Human GSTs are divided into two distinct family members: the membrane-bound microsomal and cytosolic family members. The conjugates formed are usually excreted in the bile, but can also undergo modification to mercapturic acid.
Another important GSH function is the maintenance of the intracellular redox balance and the essential thiol status of proteins [21].
The reaction with the protein is as follows:
| (1) |
The equilibrium of this reaction depends on the concentrations of GSH and GSSG. The reversible thiolation of proteins is known to regulate several metabolic processes including enzyme activity, transport activity, signal transduction and gene expression through redox-sensitive nuclear transcription factors such as AP-1, NF-kappaB (NF-kB) and p53 [21, 23]. In fact, DNA-binding activity of transcription factors often involves critical Cys residues, and the maintenance of these residues in a reduced form, at least in the nuclear compartment, is necessary [24]. AP-1 is a transcription factor related to tumor promotion [25], and its DNA-binding activity can be diminished if Cys-252 is oxidized [26]. Tumor suppressor p53, known as the “guardian of the genome,” contains 12 Cys residues in its amino acid sequence [27], and oxidation of some of these inhibits p53 function [28]. Moreover, GSH performs an antioxidant function (Figure 1).
Figure 1.
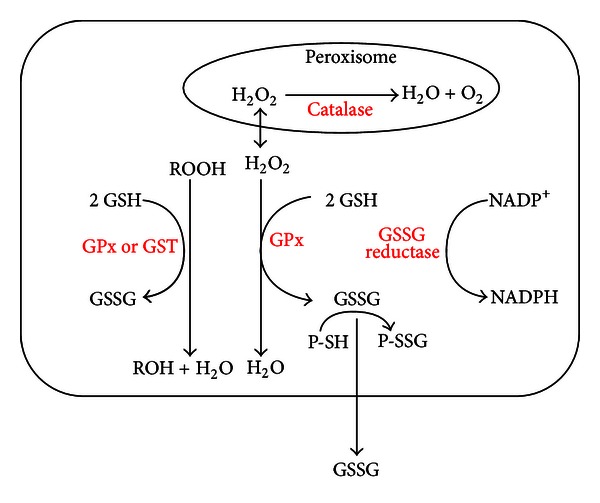
Antioxidant function of GSH. The hydrogen peroxide, produced during the aerobic metabolism, can be metabolized in the cytosol by GSH peroxidase (GPx) and catalase in peroxisomes. In order to prevent oxidative damage, the GSSG is reduced to GSH by GSSG reductase at the expense of NADPH, forming a redox cycle [17]. Organic peroxides can be reduced both by GPx and GSH-transferase (GST). In extreme conditions of oxidative stress, the ability of the cell to reduce GSSG to GSH may be less, inducing the accumulation of GSSG within the cytosol. In order to avoid a shift in the redox equilibrium, the GSSG can be actively transported out of the cell or react with protein sulfhydryl groups (PSH) and form mixed disulfides (PSSG).
In addition, storage of cysteine is one of the most important functions of GSH because cysteine is extremely unstable extracellularly and rapidly autooxidizes to cystine in a process that produces potentially toxic oxygen-free radicals [29]. The γ-glutamyl cycle allows GSH to be the main source of cysteine (Figure 2).
Figure 2.
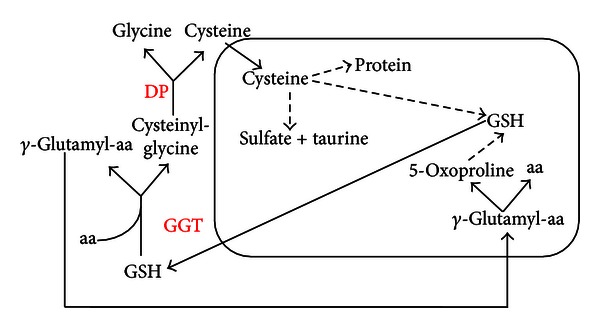
γ-Glutamyl cycle. In the γ-glutamyl cycle, GSH is released from the cell and the ectoenzyme GGT transfers the γ-glutamyl moiety of GSH to an amino acid (aa, the best acceptor being cysteine), forming γ-glutamyl-aa and cysteinyl-glycine. The γ-glutamyl-amino acid can then be transported back into the cell and once inside can be further metabolized to release the aa and 5-oxoproline, which can be converted to glutamate and used for GSH synthesis. Cysteinyl-glycine is broken down by dipeptidase (DP) to generate cysteine and glycine. Once inside the cell, the majority of cysteine is incorporated into GSH, some being incorporated into protein, and some degraded into sulfate and taurine [18].
4. Role of GSH in Regulating Cancer Development and Growth
In many normal and malignant cells, increased GSH level is associated with a proliferative response and is essential for cell cycle progression [30, 31]. The molecular mechanism of how GSH modulates cell proliferation remains largely speculative. A key mechanism for GSH's role in DNA synthesis relates to the maintenance of reduced glutaredoxin or thioredoxin, which is required for the activity of ribonucleotide reductase, the rate-limiting enzyme in DNA synthesis [32].
Furthermore, in liver cancer and metastatic melanoma cells, GSH status is correlated with growth [33–35] and it has also been demonstrated that a direct correlation between GSH levels associated with cellular proliferation and metastatic activity exists [33]. In fact, intrasplenic inoculation of B16 melanoma (B16M) cells into C57BL/6J syngenic mice induced metastatic foci formation by colonizing different organs. However, the number and size of metastases were much higher when B16M cells with high GSH content were inoculated in vivo [33]. A high percentage of tumor cells with high GSH content were able to survive in the presence of the nitrosative and oxidative stress, thereby representing the main task force in the metastatic invasion [36]. Therefore, it is plausible that maintenance of high intracellular levels of GSH could be critical for the extravascular growth of metastatic cells. Moreover, maintenance of mitochondrial GSH homeostasis may be a limiting factor for the survival of metastatic cells in the immediate period following intra-sinusoidal arrest and interaction with activated vascular endothelial cells. Mitochondrial dysfunction is a common event in the mechanism leading to cell death [37], and, recently, it has been found to be an essential step for the killing of non-small-cell lung (NSCLC) carcinomas which are resistant to conventional treatments [38]. Thus, the impairment of GSH uptake by mitochondria may be important to sensitize invasive cancer cells to prooxidant compounds capable of activating the cell death mechanism.
As previously reported, GSH is effluxed by cells through GGT-mediated metabolism, allowing a “GSH-cycle” to take place, which is implicated in tumor development [39]. In fact, GGT-positive foci were found in animals exposed to prooxidant carcinogens, suggesting the hypothesis of GGT as an early marker of neoplastic transformation [40, 41]. Moreover, increased levels of GGT have been observed in cancers of the ovaries [42], colon [43], liver [44], melanoma [45], and leukemias [46]. In studies on melanoma cells in vitro and in vivo, elevated GGT activity has been found to accompany an increased invasive growth [45, 47, 48], and a positive correlation has been described between GGT expression and unfavourable prognostic signs in human breast cancer [49].
The prooxidant activity of GGT has also recently been shown to promote the iron-dependent oxidative damage of DNA in GGT-transfected melanoma cells, thus potentially contributing to genomic instability and an increased mutation risk in cancer cells [50]. GGT/GSH-dependent prooxidant reactions has been shown to exert an antiproliferative action in ovarian cancer cells [51], while other studies in U937 lymphoma cells have shown that basal GGT-dependent production of hydrogen peroxide can represent an antiapoptotic signal [52]. The modulatory effects of GGT-mediated prooxidant reactions could contribute to the resistance phenotype of GGT-expressing cancer cells by regulating both signal transduction pathways involved in proliferation/apoptosis balance, as well as by inducing protective adaptations in the pool of intracellular antioxidants.
5. GSH Depletion as an Experimental Approach to Sensitize Tumor Cells to Therapy
Cancer cell lines containing low GSH levels have been demonstrated to be much more sensitive than control cells to the effect of irradiation [18]. In fact, GSH depletion obtained by BSO, the irreversible inhibitor of GCL, is the most frequently used approach and it is associated with many chemotherapeutic agents [53–57]. However, molecular signaling of BSO-induced apoptosis is poorly understood, and, recently, it has been demonstrated that in different leukaemia and lymphoma cells, the death receptor-mediated apoptotic pathway, induced by arsenic trioxide plus BSO, is triggered via JNK activation [58]. Moreover, in neuroblastoma cells susceptible to BSO treatment, DNA damage and apoptosis was triggered via PKC-δ activation and ROS production [59, 60].
In fact, BSO in combination with melphalan [14, 61], is currently undergoing clinical evaluation in children with neuroblastoma (NCT00002730; NCT00005835) and in patients with persistent or recurrent stage III malignant melanoma (NCT00661336). Recently, it has been demonstrated that a combination of azathioprine with BSO is useful for localized treatment of human hepatocellular carcinoma [62].
Therefore, BSO clinical use is restricted by its short half-life, with the consequent need for prolonged infusions resulting in its nonselective effect of GSH depletion on both normal and malignant cells [63].
6. Role of GSH in Chemoresistance
The increase in GSH levels, GCL activity and GCLC gene transcription is associated with drug resistance in tumor cells [64, 65].
The increase in GSH is a major contributing factor to drug resistance by binding to or reacting with, drugs, interacting with ROS, preventing damage to proteins or DNA, or by participating in DNA repair processes. In melanoma cells, GSH depletion and GGT inhibition significantly increased cytotoxicity via oxidative stress [66]. In addition, it has been demonstrated that GGT-overexpressing cells were more resistant to hydrogen peroxide [67] and chemotherapics, such as doxorubicin [68], cisplatin [64], and 5-fluorouracil [69].
Moreover, it has been found that the human multidrug resistance protein (MRP), a member of the superfamily of ATP-binding cassette membrane transporters, can lead to resistance to multiple classes of chemotherapeutic agents [70, 71]. Several studies have shown coordinated overexpression of GCLC and MRP in drug-resistant tumor cell lines, in human colorectal tumors and in human lung cancer specimens after platinum exposure [70, 71].
Three mechanisms have been proposed for the role of GSH in regulating cisplatin (CDDP) resistance: (i) GSH may serve as a cofactor in facilitating MRP2-mediated CDDP efflux in mammalian cells; (ii) GSH may serve as a redox-regulating cytoprotector based on the observations that many CDDP-resistant cells overexpress GSH and γ-GCS; and (iii) GSH may function as a copper (Cu) chelator.
Moreover, overexpression of specific GSTs can also affect chemoresistance, whereas polymorphisms that decrease GST activity are associated with a high risk of developing cancer [72]. An elevated expression of GSTs, combined with high GSH levels, can increase the rate of conjugation and detoxification of chemotherapy agents, thus reducing their effectiveness [73]. In addition to the transferase function, GSTs have been shown to form protein-protein interactions with members of the mitogen activated protein (MAP) kinases. By interacting directly with MAPKs, including c-Jun N-terminal kinase 1 (JNK1) and apoptosis signal-regulating kinase 1 (ASK1), GSTs bind the ligand in a complex structure, preventing interactions with their downstream targets [74]. Many anticancer agents induce apoptosis via activation of MAP kinases, in particular JNK and p38 [75, 76]. This novel, nonenzymatic role for GSTs has direct relevance to the GST overexpressing phenotypes of many drug-resistant tumors. As an endogenous switch for the control of intracellular signaling pathways, an elevated expression of GST can alter the balance of kinases during drug treatment, thereby causing a potential selective advantage for tumor growth.
The promoter regions encoding GSTs and γGCL possess binding sites for transcriptional regulators such as NF-kB, AP-1, AP-2, and the Nrf2/Kelch-like ECH-associated protein 1 (Keap1) system. After exposure to oxidative stimuli, Nrf2 dissociates from Keap1, its negative regulator, and translocates into the nucleus where it heterodimerizes with small Maf proteins [77] and binds to antioxidant responsive element (ARE) sequences, triggering a cytoprotective adaptative response. This response is characterised by upregulation of several cytoprotective and detoxification genes, including ferritin, GSH-S-reductase (GSR), GST, GCLM, and GCLC, phase-I drug oxidation enzyme NAD(P)H:quinone oxidoreductase 1 (NQO1), MRP, and heme oxygenase-1 (HO-1) [78, 79].
However, in numerous types of cancer, Nrf2 is upregulated and takes on a protumoral identity since the above-cited cytoprotective genes, not only give tumors an advantage, but also lead to drug resistance [80–82].
To date, numerous mutations have been found of both Keap1 and Nrf2 in various human cancers resulting in the constitutive expression of prosurvival genes. Most of Nrf2 somatic mutations have led to the impairment of their recognition site for Keap1, which then has led to the continuous activation of Nrf2. Moreover, it has been observed that the prognosis of patients with either Nrf2 or Keap1 mutations is much lower than patients with no mutation [83].
Among Nrf2-regulated genes, HO-1 is the most well known as a stress protein that can have both antioxidative and anti-inflammatory effects [84]. It catalyzes the rate-limiting step in the catabolism of the prooxidant heme to carbon monoxide, biliverdin, and free iron [85].
Recent experimental evidence has shown the involvement of HO-1 in cancer cell biology. On one hand, HO-1 protects healthy cells from transformation into neoplastic cells by counteracting ROS-mediated carcinogenesis and, on the other hand, HO-1 protects cancer cells, enhancing their survival and their resistance to anticancer treatment [86]. In addition, high levels of HO-1 have been observed in various human solid tumors, such as renal [87], prostatic [88], and pancreatic cancers [89]. Moreover, HO-1 expression in tumor cells can be further increased by anticancer treatments (chemo-, radio-, and photodynamic therapy) [90], and it has been hypothesized that HO-1 and its products may have an important role in the development of a resistant phenotype. In this context, it has been recently demonstrated that BSO and/or the inhibition of the Nrf2/HO-1 axis is able to increase the sensitivity of neuroblastoma cells to etoposide [91, 92].
7. Therapeutic Potential of GSH and GSH-Modulating Agents
The modulation of the GSH-based antioxidant redox system (GRS), the major determinant of the cellular redox status [93], might represent a promising therapeutic strategy for overcoming cancer cell progression and chemoresistance.
However, GSH itself cannot be administered clinically with any effect, and for this reason, a variety of precursors or chemically modified analogues have been generated in order to mimic glutathione's various physiological or pharmacological effects. N-acetylcysteine (NAC; Mucomyst) represents the earlier GSH analogue, and YM737, a monoester of GSH, recently discovered, has been favourably compared to NAC [94]. Another GSH analogue approach is cysteine-substituted S-nitrosoglutathione [95].
Since abundant levels of GST [96] have been identified in resistant tumors, GSH analogues, that differentially inhibit GST isoforms, have been developed. Telcyta (TLK-286) is a GSH analogue utilized in combination with cytotoxic chemotherapies such as platinum, taxanes, and anthracyclines in a variety of tumors with very high levels of glutathione S-transferase pi-1-gene (GST-P1-1) [97]. Telintra (TLK199) is another small molecule inhibitor of GST-P1-1 developed for the potential prevention of myelosuppression in blood diseases, namely, myelodysplastic syndrome [98]. After a phase 1 clinical trial [99] and a phase 2 using an oral formulation, TLK199 appeared to be well tolerated and with some efficacy in the myeloplastic syndrome treatment [100].
In addition, the implication of GGT activity in the resistance phenotype of cancer cells suggests a potential use of GGT inhibitors associated with chemotherapeutics in order to deplete intracellular GSH and/or to inhibit extracellular drug detoxification. Different GGT inhibitors are known [101–103], but, unfortunately, these molecules are toxic and cannot be used in humans.
Moreover, drugs that target S-glutathionylation have direct anticancer effects since they act on a wide range of signalling pathways [57]. Among the agents that mediate their effects through S-glutathionylation, NOV-002 has been most extensively studied, with a phase III trial (NCT00347412) completed in advanced NSCLC [104], and data available from phase II trials in breast and ovarian cancers [105]. NOV-002 is a product containing oxidized glutathione that alter the GSH : GSSG ratio and induces S-glutathionylation [106]. NOV-002-induced S-glutathionylation has been shown to have inhibitory effects on proliferation, survival, and invasion of myeloid cell lines and significantly increased the efficacy of cyclophosphamide chemotherapy in a murine model of colon cancer [107].
In a randomized phase II trial, NOV-002 in combination with standard chemotherapy has shown promising effects in patients with stage IIIb/IV of NSCLC [108]. Positive results were also obtained from a phase II trial in patients with neoadjuvant breast cancer therapy [109].
Other therapeutic agents include phenolic antioxidants (α-napthoflavone, butylated hydroxyanisole, and tert-butyl hydroquinone), synthetic antioxidants (ethoxyquin, oltipraz, and phorbol esters), triterpenoid analogue (oleanolic acid derivatives, sesquiterpenes), and isothiocyanates (sulforaphane). Sulforaphane (SF) is the strongest natural inducer of Nrf2 and phase II detoxifying enzymes and it has a potent anticarcinogenic and chemopreventive effect by inducing apoptosis and cell cycle arrest [110].
On the other hand, the inhibition of Nrf2 signaling might be employed to enhance the sensitization of chemoresistant tumors to cytotoxic agents, and in this context, it has recently been reported that brusatol, a compound found in a plant extract, acts as inhibitor of this pathway and may exhibit therapeutic utility [111]. Another effective approach to increasing cancer cell sensitivity to chemotherapeutic drugs would be to silence both Nrf2 and Keap1 simultaneously [112]. Related to Nrf2, a potential target for redox chemotherapy is HO-1. HO-1 inhibitors, including zinc protoporphyrin and more soluble pegylated derivatives (PEG-ZnPP), have been successfully used to improve chemosensitization of cancer cells [113]. Moreover, HO-1 inhibitors administered intravenously, displayed cytotoxic activity in a murine hypoxic solid tumor model [114].
Moreover, disulfiram (DSF) does not cause depletion of total GSH, but shifts the ratio of GSH/GSSG towards the oxidized state. DSF induces apoptosis of human melanoma cells [115], and this apoptogenic effect has encouraged ongoing clinical phase I/II studies in human metastatic melanoma (NCT00256230).
Arsenic trioxide (As2O3) is a prooxidant chemotherapeutic compound combined with agents that deplete cellular GSH [116]. As2O received FDA approval in 2000 for the treatment of nonacute promyelocytic leukemia and is used in patients who have relapsed or are refractory to first-line intervention using retinoid and anthracycline chemotherapy.
As2O inhibits GPx and mitochondrial respiratory function that leads to increased ROS leakage contributing to antileukemia activity. Another piece of evidence suggests that the irreversible inhibition of thioredoxin reductase is the key mechanism underlying As2O-induced breast cancer cell apoptosis [117]. Importantly, As2O3 sensitivity is associated with low levels of GSH in cancer cells and GSH depletion, obtained by BSO or ascorbate treatment, contributes to sensitizing cells toward apoptosis [118]. The potentiation of As2O chemotherapeutic efficacy using BSO was demonstrated in an orthotopic model of prostate cancer metastasis [119].
8. Conclusions
The modulation of cellular GSH is a double-edged sword, both sides of which have been exploited for potential therapeutic benefits [120]. Enhancing the capacity of GSH and its associated enzymes, in order to protect cells from redox-related changes or environmental toxins, represents a persistent aim in the search for cytoprotective strategies against cancer. On the contrary, the strategy of depleting GSH and GSH-related detoxification pathways is aimed at sensitizing cancer cells to chemotherapy, the so-called chemosensitization [121]. In this context, it has been reported that GSH and GSH enzyme-linked system may be a determining factor for the sensitivity of some tumors to various chemotherapeutic agents. In particular, GST is a relevant parameter for chemotherapy response, and it may be utilized as a useful biomarker for selecting tumors potentially responsive to chemotherapeutic regimens.
However, the attempts to deplete GSH have been limited by the nonselective effects of BSO and have stimulated the research of new GCL inhibitors.
Since it is well known that GSH depletion leads to the upregulation of antioxidant genes, many of which are under Nrf2 control and, that in several types of tumors, Nrf2 is constitutively activated [122, 123], a new and indirect approach for cancer therapy may be used to modulate the Nrf2-ARE pathway. Based on this, Nrf2 creates a new paradigm in cytoprotection, cancer prevention, and drug resistance.
In summary, the involvement of GSH in the carcinogenesis and in the drug resistance of tumor cell is clear, but further studies, aimed at understanding the GSH-driven molecular pathways, might be crucial to design new therapeutic strategies to fight cancer progression and overcome chemoresistance.
Acknowledgments
This work was supported by Grants from PRIN 2009M8FKBB_002 and Genoa University, Italy. The authors would like to thank Ms. Suzanne Patten for language editing.
References
- 1.Dröge W. Free radicals in the physiological control of cell function. Physiological Reviews. 2002;82(1):47–95. doi: 10.1152/physrev.00018.2001. [DOI] [PubMed] [Google Scholar]
- 2.Valko M, Leibfritz D, Moncol J, Cronin MTD, Mazur M, Telser J. Free radicals and antioxidants in normal physiological functions and human disease. International Journal of Biochemistry and Cell Biology. 2007;39(1):44–84. doi: 10.1016/j.biocel.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 3.Landriscina M, Maddalena F, Laudiero G, Esposito F. Adaptation to oxidative stress, chemoresistance, and cell survival. Antioxidants and Redox Signaling. 2009;11(11):2701–2716. doi: 10.1089/ars.2009.2692. [DOI] [PubMed] [Google Scholar]
- 4.Hussain SP, Hofseth LJ, Harris CC. Radical causes of cancer. Nature Reviews Cancer. 2003;3(4):276–285. doi: 10.1038/nrc1046. [DOI] [PubMed] [Google Scholar]
- 5.Cabello CM, Bair WB, III, Wondrak GT. Experimental therapeutics: targeting the redox Achilles heel of cancer. Current Opinion in Investigational Drugs. 2007;8(12):1022–1037. [PubMed] [Google Scholar]
- 6.Lecane PS, Karaman MW, Sirisawad M, et al. Motexafin gadolinium and zinc induce oxidative stress responses and apoptosis in B-cell lymphoma lines. Cancer Research. 2005;65(24):11676–11688. doi: 10.1158/0008-5472.CAN-05-2754. [DOI] [PubMed] [Google Scholar]
- 7.Wang J, Yi J. Cancer cell killing via ROS: to increase or decrease, that is a question. Cancer Biology and Therapy. 2008;7(12):1875–1884. doi: 10.4161/cbt.7.12.7067. [DOI] [PubMed] [Google Scholar]
- 8.Meister A. Glutathione metabolism. Methods in Enzymology. 1995;251:3–7. doi: 10.1016/0076-6879(95)51106-7. [DOI] [PubMed] [Google Scholar]
- 9.Sies H. Glutathione and its role in cellular functions. Free Radical Biology and Medicine. 1999;27(9-10):916–921. doi: 10.1016/s0891-5849(99)00177-x. [DOI] [PubMed] [Google Scholar]
- 10.Calvert P, Yao KS, Hamilton TC, O’Dwyer PJ. Clinical studies of reversal of drug resistance based on glutathione. Chemico-Biological Interactions. 1998;111-112:213–224. doi: 10.1016/s0009-2797(98)00008-8. [DOI] [PubMed] [Google Scholar]
- 11.Estrela JM, Ortega A, Obrador E. Glutathione in cancer biology and therapy. Critical Reviews in Clinical Laboratory Sciences. 2006;43(2):143–181. doi: 10.1080/10408360500523878. [DOI] [PubMed] [Google Scholar]
- 12.O’Brien ML, Tew KD. Glutathione and related enzymes in multidrug resistance. European Journal of Cancer A. 1996;32(6):967–978. doi: 10.1016/0959-8049(96)00051-2. [DOI] [PubMed] [Google Scholar]
- 13.Griffith OW. Mechanism of action, metabolism, and toxicity of buthionine sulfoximine and its higher homologs, potent inhibitors of glutathione synthesis. The Journal of Biological Chemistry. 1982;257(22):13704–13712. [PubMed] [Google Scholar]
- 14.O’Dwyer PJ, Hamilton TC, LaCreta FP, et al. Phase I trial of buthionine sulfoximine in combination with melphalan in patients with cancer. Journal of Clinical Oncology. 1996;14(1):249–256. doi: 10.1200/JCO.1996.14.1.249. [DOI] [PubMed] [Google Scholar]
- 15.Wu JH, Batist G. Glutathione and glutathione analogues, therapeutic potentials. Biochimica et Biophysica Acta. 2012;1830(5):3350–3353. doi: 10.1016/j.bbagen.2012.11.016. [DOI] [PubMed] [Google Scholar]
- 16.Nguyen T, Nioi P, Pickett CB. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. The Journal of Biological Chemistry. 2009;284(20):13291–13295. doi: 10.1074/jbc.R900010200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lu SC. Regulation of glutathione synthesis. Molecular Aspects of Medicine. 2009;30(1-2):42–59. doi: 10.1016/j.mam.2008.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Meister A. Glutathione deficiency produced by inhibition of its synthesis, and its reversal; applications in research and therapy. Pharmacology and Therapeutics. 1991;51(2):155–194. doi: 10.1016/0163-7258(91)90076-x. [DOI] [PubMed] [Google Scholar]
- 19.Oppenheimer L, Wellner VP, Griffith OW, Meister A. Glutathione synthetase. Purification from rat kidney and mapping of the substrate binding sites. The Journal of Biological Chemistry. 1979;254(12):5184–5190. [PubMed] [Google Scholar]
- 20.Luo JL, Hammarqvist F, Andersson K, Wernerman J. Surgical trauma decreases glutathione synthetic capacity in human skeletal muscle tissue. American Journal of Physiology. 1998;275(2, part 1):E359–E365. doi: 10.1152/ajpendo.1998.275.2.E359. [DOI] [PubMed] [Google Scholar]
- 21.Lu SC. Regulation of hepatic glutathione synthesis: current concepts and controversies. The FASEB Journal. 1999;13(10):1169–1183. [PubMed] [Google Scholar]
- 22.Meister A. On the discovery of glutathione. Trends in Biochemical Sciences. 1988;13(5):185–188. doi: 10.1016/0968-0004(88)90148-x. [DOI] [PubMed] [Google Scholar]
- 23.Townsend DM, Tew KD, Tapiero H. The importance of glutathione in human disease. Biomedicine and Pharmacotherapy. 2003;57(3):145–155. doi: 10.1016/s0753-3322(03)00043-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Morel Y, Barouki R. Repression of gene expression by oxidative stress. Biochemical Journal. 1999;342, part 3:481–496. [PMC free article] [PubMed] [Google Scholar]
- 25.Sen CK, Packer L. Antioxidant and redox regulation of gene transcription. The FASEB Journal. 1996;10(7):709–720. doi: 10.1096/fasebj.10.7.8635688. [DOI] [PubMed] [Google Scholar]
- 26.Nikitovic D, Holmgren A, Spyrou G. Inhibition of AP-1 DNA binding by nitric oxide involving conserved cysteine residues in Jun and Fos. Biochemical and Biophysical Research Communications. 1998;242(1):109–112. doi: 10.1006/bbrc.1997.7930. [DOI] [PubMed] [Google Scholar]
- 27.Rainwater R, Parks D, Anderson ME, Tegtmeyer P, Mann K. Role of cysteine residues in regulation of p53 function. Molecular and Cellular Biology. 1995;15(7):3892–3903. doi: 10.1128/mcb.15.7.3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu HH, Momand J. Pyrrolidine dithiocarbamate prevents p53 activation and promotes p53 cysteine residue oxidation. The Journal of Biological Chemistry. 1998;273(30):18898–18905. doi: 10.1074/jbc.273.30.18898. [DOI] [PubMed] [Google Scholar]
- 29.Meister A, Anderson ME. Glutathione. Annual Review of Biochemistry. 1983;52:711–760. doi: 10.1146/annurev.bi.52.070183.003431. [DOI] [PubMed] [Google Scholar]
- 30.Messina JP, Lawrence DA. Cell cycle progression of glutathione-depleted human peripheral blood mononuclear cells is inhibited at S phase. Journal of Immunology. 1989;143(6):1974–1981. [PubMed] [Google Scholar]
- 31.Lu SC, Ge JL. Loss of suppression of GSH synthesis at low cell density in primary cultures of rat hepatocytes. American Journal of Physiology. 1992;263(6, part 1):C1181–C1189. doi: 10.1152/ajpcell.1992.263.6.C1181. [DOI] [PubMed] [Google Scholar]
- 32.Holmgren A. Regulation of ribonucleotide reductase. Current Topics in Cellular Regulation. 1981;19:47–76. doi: 10.1016/b978-0-12-152819-5.50019-1. [DOI] [PubMed] [Google Scholar]
- 33.Carretero J, Obrador E, Anasagasti MJ, Martin JJ, Vidal-Vanaclocha F, Estrela JM. Growth-associated changes in glutathione content correlate with liver metastatic activity of B16 melanoma cells. Clinical and Experimental Metastasis. 1999;17(7):567–574. doi: 10.1023/a:1006725226078. [DOI] [PubMed] [Google Scholar]
- 34.Huang ZZ, Chen C, Zeng Z, et al. Mechanism and significance of increased glutathione level in human hepatocellular carcinoma and liver regeneration. The FASEB Journal. 2001;15(1):19–21. doi: 10.1096/fj.00-0445fje. [DOI] [PubMed] [Google Scholar]
- 35.Marengo B, Ciucis CD, Ricciarelli R, et al. DNA oxidative damage of neoplastic rat liver lesions. Oncology Reports. 2010;23(5):1241–1246. doi: 10.3892/or_00000756. [DOI] [PubMed] [Google Scholar]
- 36.Carretero J, Obrador E, Esteve JM, et al. Tumoricidal activity of endothelial cells: inhibition of endothelial nitric oxide production abrogates tumor cytotoxicity induced by hepatic sinusoidal endothelium in response to B16 melanoma adhesion in vitro. The Journal of Biological Chemistry. 2001;276(28):25775–25782. doi: 10.1074/jbc.M101148200. [DOI] [PubMed] [Google Scholar]
- 37.Costantini P, Jacotot E, Decaudin D, Kroemer G. Mitochondrion as a novel target of anticancer chemotherapy. Journal of the National Cancer Institute. 2000;92(13):1042–1053. doi: 10.1093/jnci/92.13.1042. [DOI] [PubMed] [Google Scholar]
- 38.Joseph B, Marchetti P, Formstecher P, Kroemer G, Lewensohn R, Zhivotovsky B. Mitochondrial dysfunction is an essential step for killing of non-small cell lung carcinomas resistant to conventional treatment. Oncogene. 2002;21(1):65–77. doi: 10.1038/sj.onc.1205018. [DOI] [PubMed] [Google Scholar]
- 39.Pompella A, Corti A, Paolicchi A, Giommarelli C, Zunino F. γ-Glutamyltransferase, redox regulation and cancer drug resistance. Current Opinion in Pharmacology. 2007;7(4):360–366. doi: 10.1016/j.coph.2007.04.004. [DOI] [PubMed] [Google Scholar]
- 40.Pompella A, De Tata V, Paolicchi A, Zunino F. Expression of γ-glutamyltransferase in cancer cells and its significance in drug resistance. Biochemical Pharmacology. 2006;71(3):231–238. doi: 10.1016/j.bcp.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 41.Roomi MW, Gaal K, Yuan QX, et al. Preneoplastic liver cell foci expansion induced by thioacetamide toxicity in drug-primed mice. Experimental and Molecular Pathology. 2006;81(1):8–14. doi: 10.1016/j.yexmp.2006.02.006. [DOI] [PubMed] [Google Scholar]
- 42.Hanigan MH, Frierson HF, Jr., Brown JE, Lovell MA, Taylor PT. Human ovarian tumors express γ-glutamyl transpeptidase. Cancer Research. 1994;54(1):286–290. [PubMed] [Google Scholar]
- 43.Murata J, Ricciardi-Castagnoli P, Mange PD, Martin F, Juillerat-Jeanneret L. Microglial cells induce cytotoxic effects toward colon carcinoma cells: measurement of tumor cytotoxicity with a gamma-glutamyl transpeptidase assay. International Journal of Cancer. 1997;70(2):169–174. doi: 10.1002/(sici)1097-0215(19970117)70:2<169::aid-ijc6>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 44.Tsutsumi M, Sakamuro D, Takada A, Zang SC, Furukawa T, Taniguchi N. Detection of a unique γ-glutamyl transpeptidase messenger RNA species closely related to the development of hepatocellular carcinoma in humans: a new candidate for early diagnosis of hepatocellular carcinoma. Hepatology. 1996;23(5):1093–1097. doi: 10.1002/hep.510230524. [DOI] [PubMed] [Google Scholar]
- 45.Supino R, Mapelli E, Sanfilippo O, Silvestro L. Biological and enzymatic features of human melanoma clones with different invasive potential. Melanoma Research. 1992;2(5-6):377–384. doi: 10.1097/00008390-199212000-00012. [DOI] [PubMed] [Google Scholar]
- 46.Tager M, Ittenson A, Franke A, Frey A, Gassen HG, Ansorge S. γ-glutamyl transpepsidase-cellular expression in populations of normal human mononuclear cells and patients suffering from leukemias. Annals of Hematology. 1995;70(5):237–242. doi: 10.1007/BF01784042. [DOI] [PubMed] [Google Scholar]
- 47.Prezioso JA, Wang N, Duty L, Bloomer WD, Gorelik E. Enhancement of pulmonary metastasis formation and γ-glutamyltranspeptidase activity in B16 melanoma induced by differentiation in vitro. Clinical and Experimental Metastasis. 1993;11(3):263–274. doi: 10.1007/BF00121169. [DOI] [PubMed] [Google Scholar]
- 48.Obrador E, Carretero J, Ortega A, et al. γ-glutamyl transpeptidase overexpression increases metastatic growth of B16 melanoma cells in the mouse liver. Hepatology. 2002;35(1):74–81. doi: 10.1053/jhep.2002.30277. [DOI] [PubMed] [Google Scholar]
- 49.Bard S, Noel P, Chauvin F, Quash G. γ-glutamyltranspeptidase activity in human breast lesions: an unfavourable prognostic sign. British Journal of Cancer. 1986;53(5):637–642. doi: 10.1038/bjc.1986.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Corti A, Duarte TL, Giommarelli C, et al. Membrane gamma-glutamyl transferase activity promotes iron-dependent oxidative DNA damage in melanoma cells. Mutation Research. 2009;669(1-2):112–121. doi: 10.1016/j.mrfmmm.2009.05.010. [DOI] [PubMed] [Google Scholar]
- 51.Perego P, Paolicchi A, Tongiani R, et al. The cell-specific anti-proliferative effect of reduced glutathione is mediated by gamma-glutamyl transpeptidase-dependent extracellular pro-oxidant reactions. International Journal of Cancer. 1997;71(2):246–250. doi: 10.1002/(sici)1097-0215(19970410)71:2<246::aid-ijc20>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 52.del Bello B, Paolicchi A, Comporti M, Pompella A, Maellaro E. Hydrogen peroxide produced during γ-glutamyl transpeptidase activity is involved in prevention of apoptosis and maintenance of proliferation in U937 cells. The FASEB Journal. 1999;13(1):69–79. doi: 10.1096/fasebj.13.1.69. [DOI] [PubMed] [Google Scholar]
- 53.Reber U, Wüllner U, Trepel M, et al. Potentiation of treosulfan toxicity by the glutathione-depleting agent buthionine sulfoximine in human malignant glioma cells. The role of BCL-2. Biochemical Pharmacology. 1998;55(3):349–359. doi: 10.1016/s0006-2952(97)00480-2. [DOI] [PubMed] [Google Scholar]
- 54.Anderson CP, Tsai JM, Meek WE, et al. Depletion of glutathione by buthionine sulfoximine is cytotoxic for human neuroblastoma cell lines via apoptosis. Experimental Cell Research. 1999;246(1):183–192. doi: 10.1006/excr.1998.4303. [DOI] [PubMed] [Google Scholar]
- 55.Friesen C, Kiess Y, Debatin KM. A critical role of glutathione in determining apoptosis sensitivity and resistance in leukemia cells. Cell Death and Differentiation. 2004;11(supplement 1):S73–S85. doi: 10.1038/sj.cdd.4401431. [DOI] [PubMed] [Google Scholar]
- 56.D’Alessio M, Cerella C, Amici C, et al. Glutathione depletion up-regulates Bcl-2 in BSO-resistant cells. The FASEB Journal. 2004;18(13):1609–1611. doi: 10.1096/fj.04-1813fje. [DOI] [PubMed] [Google Scholar]
- 57.Wondrak GT. Redox-directed cancer therapeutics: molecular mechanisms and opportunities. Antioxidants and Redox Signaling. 2009;11(12):3013–3069. doi: 10.1089/ars.2009.2541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chen D, Chan R, Waxman S, Jing Y. Buthionine sulfoximine enhancement of arsenic trioxide-induced apoptosis in leukemia and lymphoma cells is mediated via activation of c-Jun NH 2-terminal kinase and up-regulation of death receptors. Cancer Research. 2006;66(23):11416–11423. doi: 10.1158/0008-5472.CAN-06-0409. [DOI] [PubMed] [Google Scholar]
- 59.Domenicotti C, Marengo B, Verzola D, et al. Role of PKC-δ activity in glutathione-depleted neuroblastoma cells. Free Radical Biology and Medicine. 2003;35(5):504–516. doi: 10.1016/s0891-5849(03)00332-0. [DOI] [PubMed] [Google Scholar]
- 60.Marengo B, de Ciucis C, Ricciarelli R, et al. PKCδ sensitizes neuroblastoma cells to L-buthionine-sulfoximine and etoposide inducing reactive oxygen species overproduction and DNA damage. PLoS ONE. 2011;6(2) doi: 10.1371/journal.pone.0014661.e14661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bailey HH, Ripple G, Tutsch KD, et al. Phase I study of continuous-infusion L-S,R-buthionine sulfoximine with intravenous melphalan. Journal of the National Cancer Institute. 1997;89(23):1789–1796. doi: 10.1093/jnci/89.23.1789. [DOI] [PubMed] [Google Scholar]
- 62.Hernandez-Breijo B, Monserrat J, Ramirez-Rubio S, et al. Preclinical evaluation of azathioprine plus buthionine sulfoximine in the treatment of human hepatocarcinoma and colon carcinoma. World Journal of Gastroenterology. 2011;17(34):3899–3911. doi: 10.3748/wjg.v17.i34.3899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hamilton D, Jian HW, Batist G. Structure-based identification of novel human γ-glutamylcysteine synthetase inhibitors. Molecular Pharmacology. 2007;71(4):1140–1147. doi: 10.1124/mol.106.024778. [DOI] [PubMed] [Google Scholar]
- 64.Godwin AK, Meister A, O’Dwyer PJ, Chin Shiou Huang, Hamilton TC, Anderson ME. High resistance to cisplatin in human ovarian cancer cell lines is associated with marked increase of glutathione synthesis. Proceedings of the National Academy of Sciences of the United States of America. 1992;89(7):3070–3074. doi: 10.1073/pnas.89.7.3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mulcahy RT, Untawale S, Gipp JJ. Transcriptional up-regulation of γ-glutamylcysteine synthetase gene expression in melphalan-resistant human prostate carcinoma cells. Molecular Pharmacology. 1994;46(5):909–914. [PubMed] [Google Scholar]
- 66.Benlloch M, Ortega A, Ferrer P, et al. Acceleration of glutathione efflux and inhibition of γ- glutamyltranspeptidase sensitize metastatic B16 melanoma cells to endothelium-induced cytotoxicity. The Journal of Biological Chemistry. 2005;280(8):6950–6959. doi: 10.1074/jbc.M408531200. [DOI] [PubMed] [Google Scholar]
- 67.Shi M, Gozal E, Choy HA, Forman HJ. Extracellular glutathione and γ-glutamyl transpeptidase preent H2O2-induced injury by 2,3-dimethoxy-1,4-naphthoquinone. Free Radical Biology and Medicine. 1993;15(1):57–67. doi: 10.1016/0891-5849(93)90125-e. [DOI] [PubMed] [Google Scholar]
- 68.Hochwald SN, Rose DM, Brennan MF, Burt ME. Elevation of glutathione and related enzyme activities in high-grade and metastatic extremity soft tissue sarcoma. Annals of Surgical Oncology. 1997;4(4):303–309. doi: 10.1007/BF02303579. [DOI] [PubMed] [Google Scholar]
- 69.Lewis AD, Hayes JD, Wolf CR. Glutathione and glutathione-dependent enzymes in ovarian adenocarcinoma cell lines derived from a patient before and after the onset of drug resistance: intrinsic differences and cell cycle effects. Carcinogenesis. 1988;9(7):1283–1287. doi: 10.1093/carcin/9.7.1283. [DOI] [PubMed] [Google Scholar]
- 70.Kuo MT, Bao JJ, Furuichi M, et al. Frequent coexpression of MRP/GS-X pump and γ-glutamylcysteine synthetase mRNA in drug-resistant cells, untreated tumor cells, and normal mouse tissues. Biochemical Pharmacology. 1998;55(5):605–615. doi: 10.1016/s0006-2952(97)00494-2. [DOI] [PubMed] [Google Scholar]
- 71.Oguri T, Fujiwara Y, Isobe T, Katoh O, Watanabe H, Yamakido M. Expression of γ-glutamylcysteine synthetase (γ-GCS) and Multidrug Resistance-associated Protein (MRP), but not human canalicular Multispecific Organic Anion Transporter (cMOAT), genes correlates with exposure of human lung cancers to platinum drugs. British Journal of Cancer. 1998;77(7):1089–1096. doi: 10.1038/bjc.1998.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Balendiran GK, Dabur R, Fraser D. The role of glutathione in cancer. Cell Biochemistry and Function. 2004;22(6):343–352. doi: 10.1002/cbf.1149. [DOI] [PubMed] [Google Scholar]
- 73.McLellan LI, Wolf CR. Glutathione and glutathione-dependent enzymes in cancer drug resistance. Drug Resistance Updates. 1999;2(3):153–164. doi: 10.1054/drup.1999.0083. [DOI] [PubMed] [Google Scholar]
- 74.Adler V, Yin Z, Fuchs SY, et al. Regulation of JNK signaling by GSTp. The EMBO Journal. 1999;18(5):1321–1334. doi: 10.1093/emboj/18.5.1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ono K, Han J. The p38 signal transduction pathway activation and function. Cellular Signalling. 2000;12(1):1–13. doi: 10.1016/s0898-6568(99)00071-6. [DOI] [PubMed] [Google Scholar]
- 76.Marengo B, De Ciucis CG, Ricciarelli R, et al. p38MAPK inhibition: a new combined approach to reduce neuroblastoma resistance under etoposide treatment. Cell Death and Disease. 2013;4, article e589 doi: 10.1038/cddis.2013.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Dhakshinamoorthy S, Jaiswal AK. Small Maf (MafG and MafK) proteins negatively regulate antioxidant response element-mediated expression and antioxidant induction of the NAD(P)H:quinone oxidoreductase1 gene. The Journal of Biological Chemistry. 2000;275(51):40134–40141. doi: 10.1074/jbc.M003531200. [DOI] [PubMed] [Google Scholar]
- 78.Itoh K, Chiba T, Takahashi S, et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochemical and Biophysical Research Communications. 1997;236(2):313–322. doi: 10.1006/bbrc.1997.6943. [DOI] [PubMed] [Google Scholar]
- 79.Rushworth SA, MacEwan DJ, O’Connell MA. Lipopolysaccharide-induced expression of NAD(P)H:quinone oxidoreductase 1 and heme oxygenase-1 protects against excessive inflammatory responses in human monocytes. Journal of Immunology. 2008;181(10):6730–6737. doi: 10.4049/jimmunol.181.10.6730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ohta T, Iijima K, Miyamoto M, et al. Loss of Keap1 function activates Nrf2 and provides advantages for lung cancer cell growth. Cancer Research. 2008;68(5):1303–1309. doi: 10.1158/0008-5472.CAN-07-5003. [DOI] [PubMed] [Google Scholar]
- 81.Jiang T, Chen N, Zhao F, et al. High levels of Nrf2 determine chemoresistance in type II endometrial cancer. Cancer Research. 2010;70(13):5486–5496. doi: 10.1158/0008-5472.CAN-10-0713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Shibata T, Kokubu A, Gotoh M, et al. Genetic alteration of Keap1 confers constitutive Nrf2 activation and resistance to chemotherapy in gallbladder cancer. Gastroenterology. 2008;135(4):1358.e4–1368.e4. doi: 10.1053/j.gastro.2008.06.082. [DOI] [PubMed] [Google Scholar]
- 83.Shibata T, Ohta T, Tong KI, et al. Cancer related mutations in NRF2 impair its recognition by Keap1-Cul3 E3 ligase and promote malignancy. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(36):13568–13573. doi: 10.1073/pnas.0806268105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Otterbein LE, Zuckerbraun BS, Haga M, et al. Carbon monoxide suppresses arteriosclerotic lesions associated with chronic graft rejection and with balloon injury. Nature Medicine. 2003;9(2):183–190. doi: 10.1038/nm817. [DOI] [PubMed] [Google Scholar]
- 85.Otterbein LE, Soares MP, Yamashita K, Bach FH. Heme oxygenase-1: unleashing the protective properties of heme. Trends in Immunology. 2003;24(8):449–455. doi: 10.1016/s1471-4906(03)00181-9. [DOI] [PubMed] [Google Scholar]
- 86.Jozkowicz A, Was H, Dulak J. Heme oxygenase-1 in tumors: is it a false friend? Antioxidants and Redox Signaling. 2007;9(12):2099–2117. doi: 10.1089/ars.2007.1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Goodman AI, Choudhury M, Da Silva JL, Schwartzman ML, Abraham NG. Overexpression of the heme oxygenase gene in renal cell carcinoma. Proceedings of the Society for Experimental Biology and Medicine. 1997;214(1):54–61. doi: 10.3181/00379727-214-44069. [DOI] [PubMed] [Google Scholar]
- 88.Maines MD, Abrahamsson PA. Expression of heme oxygenase-1 (HSP32) in human prostate: normal, hyperplastic, and tumor tissue distribution. Urology. 1996;47(5):727–733. doi: 10.1016/s0090-4295(96)00010-6. [DOI] [PubMed] [Google Scholar]
- 89.Berberat PO, Dambrauskas Z, Gulbinas A, et al. Inhibition of heme oxygenase-1 increases responsiveness of pancreatic cancer cells to anticancer treatment. Clinical Cancer Research. 2005;11(10):3790–3798. doi: 10.1158/1078-0432.CCR-04-2159. [DOI] [PubMed] [Google Scholar]
- 90.Nowis D, Legat M, Grzela T, et al. Heme oxygenase-1 protects tumor cells against photodynamic therapy-mediated cytotoxicity. Oncogene. 2006;25(24):3365–3374. doi: 10.1038/sj.onc.1209378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Marengo B, Balbis E, Patriarca S, et al. GSH loss per se does not affect neuroblastoma survival and is not genotoxic. International Journal of Oncology. 2008;32(1):121–127. [PubMed] [Google Scholar]
- 92.Furfaro AL, Macay JR, Marengo B, et al. Resistance of neuroblastoma GI-ME-N cell line to glutathione depletion involves Nrf2 and heme oxygenase-1. Free Radical Biology and Medicine. 52(2):488–496. doi: 10.1016/j.freeradbiomed.2011.11.007. [DOI] [PubMed] [Google Scholar]
- 93.Schafer FQ, Buettner GR. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radical Biology and Medicine. 2001;30(11):1191–1212. doi: 10.1016/s0891-5849(01)00480-4. [DOI] [PubMed] [Google Scholar]
- 94.Shibata S, Tominaga K, Watanabe S. Glutathione protects against hypoxic/hypoglycemic decreases in 2-deoxyglucose uptake and presynaptic spikes in hippocampal slices. European Journal of Pharmacology. 1995;273(1-2):191–195. doi: 10.1016/0014-2999(94)00727-o. [DOI] [PubMed] [Google Scholar]
- 95.Chakrapani H, Kalathur RC, Maciag AE, et al. Synthesis, mechanistic studies, and anti-proliferative activity of glutathione/glutathione S-transferase-activated nitric oxide prodrugs. Bioorganic and Medicinal Chemistry. 2008;16(22):9764–9771. doi: 10.1016/j.bmc.2008.09.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Batist G, Tulpule A, Sinha BK. Overexpression of a novel anionic glutathione transferase in multidrug-resistent human breast cancer cells. The Journal of Biological Chemistry. 1986;261(33):15544–15549. [PubMed] [Google Scholar]
- 97.Wu JH, Miao W, Hu LG, Batist G. Identification and characterization of novel Nrf2 inducers designed to target the intervening region of keap1. Chemical Biology and Drug Design. 2010;75(5):475–480. doi: 10.1111/j.1747-0285.2010.00955.x. [DOI] [PubMed] [Google Scholar]
- 98.Hamilton D, Batist G. TLK-199 Telik. IDrugs. 2005;8(8):662–669. [PubMed] [Google Scholar]
- 99.Raza A, Galili N, Smith S, et al. Phase 1 multicenter dose-escalation study of ezatiostat hydrochloride (TLK199 tablets), a novel glutathione analog prodrug, in patients with myelodysplastic syndrome. Blood. 2009;113(26):6533–6540. doi: 10.1182/blood-2009-01-176032. [DOI] [PubMed] [Google Scholar]
- 100.Raza A, Galili N, Smith SE, et al. A phase 2 randomized multicenter study of 2 extended dosing schedules of oral ezatiostat in low to intermediate-1 risk myelodysplastic syndrome. Cancer. 2012;118(8):2138–2147. doi: 10.1002/cncr.26469. [DOI] [PubMed] [Google Scholar]
- 101.Tate SS, Meister A. Serine-borate complex as a transition-state inhibitor of γ-glutamyl transpeptidase. Proceedings of the National Academy of Sciences of the United States of America. 1978;75(10):4806–4809. doi: 10.1073/pnas.75.10.4806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.London RE, Gabel SA. Development and evaluation of a boronate inhibitor of γ-glutamyl transpeptidase. Archives of Biochemistry and Biophysics. 2001;385(2):250–258. doi: 10.1006/abbi.2000.2169. [DOI] [PubMed] [Google Scholar]
- 103.Han L, Hiratake J, Kamiyama A, Sakata K. Design, synthesis, and evaluation of γ-phosphono diester analogues of glutamate as highly potent inhibitors and active site probes of γ-glutamyl transpeptidase. Biochemistry. 2007;46(5):1432–1447. doi: 10.1021/bi061890j. [DOI] [PubMed] [Google Scholar]
- 104.Fidias P, Novello S. Strategies for prolonged therapy in patients with advanced non-small-cell lung cancer. Journal of Clinical Oncology. 2010;28(34):5116–5123. doi: 10.1200/JCO.2010.30.7074. [DOI] [PubMed] [Google Scholar]
- 105.Matulonis UA, Horowitz NS, Campos SM, et al. Phase II study of carboplatin and pemetrexed for the treatment of platinum-sensitive recurrent ovarian cancer. Journal of Clinical Oncology. 2008;26(35):5761–5766. doi: 10.1200/JCO.2008.17.0282. [DOI] [PubMed] [Google Scholar]
- 106.Townsend DM, Pazoles CJ, Tew KD. NOV-002, a mimetic of glutathione disulfide. Expert Opinion on Investigational Drugs. 2008;17(7):1075–1083. doi: 10.1517/13543784.17.7.1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Montero AJ, Jassem J. Cellular redox pathways as a therapeutic target in the treatment of cancer. Drugs. 2011;71(11):1385–1396. doi: 10.2165/11592590-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 108.Townsend DM, Tew KD. Pharmacology of a mimetic of glutathione disulfide, NOV-002. Biomedicine and Pharmacotherapy. 2009;63(2):75–78. doi: 10.1016/j.biopha.2008.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Montero AJ, Diaz-Montero CM, Deutsch YE, et al. Phase 2 study of neoadjuvant treatment with NOV-002 in combination with doxorubicin and cyclophosphamide followed by docetaxel in patients with HER-2 negative clinical stage II-IIIc breast cancer. Breast Cancer Research and Treatment. 2012;132(1):215–223. doi: 10.1007/s10549-011-1889-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zhang Y, Talalay P, Cho CG, Posner GH. A major inducer of anticarcinogenic protective enzymes from broccoli: isolation and elucidation of structure. Proceedings of the National Academy of Sciences of the United States of America. 1992;89(6):2399–2403. doi: 10.1073/pnas.89.6.2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ren D, Villeneuve NF, Jiang T, et al. Brusatol enhances the efficacy of chemotherapy by inhibiting the Nrf2-mediated defense mechanism. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(4):1433–1438. doi: 10.1073/pnas.1014275108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Zhan L, Zhang H, Zhang Q, et al. Regulatory role of KEAP1 and NRF2 in PPARgamma expression and chemoresistance in human non-small-cell lung carcinoma cells. Free Radical Biology and Medicine. 2012;53(4):758–768. doi: 10.1016/j.freeradbiomed.2012.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Fang J, Sawa T, Akaike T, Greish K, Maeda H. Enhancement of chemotherapeutic response of tumor cells by a heme oxygenase inhibitor, pegylated zinc protoporphyrin. International Journal of Cancer. 2004;109(1):1–8. doi: 10.1002/ijc.11644. [DOI] [PubMed] [Google Scholar]
- 114.Fang J, Sawa T, Akaike T, et al. In vivo antitumor activity of pegylated zinc protoporphyrin: targeted inhibition of heme oxygenase in solid tumor. Cancer Research. 2003;63(13):3567–3574. [PubMed] [Google Scholar]
- 115.Cen D, Brayton D, Shahandeh B, Meyskens FL, Farmer PJ. Disulfiram facilitates intracellular Cu uptake and induces apoptosis in human melanoma cells. Journal of Medicinal Chemistry. 2004;47(27):6914–6920. doi: 10.1021/jm049568z. [DOI] [PubMed] [Google Scholar]
- 116.Fruehauf JP, Meyskens FL., III Reactive oxygen species: a breath of life or death? Clinical Cancer Research. 2007;13(3):789–794. doi: 10.1158/1078-0432.CCR-06-2082. [DOI] [PubMed] [Google Scholar]
- 117.Lu J, Chew EH, Holmgren A. Targeting thioredoxin reductase is a basis for cancer therapy by arsenic trioxide. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(30):12288–12293. doi: 10.1073/pnas.0701549104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Dai J, Weinberg RS, Waxman S, Jing Y. Malignant cells can be sensitized to undergo growth inhibition and apoptosis by arsenic trioxide through modulation of the glutathione redox system. Blood. 1999;93(1):268–277. [PubMed] [Google Scholar]
- 119.Maeda H, Hori S, Ohizumi H, et al. Effective treatment of advanced solid tumors by the combination of arsenic trioxide and L-buthionine-sulfoximine. Cell Death and Differentiation. 2004;11(7):737–746. doi: 10.1038/sj.cdd.4401389. [DOI] [PubMed] [Google Scholar]
- 120.Lushchak VI. Glutathione homeostasis and functions: potential targets for medical interventions. Journal of Amino Acids. 2012;2012:26 pages. doi: 10.1155/2012/736837.736837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Cowan KH, Batist G, Tulpule A. Similar biochemical changes associated with multidrug resistance in human breast cancer cells and carcinogen-induced resistance to xenobiotics in rats. Proceedings of the National Academy of Sciences of the United States of America. 1986;83(24):9328–9332. doi: 10.1073/pnas.83.24.9328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Hayes JD, McMahon M. NRF2 and KEAP1 mutations: permanent activation of an adaptive response in cancer. Trends in Biochemical Sciences. 2009;34(4):176–188. doi: 10.1016/j.tibs.2008.12.008. [DOI] [PubMed] [Google Scholar]
- 123.Maher JM, Dieter MZ, Aleksunes LM, et al. Oxidative and electrophilic stress induces multidrug resistance-associated protein transporters via the nuclear factor-E2-related factor-2 transcriptional pathway. Hepatology. 2007;46(5):1597–1610. doi: 10.1002/hep.21831. [DOI] [PubMed] [Google Scholar]