

Abstract
TatCN21 is a membrane permeable CaMKII inhibitor derived from the inhibitor protein CaMKIIN. TatCN21 has been used to demonstrate the involvement of CaMKII in a variety of physiological and pathological phenomena, and it also limits excitotoxic damage in neurons. Here we use preembedding immunogold electron microscopy to examine the effect of tatCN21 on the redistribution of CaMKII in cultured hippocampal neurons. Incubation of cultures with tatCN21 (20 μM for 20 min) prior to exposure to NMDA (50 μM for 2 min) inhibited both the accumulation of CaMKII at postsynaptic densities (PSDs) and CaMKII clustering in the dendrites. Under these conditions, CaMKII also formed morphologically distinct aggregates with polyribosomes near the PSD and in dendrites. Formation of these CaMKII-polyribosome aggregates requires the presence of both tatCN21 and calcium, and was augmented upon exposure to high K+ or NMDA. CaMKII-polyribosome aggregates formed consistently with 20 μM tatCN21, but minimally or not at all with 5 μM. However, these aggregates are not induced by another CaMKII inhibitor, KN93. Formation of CaMKII-polyribosome aggregates was completely reversible within 1 h after washout of tatCN21. Effects of tatCN21 were largely restricted to dendrites, with minimal effect in the soma. The effects of tatCN21 on CaMKII distribution can be used to dissect the mechanism of CaMKII involvement in cellular events.
Keywords: synapse, CaMKII inhibitor, tatCN21, polyribosomes, electron microscopy, PSD
The calcium/calmodulin-dependent protein kinase II (CaMKII) is an abundant protein in brain and is involved in synaptic plasticity and memory formation (reviewed in: Lisman et al., 2002; Coultrap and Bayer, 2012). TatCN21 is a cell permeable specific inhibitor of CaMKII, derived from a naturally occurring CaMKII inhibitory protein CaM-KIIN (Chang et al., 1998; Vest et al., 2007), and inhibits stimulated and “autonomous” CaMKII activity with equal potency (Buard et al., 2010). TatCN21 has been used to study the involvement of CaMKII in a variety of phenomena in neurons, including LTP (Buard et al., 2010; Sanhueza et al., 2011) and excitotoxic insult (Vest et al., 2010; Ashpole and Hudmon, 2011; Ashpole et al., 2012). The effects of tatCN21 in blocking specific cellular events are usually attributed to inhibition of CaMKII activity. This proved to be true for LTP induction (Buard et al., 2010) and glutamate-induced cell death (Vest et al., 2010; Ashpole and Hudman, 2011). On the other hand, certain other effects of the peptide appear to involve an additional mechanism.
TatCN21 reverses LTP and also decreases basal synaptic strength (Sanhueza et al., 2011) at a concentration (20 μM) higher than that (5 μM) sufficient for the inhibition of kinase activity and LTP induction. Fluorescence microscopy experiments using the 20 μM dose of tatCN21 in organotypic slice cultures showed a decrease in the bound pool of CaMKII in spines and co-immunoprecipitation experiments showed disruption of CaMKII-NR2B binding (Sanhueza et al., 2011). These observations suggest that 20 μM tatCN21 could interfere with the accumulation of CaMKII at the postsynaptic density (PSD) and thereby reduce synaptic strength. However, direct evidence for an effect of tatCN21 on CaMKII accumulation at the PSD is still lacking.
Electron microscopy can resolve the regional distribution of CaMKII in the spine as well as the CaMKII accumulation at the PSD. Immuno electron microscopy shows transient translocation of CaMKII to the PSD under excitatory conditions (Dosemeci et al., 2001; 2002), and persistent translocation following the induction of LTP (Otmakhov et al., 2004). Additionally, excitatory and ischemic conditions induce formation of CaMKII clusters at locations not associated with synapses (Dosemeci et al., 2000; Tao-Cheng et al., 2001). In the present study we use immuno electron microscopy to examine how tatCN21 affects the redistribution of CaMKII under excitatory conditions. The activity-dependent translocation of CaMKII to PSD and the formation of extrasynaptic CaMKII clusters were assessed under various experimental conditions in the presence of different doses of tatCN21.
EXPERIMENTAL PROCEDURES
1.1 Materials and antibodies
The membrane-permeable peptide CaMKII inhibitor used in this study, tatCN21 (Vest et al., 2007), contains a cell-penetrating “tat” sequence and a 21-amino acid peptide (CN21, amino acid sequence KRPPKLGQIGRSKRVVIEDDR) derived from CaMKIIN (Chang et al.,1998). The control peptide contains the tat sequence fused to a scrambled sequence (VKEPRIDGKPVRLRGQKSDRI) of CN21 (Vest et al., 2010). KN93, another cell-permeable CaMKII inhibitor, is from Calbiochem (Millipore, Billericam MA). N-methyl-D-asparic acid (NMDA), APV (an NMDA antagonist) are from Tocris (Ellisville, MO).
Mouse monoclonal antibody against α-CaMKII [clone 6G9(2)] is from Millipore (Billerica, MA); rabbit polyclonal antibody against the phosphorylated (T-286) form of CaMKII is from Promega (Madison, WI); rabbit polyclonal antibody against RS18 (a ribosomal marker) is from Epitomics (Burlingame, CA).
1.2 Dissociated hippocampal neuronal cultures and treatments
The animal protocol was approved by the NIH Animal Use and Care Committee and conforms to NIH guidelines. Hippocampal cells from 21-day embryonic Sprague-Dawley rats were dissociated and grown on a feeder layer of glial cells (Lu et al., 1998) for 19 – 21 days.
During experiments, culture dishes were placed on a floating platform in a water bath maintained at 37°C. Control incubation medium was: 124 mM NaCl, 2mM KCl, 1.24 mM KH2PO4, 1.3 mM MgCl2, 2.5 mM CaCl2, 30 mM glucose in 25 mM Hepes at pH 7.4. Whenever indicated, control medium was modified to include 5–20 μM tatCN21 or tat-control peptide, 20–40 μM KN93, 30–50 μM NMDA, 50μM APV, 90 mM KCl (compensated by reducing the concentration of NaCl) or 1mM EGTA with no CaCl2. Cell cultures were washed with control medium and preincubated for 20 min in control medium, in medium containing tatCN21 or control peptide or KN93, as indicated. Samples were then treated for indicated intervals with experimental media - Control, NMDA or high K+ media with the same additions (none, tatCN21, tat-control peptide or KN93) as preincubation. For recovery experiments, samples were processed as above and then washed with control medium (5 times within 2 min), then left in control medium for one hour. Some of the recovered samples were further treated with high K+ or NMDA. In experiments with APV or EGTA, respective media were used both in preincubation and treatment intervals.
1.3 Preembedding immunogold labeling and electron microscopy
Samples were fixed with 4% paraformaldehyde (EMS, Fort Washington, PA) in PBS for 30–45 min, washed, and then blocked and permeablized with 5% normal goat serum and 0.1% saponin for 40–60 min. Samples were incubated with primary and secondary antibodies (Nanogold, Nanoprobes, Yaphand, NY) for 1–2 hr, fixed with 2% glutaraldehyde in PBS for 30 min- overnight, silver enhanced (HQ kit, Nanoprobes), en block mordanted with 0.25–0.5% uranyl acetate in acetate buffer for 1 hr, treated with 0.2% osmium tetroxide in 0.1M phosphate buffer for 30 min, dehydrated in graded ethanols, and embedded in epoxy resin. The primary antibody was omitted in some experiments to control for nonspecific labeling by the secondary antibody.
1.4. Sampling of synapses and classification of synaptic profiles
Synapses are identified by their typical structural characteristics: clustered synaptic vesicles in the presynaptic terminals, rigidly apposed synaptic cleft, and prominent postsynaptic density (PSD; cf. Fig. 1). At least five randomly chosen grid openings from each thin-sectioned sample were sampled. Every cross-sectioned synaptic profile encountered was photographed with a digital CCD camera (AMT XR-100, Danvers, MA, USA).
Fig. 1. The three major patterns of CaMKII distribution in the postsynaptic compartment.
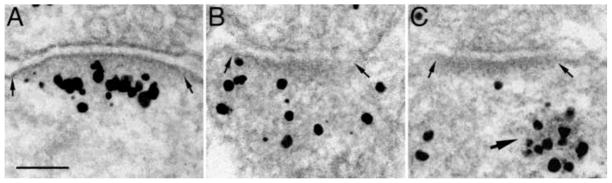
Cultures were immunogold labeled with an antibody to α-CaMKII. Silver enhanced gold label appears as black grains of variable size. (A) CaMKII label accumulated at PSD (between two arrows), indicative of an excitatory state. (B) Label dispersed in the cytoplasm, indicative of a basal, unstimulated state. (C) Label not at the PSD (between two small arrows), but aggregated in a nearby structure (large arrow) containing dark particles, a pattern never seen in the absence of tatCN21. Scale bar = 0.1 μm.
PSD-associated CaMKII labels are reliable indicators of the activity state of a synapse. In control samples where synapses are at their basal states, CaMKII labels are dispersed in the cytoplasm. In contrast, in high K+ or NMDA-treated samples where synapses become activated, CaMKII label accumulates at the PSD (Dosemeci et al., 2001; 2002). In the present study, thin-sectioned synaptic profiles are categorized into 4 groups based on the CaMKII distribution pattern in the postsynaptic dendritic compartments: (1) Accumulated at PSD – CaMKII label accumulates at the PSD more than the surrounding cytoplasm. The great majority of synapses in this category have distinct translocation of CaMKII (cf. Fig. 1A), while approximately 5–20% of synapses in this category has slight to moderate accumulations (picture not shown). (2) Dispersed – CaMKII label is dispersed in the cytoplasm near the PSD (cf. Fig. 1B). (3) Polyribosome aggregate – This category is unique to samples treated with CaMKII inhibitor, tatCN21. CaMKII label is not associated with the PSD but is concentrated in polyribosome aggregate up to 300 nm away (Fig. 1C). (4) A small percentage (0–10%) of synaptic profiles had no CaMKII label (picture not shown, data omitted in Table 1).
Table 1.
Frequency of Different Types of Synapses Categorized by CaMKII Distribution Pattern in Postsynaptic Dendritic Compartments under Various Conditions
| Frequency (%) | ||||
|---|---|---|---|---|
| Condition Preincubation / treatment | Accumulated at PSD | Dispersed | Polyribosome aggregate | |
| Distinct | Slight to moderate | |||
| 1. cont. medium / cont. medium (n=49–90) | 2.5 ± 1.3 | 14.8 ± 4.1 | 81.7 ± 3.1 | 0 |
| 2. cont. medium / NMDA (n=58–68) | 67.8 ± 13.7 | 12.7 ± 4.7 | 12.1 ± 8.7 | 0 |
| 3. tatCN21 / NMDA (n=47–89) | 9.1 ± 2.2 | 13.4 ± 4.0 | 39.5 ± 4.6 | 30.0 ± 3.4 |
| 4. tat-control peptide/NMDA (n=42–76) | 63.3 ± 10.9 | 19.3 ± 4.3 | 15.3 ± 9.0 | 0 |
| 5. tatCN21 / cont. medium (n=42–67) | 3.7 ± 2.9 | 3.4 ± 2.1 | 81.3 ± 2.5 | 5.9 ± 3.0 |
Cell cultures were preincubated for 20 min followed by 2 min treatment.
[tatCN21] and [tat-control peptide]: 20 μM, [NMDA]: 50 μM.
n=Number of synaptic profiles sampled. Frequency (%) is mean ± S.E.M. from three experiments.
The frequency of synapses with CaMKII accumulated at PSD was signigicantly different between the following groups: 1 & 2, 2 & 3, 3 & 4; P<0.005 ANOVA with Tukey HSD test.
1.5 Number of CaMKII-polyribosome aggregates per unit area
For each sample, at least six randomly chosen grid openings of thin sections mounted on 400 mesh, thin-bar grids were thoroughly examined for presence of CaMKII-polyribosome aggregates. Every CaMKII-polyribosome aggregate encountered was photographed and later counted and normalized as number of aggregates per 10,000 μm2. Statistics were carried out as paired t test or ANOVA with Tukey HSD test (Kaleidagraph, Synergy Software, Reading, PA).
RESULTS
2.1 tatCN21 inhibits the activity-dependent translocation of CaMKII to the PSD
Dissociated hippocampal neurons were preincubated for 20 min with a CaMKII inhibitor (tatCN21 at 20 μM) or with control medium, then treated for 2 min with NMDA (50 μM) or control medium, and then immunogold labeled to examine the distribution of CaMKII. Every synaptic profile encountered was classified into one of the defined categories (section 1.4.2). The frequencies of the different categories under different experimental conditions are listed in Table 1.
Under basal conditions (#1 in Table 1), about 80% of the synapses had a dispersed pattern of CaMKII labeling (cf. Fig. 1B), and only ~3% had distinct CaMKII label accumulated at the PSD (cf. Fig. 1A). After NMDA treatment (#2 in Table 1), only 12% of the synapses had a dispersed pattern of CaMKII while 68% of the synapses had CaMKII label distinctly accumulated at the PSD. In samples preincubated with tatCN21 (#3 in Table 1), the NMDA-induced CaMKII translocation onto the PSD was substantially reduced, and less than 10% of the synapses had distinct CaMKII label accumulated at the PSD. The specificity of the tatCN21 inhibition was tested by preincubation with a tat sequence fused control peptide (#4 in Table 1). This control peptide had no inhibitory effect.
The degree of tatCN21 inhibition of NMDA-induced CaMKII accumulation at the PSD is dependent on tatCN21 dosage. In samples preincubated at a lower concentration (5 μM) of tatCN21 before the addition of NMDA, the percentage of synapses with distinct CaMKII accumulation at PSD was 37.1 ± 3.1 % (3 experiments), significantly lower than in the matching samples that were treated with NMDA without preincubation with the inhibitor (72.8 ± 8.4%, P<0.01, ANOVA with Tukey HSD test), but higher than samples preincubated with 20 μM tatCN21 (9.1 ± 2.2%, P<0.05, ANOVA with Tukey HSD test).
2.2 tatCN21 causes formation of CaMKII-polyribosome aggregates that are morphologically distinct from CaMKII clusters
Interestingly, preincubation in tatCN21 produced aggregates of particles that heavily labeled for CaMKII (large arrow in Fig. 1C, Fig. 2A, B). In synaptic profiles that had these aggregates, CaMKII label was typically concentrated within the aggregates and not associated with the nearby PSD (Fig. 2A, B). This pattern of CaMKII distribution was never seen in samples without tatCN21 preincubation, and was more prevalent after treatment with NMDA (Table 1, last column).
Fig. 2. CaMKII-polyribosome aggregates near PSD and in dendrites.

In the presence of the CaMKII inhibitor, tatCN21 (20 μM), CaMKII label is sometimes aggregated within a structure containing dark particles (large arrows in A & B) rather than associated with the PSD (area between small arrows in A & B). These aggregates are also present in dendrites, and are labeled with antibodies specific to T286-phospho CaMKII (C), and to a ribosomal marker, RS18 (D). Insets in C & D were taken from aggregates positively labeled for CaMKII and RS18, respectively. Individual particles in aggregates (small arrows) have the typical size and shape of individual ribosomes, and are identical in the aggregates labeled for CaMKII (inset in C) or RS18 (inset in D). Labels omitted in insets in order not to obscure the underlying ribosomal structure. Scale bar = 0.1 μm for A–D, and 0.05 μm for insets.
In samples preincubated with tatCN21, CaMKII-labeled aggregates were not restricted to synapses, but were also present in dendrites. These CaMKII-labeled aggregates were highly variable in size (100–600 nm), irregular in shape (spherical or elongated), and also labeled with an antibody specific for T286-phospho form of CaMKII (Fig. 2C), consistent with a previous report that tatCN21 does not completely block phosphorylation at this site (Vest et al., 2007).
These aggregates contained tightly packed dark particles identified as ribosomes based on their homogenous size (~25 nm) and an appearance previously described for ribosomes (small arrows in Fig. C & D insets). Polyribosomes are prevalent throughout dendritic shafts and spines in dissociated hippocampal cultures (Tao-Cheng, 2012). The location and occurrence frequency of the CaMKII-labeled aggregates in tatCN21-treated samples (Fig. 2A–C) were consistent with those of polyribosomes in untreated samples (Tao-Cheng, 2012). Finally, an antibody against RS18 (a ribosomal marker) also specifically labeled these aggregates (Fig. 2D), further confirming their identity as polyribosomes. We now term these structures CaMKII-polyribosome aggregates.
These aggregates are morphologically distinct from the spherical CaMKII clusters formed under excitatory conditions (Dosemeci et al., 2000; Tao-Cheng et al., 2001; Tao-Cheng et al., 2007). CaMKII clusters are of uniform size (~110 nm in diameter), and frequently occur in neuronal somas and dendrites, but never in spines or near PSDs, and do not contain ribosomes (Fig. 3B, C, D). Thus, CaMKII clusters are different from CaMKII-polyribosome aggregates in size, shape, location and composition (Fig. 3A vs. 3 B, C & D).
Fig. 3. CaMKII-polyribosome aggregates are different from CaMKII clusters.

A–C show dissociated hippocampal cultures labeled for CaMKII, and D a mouse brain fixed with a delayed perfusion (Tao-Cheng et al., 2007) without CaMKII labeling. (A) CaMKII-polyribosome aggregates can be as large as 600 nm in diameter, and contain ribosomes (double arrow points to an individual ribosome). (B–D) CaMKII clusters (arrows) are uniform in size at ~ 110 nm, have a distinct texture (D), and do not contain ribosomes. Asterisks in B & C mark areas of polyribosomes in nearby cytoplasm lacking CaMKII labeling. Optimal structural preservation in D allows clear differentiation between CaMKII clusters (arrows) and nearby polyribosomes (double arrows). A–C were preincubated with tatCN21 followed by high K+ (A, B) or NMDA (C). A is from a dendrite, B and C are from neuronal somas. D is from a neuronal soma in the hippocampal CA1 region. Scale bar = 0.1 μm.
2.3 tatCN21 inhibits activity-dependent formation of extrasynaptic CaMKII clusters in dendrites
In the absence of tatCN21, a dendrite under basal control conditions typically had CaMKII label dispersed throughout its cytoplasm (Fig. 4A). After high K+ or NMDA treatment, CaMKII clusters (arrowheads in Fig. 4B) sometimes form in dendrites at extrasynaptic locations. In contrast, in the presence of 20 μM tatCN21 for 20 min followed by exposure to control, high K+ or NMDA-containing medium, CaMKII clusters were no longer detected in the dendrites, and CaMKII label was often concentrated into CaMKII-polyribosome aggregates instead (arrows in Fig. 4C). The frequency of CaMKII-polyribosome aggregates was highly variable among different experimental conditions as well as among different dendrites within the same sample. Some dendrites contained dispersed CaMKII label, while others contained variable amounts of CaMKII-polyribosome aggregates. However, no CaMKII clusters were detected anywhere in dendrites in the presence of tatCN21. Thus, tatCN21 effectively blocks the activity-dependent formation of CaMKII clusters in dendrites but induces the formation of the newly described CaMKII-polyribosome aggregates.
Fig. 4. tatCN21 inhibits formation of CaMKII clusters and promotes formation of CaMKII-polyribosome aggregates in dendrites upon excitation.

In the absence of tatCN21 under basal conditions, CaMKII label is dispersed in the cytoplasm and not associated with polyribosomes (asterisks in A). After NMDA treatment, CaMKII clusters (arrowheads in B) often form in dendrites. CaMKII clusters are no longer present in the presence of tatCN21, and CaMKII label is often concentrated in CaMKII-polyribosome aggregates (arrows in C), especially upon excitation. Both the CaMKII clusters (B) and aggregates (C) typically lie near endoplasmic reticulum (ER) or mitochondria (mit). Scale bars = 0.1 μm.
2.4 Formation of CaMKII-polyribosome aggregates is enhanced by excitation and requires calcium
To test the role of excitation in the formation of CaMKII-polyribosome aggregates, the number of aggregates per unit area were compared in samples preincubated with tatCN21 (20 μM for 20 min) followed by treatment in control medium, high K+ depolarization (90 mM for 2 min), or NMDA (50 μM for 2 min). The number of CaMKII-polyribosome aggregates increased significantly after high K+ depolarization (27.5 ± 7.6 vs. 172.3 ± 26.2 per 10,000 μm2, P<0.05, paired t test, 4 experiments) as well as NMDA treatment (22.0 ± 7.8 vs. 244.0 ± 52.3 per 10,000 μm2, P<0.05, paired t test, 4 experiments). These data demonstrate that the formation of CaMKII-polyribosome aggregates is greatly augmented by excitatory conditions following tatCN21 incubation.
When EGTA, a calcium chelating agent, is included in the tatCN21 preincubation and treatment reagents, no CaMKII-polyribosome aggregate was detected either under basal conditions or upon high K+ depolarization. Thus, the formation of CaMKII-polyribosome aggregates requires calcium.
To test the role of [Ca2+] rise induced by NMDA in the formation of CaMKII-polyribosome aggregates, samples were preincubated with tatCN21 (20 μM) in the presence of NMDA receptor antagonist, APV (50 μM). Under basal conditions at which CaMKII-polyribosome aggregates consistently formed, APV reduced the number of aggregates to half (45.6 ± 9.2%, P<0.05, paired t test, 3 experiments) but did not completely block aggregate formation. Thus, Ca2+ from sources other than NMDA receptor may also contribute to the formation of CaMKII-polyribosome aggregates.
2.5 Formation of CaMKII-polyribosome aggregates is tatCN21 dose dependent and minimal at 5 μM
To test whether the number of aggregates per unit area depends on the concentration of tatCN21, samples were preincubated with 5, 10, or 20 μM tatCN21 for 20 min and then treated with either control medium or NMDA. Fig. 5 shows the results from one experiment in which all conditions were tested in parallel. Under basal conditions, the number of aggregates per unit area was minimal at 5 μM tatCN21, but gradually increased with dose. As expected, treatment with NMDA consistently induced higher number of aggregates.
Fig. 5. Formation of CaMKII-labeled polyribosome aggregates is tatCN21 dose-dependent and promoted by NMDA.
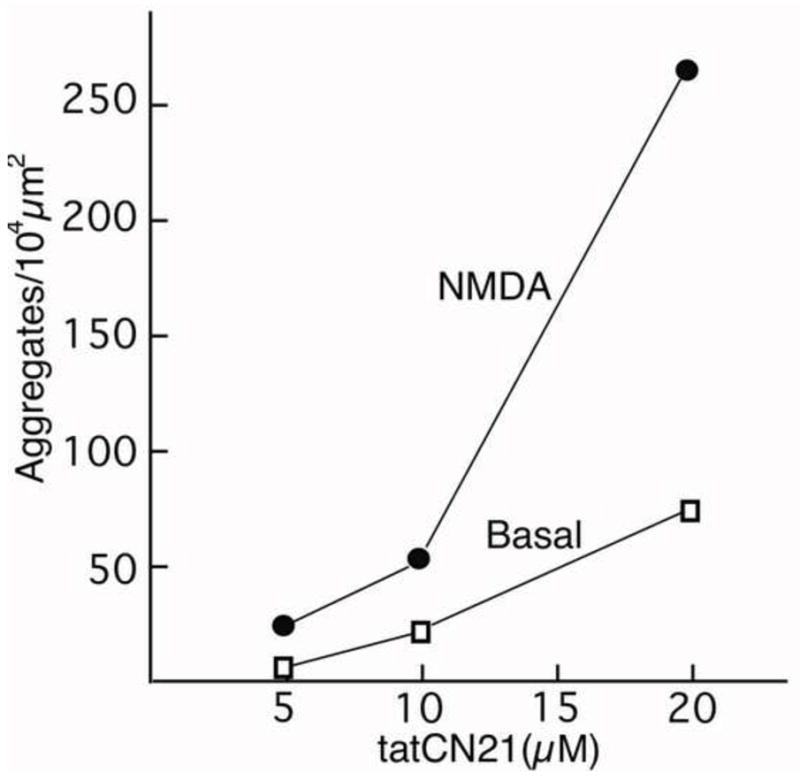
Dissociated hippocampal cultures were preincubated in media containing different concentrations of tatCN21 before exposure for 2 min to control medium (basal) or 50 μM NMDA.
In other experiments that tested 5 μM tatCN21, there was no formation at all of CaMKII-polyribosome aggregates, neither under basal conditions nor after stimulation (3 experiments each with high K+ depolarization or NMDA). In contrast, when 20 μM tatCN21 was tested, formation of CaMKII-polyribosome-aggregates occurred in all 12 experiments under basal conditions, and was consistently more frequent after stimulation. These data further support a strong dependence of CaMKII-polyribosome aggregate formation on the dose of tatCN21, with doses higher than 5 μM tatCN21 needed for robust induction of the aggregates.
To test if formation of these aggregates is specific to tatCN21, KN93, a non-peptide CaMKII inhibitor, was used (20 min preincubation at 20–40 μM, followed by 2 min NMDA). The KN93 concentration used here is much higher than typically used to inhibit CaMKII in cell cultures (cf. 10 μM in Vest et al., 2010; 1 μM in Ashpole and Hudmon, 2011). No CaMKII-polyribosome aggregates were detected in any of the samples treated with KN93 (2 experiments, 3 samples examined in great detail over 20,000 μm2 of thin sectioned area for each sample), while parallel samples treated with tatCN21 had abundant aggregates. Thus, CaMKII-polyribosome aggregates formation is induced by tatCN21 and not by KN93.
2.6 CaMKII-polyribosome aggregates formation is completely reversible after washout of tatCN21
When samples were preincubated with 20 μM tatCN21 followed by excitation, conditions that consistently and robustly produce aggregates, and then thoroughly washed and allowed to recover in control medium for one hour, no CaMKII-polyribosome aggregates remained (one experiment with high K+ depolarization, three experiments with NMDA treatment). Thus, formation of CaMKII-polyribosome aggregates is completely reversible.
After the recovered samples were stimulated again with NMDA, there were no CaMKII-polyribosome aggregates in the re-stimulated samples, but CaMKII clusters appeared in somas and dendrites, and CaMKII label accumulated at the PSDs. Thus, after a one-hour wash out, CaMKII distribution under basal conditions or upon excitation resembles that in the absence of tatCN21.
2.7 CaMKII distribution in hippocampal neuronal somas is minimally affected by tatCN21
CaMKII distribution at excitatory synapses located on neuronal somas was generally unaffected by tatCN21. After tatCN21 preincubation and NMDA or high K+ treatment, most PSDs on somas accumulated CaMKII label (Fig. 6B), and none had CaMKII-polyribosome aggregates nearby. In general, CaMKII-polyribosome aggregates were rare in neuronal somas in samples treated with tatCN21. Even upon stimulation, CaMKII-polyribosome aggregates were typically absent in somas, but CaMKII clusters formed (Fig. 4B, C). In the same samples, CaMKII-polyribosome aggregates could be detected in a primary dendrite (Fig. 6A, enlarged in 6D), but not in the cytoplasm of the soma (Fig. 6A, enlarged in 6C). These results might indicate that hippocampal neuronal somas are less affected by tatCN21 under our experimental conditions.
Fig. 6. Neuronal somas are minimally affected by tatCN21.
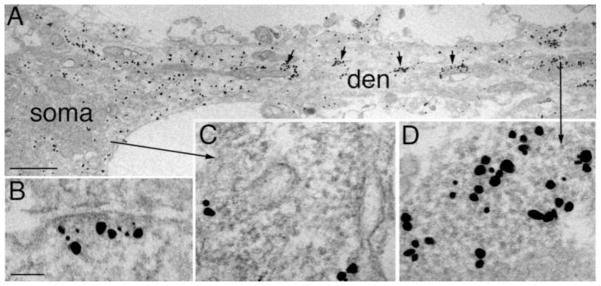
(A) Soma and dendrite from a culture preincubated with tatCN21 followed by NMDA. A primary dendrite (den) contains CaMKII-polyribosome aggregates (arrows in A, enlarged in D), while the soma has dispersed CaMKII label. Polyribosomes in neuronal soma (enlarged in C) are not labeled for CaMKII. An excitatory synapse located on a soma is enlarged in B, showing CaMKII label accumulated at the PSD upon high K+ depolarization. Scale bars = 1 μm in A, 0.1 μm in B–D.
DISCUSSION
Excitation-dependent regulation of CaMKII involves not only stimulation of its kinase activity but also changes in its cellular distribution. A recent study (Sanhueza et al., 2011) indicated that high concentrations of the CaMKII inhibitor peptide tatCN21 (20 μM) interfere with both aspects of CaMKII regulation: kinase activity as well as redistribution. Here, immuno electron microscopy provided direct evidence that tatCN21 inhibits activity-induced accumulation of CaMKII at the PSD, extending conclusions from the previous study using fluorescence microscopy, which showed reduced levels of CaMKII in spines upon incubation with tatCN21 (Sanhueza et al., 2011). However, the present data also reveal that in the presence of tatCN21, CaMKII can aggregate with polyribosomes near PSDs. Thus, in the presence of tatCN21, the bound fraction of CaMKII in spines, as defined by fluorescence microscopy (Sanhueza et al., 2011), cannot be directly equated to be PSD-associated CaMKII.
The inhibitory effect of tatCN21 on NMDA-induced CaMKII accumulation at PSDs is dose-dependent – the frequency of synapses with distinct accumulation of CaMKII dropped from over sixty percent in samples without tatCN21, to forty percent in the presence of 5 μM tatCN21, and to nine percent in the presence of 20 μM tatCN21. The dose-dependent effect of tatCN21 on CaMKII accumulation at the PSD is in line with its dose dependent effect on the maintenance of synaptic strength (Sanhueza et al., 2011). However, the same applied concentrations of the inhibitor would not necessarily result in the same intracellular concentrations in the dissociated cultures used here and in the organotypic slice cultures or acute slices used by Sanhueza et al., 2011.
Immuno electron microscopy revealed a hitherto unsuspected effect of tatCN21 on the redistribution of CaMKII in neurons. Under excitatory conditions, tatCN21 promotes formation of CaMKII-polyribosome aggregates that sequester cytoplasmic CaMKII within dendrites and spines away from the PSD. This observation indicates a preference, in the presence of tatCN21, for CaMKII to bind to partners on polyribosome aggregates rather than those at PSD. However, in the same tatCN21 and NMDA treated samples, 40% of synapses still have dispersed CaMKII label in the postsynaptic cytoplasm. This percentage is higher than the 12–15% of synapses with dispersed CaMKII from samples treated with NMDA in the absence of tatCN21. These data suggest that tatCN21 directly decreases affinity of CaMKII for binding partners at the PSD, independent of the additional aggregation with polyribosomes.
Formation of CaMKII-polyribosome aggregates reverses within an hour after the removal of tatCN21 and could underlie the temporary effect of the inhibitor on synaptic strength (Sanhueza et al 2011). The smaller but persistent decrease in synaptic strength, on the other hand, could be due to the dissociation of CaMKII from PSDs.
The formation of CaMKII-polyribosome aggregates is Ca2+-dependent, in agreement with the known binding requirements of tatCN21 to CaMKII. The peptide binds to the designated ‘T’ site of CaMKII (Vest et al., 2007), which would become accessible in the presence of Ca2+ (review by Coultrap and Bayer, 2012). It is possible that upon binding to CaMKII, the positively charged tatCN21 mediates the interaction with negative charges on the polyribosomes. Although the formation of CaMKII-polyribosome aggregates is dependent on tatCN21, the process does not appear to be a simple consequence of CaMKII inhibition. Indeed, the peptide at a concentration reported to be sufficient for full CaMKII inhibition causes very little, if any, aggregate formation. Furthermore, KN93, a non-peptide CaMKII inhibitor, does not induce any CaMKII-polyribosome aggregate formation even at high concentrations.
CaMKII assembles into CaMKII clusters of uniform diameter (Dosemeci et al., 2000; Tao-Cheng et al., 2001 Tao-Cheng et al., 2007) under certain excitatory conditions. However, after tatCN21 incubation, CaMKII clusters are absent in dendrites. Instead, label for CaMKII often is sequestered in CaMKII-polyribosome aggregates. Thus, in the dendritic compartment, tatCN21 induces CaMKII to form aggregates with polyribosomes instead of self-clustering. Interestingly, formation of both CaMKII clusters (Tao-Cheng et al., 2001) and CaMKII-polyribosome aggregates are calcium dependent, and both are located near endoplasmic reticulum or mitochondria, two organelles involved in Ca2+ regulation.
In contrast to the dendrites, neuronal somas are minimally affected by tatCN21 with respect to both inhibition of CaMKII accumulation at the PSD and to CaMKII-polyribosome aggregate formation. It is evident that [Ca2+] did rise in the soma upon excitation in the presence of tatCN21, as CaMKII translocation to the PSD as well as CaMKII cluster formation, two calcium-dependent phenomenon, indeed occurred. The differential tatCN21 effect on soma and dendrites may result from different effective concentrations of tatCN21 in these two compartments. The large volume of cytoplasm in soma may impede tatCN21 from reaching critical concentration. Alternatively, some components unique to or more prevalent in soma may buffer tatCN21.
In neurons, polyribosomes are present throughout the dendrites and are involved in local protein synthesis (reviewed in Schuman et al., 2006). Polyribosomes are also associated with large spines after LTP (Ostroff et al., 2002). Although some ribosomal proteins are substrates for CaMKII (Mishra-Gorur et al., 2002), aggregating CaMKII with polyribosomes in the presence of tatCN21 should not lead to phosphorylation of ribosomal proteins because the enzymatic activity of CaMKII should be completely blocked at concentrations of the inhibitor necessary to form CaMKII-polyribosome aggregates (Vest et al., 2007; Buard et al., 2010).
tatCN21 effectively inhibits CaMKII autophophorylation on Thr305/6, but to a much lesser degree on Thr286 (Vest et al., 2007). CaMKII autophosphorylation on Thr305/6 blocks calmodulin binding (Patton et al, 1990), while autophosphorylation at Thr286 causes a significant increase in the affinity of CaMKII for calmodulin (Meyer et al., 1992). Thus, it is expected that under excitatory conditions, tatCN21 will maximize calmodulin binding of CaMKII. Considering the fact that CaMKII is present in huge quantities in the brain (Erondu and Kennedy, 1985), the CaMKII-polyribosome aggregates would function as calmodulin sinks. Such a mechanism could contribute to the neuroprotective effect of tatCN21. While tatCN21 was neuroprotective already at 5 μM (Vest et al., 2010), higher concentrations that result in formation of CaMKII-polyribosome aggregates may provide additional protection after stronger insults. Because prolonged (several hours) exposure of neurons to tatCN21 or other CaMKII inhibitors such as KN93 causes sensitization for neuronal cell death (Ashpole and Hudmon, 2011; Ashpole et al., 2012), it will be interesting to determine whether tatCN21 at a concentration that causes formation of CaMKII-polyribosome aggregates enhances or reduces the long-term deleterious effect of CaMKII inhibition.
Finally, it should be noted that tatCN21 peptide is derived from a naturally occurring CaMKII inhibitor protein expressed in brain, CaM-KIIN (Chang et al., 1998). Although the overall intracellular concentration of the endogenous inhibitor is thought to be low (Chang et al., 2001), conditions that lead to temporal or local increases may occur. Indeed Chang et al. (2001) speculated that the decrease in CaMKII activity without a decrease in actual CaMKII concentration or phosphorylation levels following ischemia could be due to a higher expression of the inhibitor. Also, expression of specific subtypes of the inhibitor is assumed to be up-regulated in certain memory models (Lepicard et al., 2006; Radwańska et al., 2010). Our results indicate that the basal expression level of endogenous CaM-KIIN in our hippocampal cultures is not sufficient to induce the CaMKII-polyribosome aggregates described here. However, it is possible that in instances where the endogenous inhibitor reaches sufficiently high concentrations, similar effects to that of tatCN21 on the redistribution of CaMKII may occur.
The CaMKII inhibitor, tatCN21, blocks NMDA-induced accumulation of CaMKII at the PSD.
tatCN21 blocks activity-induced CaMKII clusters in dendrites.
tatCN21 induces formation of CaMKII-polyribosome aggregate near PSD and in dendrites.
Formation of aggregates is enhanced by excitation and depends on the dose of tatCN21.
Formation of CaMKII-polyribosome aggregates is calcium dependent and reversible.
Acknowledgments
We thank Christine A. Winters for hippocampal neuronal cultures, Virginia Crocker and Rita Azzam for expert EM technical support, and Dr. Paul Gallant for helpful discussions and the perfusion-fixed brain in Fig. 3D. Supported by the Intramural Research Program of the NIH, NINDS, and by R01NS052644 (to K.U.B.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ashpole NM, Hudmon A. Excitotoxic neuroprotection and vulnerability with CaMKII inhibition. Mol Cell Neurosci. 2011;46:720–730. doi: 10.1016/j.mcn.2011.02.003. [DOI] [PubMed] [Google Scholar]
- Ashpole NM, Song W, Brustovetsky T, Engleman EA, Brustovetsky N, Cummins TR, Hudmon A. Calcium/calmodulin-dependent protein kinase II (CaMKII) inhibition induces neurotoxicity via dysregulation of glutamate/calcium signaling and hyperexcitability. J Biol Chem. 2012;287:8495–8506. doi: 10.1074/jbc.M111.323915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buard I, Coultrap SJ, Freund RK, Lee YS, Dell’Acqua ML, Silva AJ, Bayer KU. CaMKII “autonomy” is required for initiating but not for maintaining neuronal long-term information storage. J Neurosci. 2010;30:8214–8220. doi: 10.1523/JNEUROSCI.1469-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang BH, Mukherji S, Soderling TR. Characterization of a calmodulin kinase II inhibitor protein in brain. Proc Natl Acad Sci U S A. 1998;95:10890–10895. doi: 10.1073/pnas.95.18.10890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang BH, Mukherji S, Soderling TR. Calcium/calmodulin-dependent protein kinase II inhibitor protein: localization of isoforms in rat brain. Neuroscience. 2001;102:767–777. doi: 10.1016/s0306-4522(00)00520-0. [DOI] [PubMed] [Google Scholar]
- Coultrap SJ, Bayer KU. CaMKII regulation in information processing and storage. Trends Neurosci. 2012;35:607–618. doi: 10.1016/j.tins.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dosemeci A, Reese TS, Petersen J, Tao-Cheng J-H. A novel particulate form of Ca(2+)/calmodulin-dependent protein kinase II in neurons. J Neurosci. 2000;20:3076–3084. doi: 10.1523/JNEUROSCI.20-09-03076.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dosemeci A, Tao-Cheng J-H, Vinade L, Winters CA, Pozzo-Miller L, Reese TS. Glutamate-induced transient modification of the postsynaptic density. Proc Natl Acad Sci U S A. 2001;98:10428–10432. doi: 10.1073/pnas.181336998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dosemeci A, Vinade L, Winters CA, Reese TS, Tao-Cheng J-H. Inhibition of phosphatase activity prolongs NMDA-induced modification of the postsynaptic density. J Neurocytol. 2002;31:605–612. doi: 10.1023/a:1025735410738. [DOI] [PubMed] [Google Scholar]
- Erondu NE, Kennedy MB. Regional distribution of type II Ca2+/ calmodulin-dependent protein kinase in rat brain. J Neurosci. 1985;5:3270–3277. doi: 10.1523/JNEUROSCI.05-12-03270.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepicard EM, Mizuno K, Antunes-Martins A, Von Hertzen LS, Giese KP. An endogenous inhibitor of calcium/calmodulin-dependent kinase II is up-regulated during consolidation of fear memory. Eur J Neurosci. 2006;23:3063–3070. doi: 10.1111/j.1460-9568.2006.04830.x. [DOI] [PubMed] [Google Scholar]
- Lisman J, Schulman H, Cline H. The molecular basis of CaMKII function in synaptic and behavioral memory. Nat Rev Neurosci. 2002;3:175–190. doi: 10.1038/nrn753. [DOI] [PubMed] [Google Scholar]
- Lu Z, McLaren RS, Winters CA, Ralston E. Ribosome association contributes to restricting mRNAs to the cell body of hippocampal neurons. Mol Cell Neurosci. 1998;12:363–375. doi: 10.1006/mcne.1998.0723. [DOI] [PubMed] [Google Scholar]
- Meyer T, Hanson PI, Stryer L, Schulman H. Calmodulin trapping by calcium-calmodulin-dependent protein kinase. Science. 1992;256:1199–1202. doi: 10.1126/science.256.5060.1199. [DOI] [PubMed] [Google Scholar]
- Mishra-Gorur K, Singer HA, Castellot JJ., Jr The S18 ribosomal protein is a putative substrate for Ca2+/calmodulin-activated protein kinase II. J Biol Chem. 2002;277:33537–33540. doi: 10.1074/jbc.C200342200. [DOI] [PubMed] [Google Scholar]
- Ostroff LE, Fiala JC, Allwardt B, Harris KM. Polyribosomes redistribute from dendritic shafts into spines with enlarged synapses during LTP in developing rat hippocampal slices. Neuron. 2002;35:535–545. doi: 10.1016/s0896-6273(02)00785-7. [DOI] [PubMed] [Google Scholar]
- Otmakhov N, Tao-Cheng JH, Carpenter S, Asrican B, Dosemeci A, Reese TS, Lisman J. Persistent accumulation of calcium/calmodulin-dependent protein kinase II in dendritic spines after induction of NMDA receptor-dependent chemical long-term potentiation. J Neurosci. 2004;24:9324–9331. doi: 10.1523/JNEUROSCI.2350-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patton BL, Miller SG, Kennedy MB. Activation of type II calcium/calmodulin-dependent protein kinase by Ca2+/calmodulin is inhibited by autophosphorylation of threonine within the calmodulin-binding domain. J Biol Chem. 1990;265:11204–11212. [PubMed] [Google Scholar]
- Radwańska K, Tudor-Jones AA, Mizuno K, Pereira GS, Lucchesi W, Alfano I, Łach A, Kaczmarek L, Knapp S, Giese KP. Differential regulation of CaMKII inhibitor beta protein expression after exposure to a novel context and during contextual fear memory formation. Genes Brain Behav. 2010;9:648–657. doi: 10.1111/j.1601-183X.2010.00595.x. [DOI] [PubMed] [Google Scholar]
- Sanhueza M, Fernandez-Villalobos G, Stein IS, Kasumova G, Zhang P, Bayer KU, Otmakhov N, Hell JW, Lisman J. Role of the CaMKII/NMDA receptor complex in the maintenance of synaptic strength. J Neurosci. 2011;31:9170–9178. doi: 10.1523/JNEUROSCI.1250-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuman EM, Dynes JL, Steward O. Synaptic regulation of translation of dendritic mRNAs. J Neurosci. 2006;26:7143–7146. doi: 10.1523/JNEUROSCI.1796-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao-Cheng J-H. Activity-Induced Fine Structural Changes of Synapses in Mammalian Central Nervous System. In: Pickel V, Segal M, editors. Structure and Function of the Synapse. Neuroscience-net; 2012. [Google Scholar]
- Tao-Cheng J–H, Vinade L, Smith C, Winters CA, Ward R, Brightman MW, Reese TS, Dosemeci A. Sustained elevation of calcium induces Ca(2+)/calmodulin-dependent protein kinase II clusters in hippocampal neurons. Neuroscience. 2001;106:69–78. doi: 10.1016/s0306-4522(01)00262-7. [DOI] [PubMed] [Google Scholar]
- Tao-Cheng JH, Gallant PE, Brightman MW, Dosemeci A, Reese TS. Structural changes at synapses after delayed perfusion fixation in different regions of the mouse brain. J Comp Neurol. 2007;501:731–740. doi: 10.1002/cne.21276. [DOI] [PubMed] [Google Scholar]
- Vest RS, Davies KD, O’Leary H, Port JD, Bayer KU. Dual mechanism of a natural CaMKII inhibitor. Mol Biol Cell. 2007;18:5024–5033. doi: 10.1091/mbc.E07-02-0185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vest RS, O’Leary H, Coultrap SJ, Kindy MS, Bayer KU. Effective post-insult neuroprotection by a novel Ca(2+)/ calmodulin-dependent protein kinase II (CaMKII) inhibitor. J Biol Chem. 2010;285:20675–20682. doi: 10.1074/jbc.M109.088617. [DOI] [PMC free article] [PubMed] [Google Scholar]