

Abstract
β-Cells of the islet of Langerhans produce insulin to maintain glucose homeostasis. Self-replication of β-cells is the predominant mode of postnatal β-cell production in mammals, with about 20% of rodent β cells dividing in a 24-hour period. However, replicating β-cells are rare in adults. Induction of self-replication of existing β-cells is a potential treatment for diabetes. In zebrafish larvae, β-cells rarely self-replicate, even under conditions that favor β-cell genesis such overnutrition and β-cell ablation. It is not clear why larval β-cells are refractory to replication. In this study, we tested the hypothesis that insufficient activity of cyclin-dependent kinase 4 may be responsible for the low replication rate by ectopically expressing in β-cells a mutant CDK4 (CDK4R24C) that is insensitive to inhibition by cyclin-dependent kinase inhibitors. Our data show that expression of CDK4R24C in β-cells enhanced β-cell replication. CDK4R24C also dampened compensatory β-cell neogenesis in larvae and improved glucose tolerance in adult zebrafish. Our data indicate that CDK4 inhibition contributes to the limited β-cell replication in larval zebrafish. To our knowledge, this is the first example of genetically induced β-cell replication in zebrafish.
Introduction
Controlled genesis of β-cells in situ is a potential treatment for diabetes, as β-cell deficit underlies both type 1 and type 2 diabetes.1–4 Physiologically, β-cells are generated in three ways, neogenesis from precursor cells, self-replication, and transdifferentiation from other cell types. Understanding the molecular mechanisms that control β-cell genesis is essential to harness the capacity for cell-based therapy.3,5–7 In early stage mammalian embryos, β-cells are generated primarily through neogenesis, and self-replication is rare due to high levels of expression of inhibitors of cyclin-dependent kinases.8 In contrast, in perinatal and postnatal rodents, β-cell replication is the major contributor to β-cell mass expansion. β-Cell replication is highest during the perinatal period, occurring in about 20% of β-cells per day in rodents.9,10 In prenatal human embryos, 3.4% of β-cells were found to be positive for Ki67,11 a proliferation marker for all phases of cell cycle. The fraction of dividing β-cells per day may be much higher since the cell cycle length is likely shorter than 24 hours. Replicating β-cells decline precipitously to about 0.07%–3% in adults.9,10,12,13 The decline may result from the increased expression of cell cycle inhibitors and epigenetic modifications of key genes involved in cell cycle regulation.14,15 Under metabolic stress such as obesity and insulin resistance, some β-cells can re-enter the cell cycle to compensate for the increased insulin demand.16,17 However, the molecular underpinnings regulating self-replication of β-cells are not well understood.
Several groups have started to use zebrafish to study β-cell neogenesis, transdifferentiation, and replication.18–23 The zebrafish pancreatic islet shares morphological and physiological similarities with that of mammals.24 Many of the signaling pathways and transcription factors regulating β-cell development in zebrafish are homologous to mammals.24–26 However, unlike rodents, β-cell replication is rare in newly hatched embryos and free feeding larvae. Several groups reported that replicating β-cells are very rare in larvae at 3 dpf,27 4–6 dpf,22 and 6–8 dpf18 using BrdU incorporation as a measure of proliferation. Using PCNA expression as a measurement, Moro and associates found only 3 of 57 larvae at 7 dpf have a single PCNA-positive β-cell.20 Even under metabolic stress that promotes β-cell expansion, such as overnutrition or after β-cell ablation, replicating β-cells are still rarely observed.18,22 Nevertheless, certain adenosine receptor agonists, such as NECA, can stimulate β-cell replication after ablation, suggesting that larval β-cells have the capacity to replicate.22 Why larval β-cells are refractory to self-replication is not known.
A possible reason for the inability of larval β-cells to self-replicate is insufficient activity of cyclin-dependent kinases (CDKs), master regulators of proliferation. In addition to cyclins that activate CDKs, CDK inhibitors (CKI) that inactivate CDKs are also critical for regulation of CDK activity. There are two families of CKIs, the CIP family (p21, p27, and p57) and INK4 family (p15, p16, p18, and p19).28 The first step in proliferation stimulated by extracellular mitogens is induction of D-type cyclins which in turn activates CDK4.28 Active CDK4 phosphorylates and inactivates retinoblastoma (Rb), initiating G1-S progression.28 A mutant CDK4 found in human cancers, CDK4R24C, promotes tumorigenesis because it is insensitive to INK4 CKIs,29,30 the principal negative regulators of CDK4.28 In mice, knock-in of CDK4R24C at the Cdk4 locus results in expansion of endocrine progenitors and β-cell precursors during development and increases β-cell replication in adults.31–34 Moreover, mice expressing Cdk4R24C in β-cells have a 14.3-fold increase in the β-cell mass. These β-cells are functional, as the mice have improved glucose tolerance.35 These results suggest that CKIs may have a key role in controlling β-cell neogenesis and replication.
To determine whether insufficient CDK4 activity contributes to the hindrance of β-cell replication in larval zebrafish, we developed transgenic lines that express the CKI-insensitive CDK4R24C under the control of insulin promoter. Our data indicate that insufficient CDK4 activity, presumably through inhibition by CKIs, contributes to the lack of β-cell self-replication in zebrafish larvae.
Materials and Methods
Zebrafish strains, maintenance, and nutrient treatment
Adult zebrafish were raised in Aquatic Habitats systems on a 14-/10-h light/dark cycle. Embryos were obtained from natural crossing. Fertilized eggs were reared in 0.3× Danieau solution at 28.5°C in an incubator with lights on a 14-/10-h light/dark cycle, and staged following standard methods.36 For egg yolk feeding, chicken eggs were obtained from local grocery stores, the yolk separated and diluted to 5% by volume with 0.3× Danieau solution.37 Larvae were cultured in the egg yolk solution according to Maddison et al.18 The transgenic lines used for this study were Tg(−1.2ins:H2BmCherry)VU506 18. All procedures have been approved by the Vanderbilt University IACUC Committee.
Generation of transgenic zebrafish
We generated a miniTol2-based transgene consisting of human CDK4R24C cDNA38 under the control of the zebrafish insulin gene promoter, using a 1.2 kb fragment from upstream of the initiation ATG and tagRFP under the control of 0.9 kb promoter of zebrafish αA crystalline gene (Fig. 1A). The transgene, Tg(−1.2ins:CDK4R24C; −0.9cryaa:tagRFP)VU508 or Tg(−1.2ins:CDK4R24C; LR)VU508 for short, was mixed with Tol2 transposase mRNA (25 ng/μL each) and injected into zebrafish embryos at the one-cell stage using standard protocol as described.39,40 Embryos with tagRFP expression in lens were selected at 4 dpf after injection, and grown to adulthood. Individual F0 founders were outcrossed to the wild-type AB line, and their F1 progeny were screened for tagRFP expression in the lens and confirmed by PCR using primers zINSGTF:5′-CCACCATTCCTCGCCTCTGCTTC-3′ and hCDK4NR: 5′-CAGATATGTCCTTAGGTCCTGGTC-3′. Initial analyses were performed in two independent lines and similar results were obtained. All results reported here were from F2 or F3 fish of one line that has stronger red fluorescence in the lens, Tg(−1.2ins:CDK4R24C; LR)VU508.
FIG. 1.
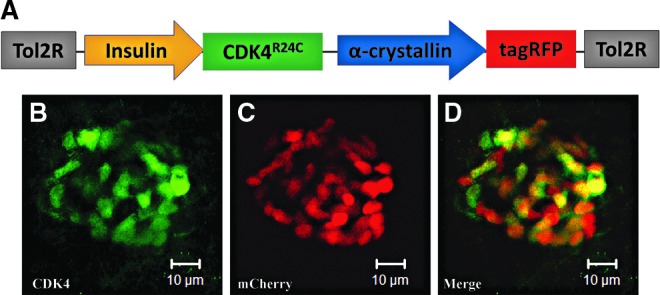
Schematic representation of the Tg(−1.2ins:CDK4R24C; LR) transgenic line and the expression pattern. (A) Expression of a human CDK4 mutant (CDK4R24C) was controlled by the zebrafish insulin promoter with α-crystallin driven tagRFP used as an indication of transgene carriers. (B, C, D) Confocal projections of CDK4R24C-expressing cells in double-transgenic Tg(−1.2ins:CDK4R24C; LR)VU508;Tg(−1.2ins:H2BmCherry)VU506 fish, with CDK4R24C-expressing cells labeled by an antibody against human specific CDK4 protein (B). β-Cells are indicated by nuclear mCherry protein (C).
Counting of β-cells
After fixation in 4% paraformaldehyde (PFA) overnight in 4°C, larvae were washed with 1x PBS plus 0.1% Tween-20 (PBST) and flat mounted in Aqua-Mount (Richard-Allan Scientific) with their right side facing the coverslip. The larvae were flattened sufficiently to disrupt the islet slightly to allow better resolution of individual nuclei. The β-cell number was counted according to the nuclear mCherry signal using a Zeiss AxioImager under a 40x lens or using confocal projections taken by Zeiss LSM510 under a 40x lens (Carl Zeiss).
Whole mount immunofluorescence
The larval zebrafish or dissected pancreata from Tg(ins:CDK4R24C; LR)VU508 or wild-type zebrafish were fixed in 4% PFA overnight at 4°C and then transferred to 100% ethanol and stored at −20°C. Larvae were rehydrated, permeabilized in acetone for 30 min at −20°C, washed in PBST, and nonspecific binding blocked with PBST plus 1.0% DMSO, 1% BSA, and 5% normal goat serum. Subsequently, antibodies for Cdk441 (1:50, Santa Cruz sc-260) or insulin (1:1000, DakoCytomation A0564) antibodies were diluted in blocking buffer and incubated overnight at 4°C. Antibodies were detected using the appropriate Alexa Fluor 488-conjugated secondary antibodies (1:2000, Invitrogen). Slides were imaged on a LSM 510 confocal microscope (Carl Zeiss).
Proliferation analysis
Proliferation was analyzed using the Click-iT EdU Alexa Fluor 488 Imaging Kit (C10337; Invitrogen). For 24 hours labeling, 5 dpf zebrafish larvae were injected pericardially with 3 nL of 100 mmol/L 5-ethynyl-2- deoxyuridine (EdU) and harvested after 24 hours. For long-term incubation, 2 dpf embryos was incubated with 100 μmol/L EdU, and harvested at 6 dpf. EdU was detected according published protocols.18 All images were collected using a Zeiss LSM510 or Zeiss LSM710 (Carl Zeiss).
Glucose tolerance testing
Glucose tolerance was determined in adult fish based on published protocols with the following modifications.42,43 The glucose solution for injection was prepared with Hanks Buffered Salt Solution (Cellgro). Fish were anesthetized using ice water, injected intraperitonially42 with 0.5 mg glucose/g fish weight and allowed to recover for the 30, 90, or 180 min after injection. For blood collection, fish were fully anesthetized in ice water, dried with a Kimwipe to remove as much water as possible. An incision was made just posterior to the gill operculum, resulting in puncture of the heart. Blood was collected into heparinized hematocrit tubes (IRIS Sample Processing) and subsequently transferred into microcentrifuge tubes and frozen at −80°C. Glucose concentration in the samples was determined using Amplex Red Glucose Assay (Life Technologies). The glucose concentration was then normalized to hemoglobin content, which was determined using Drabkin's reagent (Sigma). In our hands, this assay is more sensitive and less variable than the OneTouch Ultra Glucose meter (LifeScan).
Beta-cell area
Adult fish were euthanized and immediately injected with 4% PFA into the peritoneal cavity to ensure rapid fixation of internal organs. The fish were then placed in 4% PFA for 2 days at 4°C. The gastrointestinal tract was removed from each fish and rinsed in PBS+ 0.1% Tween-20, dehydrated in methanol and stored at −20°C. After insulin immunofluorescence using the protocol outlined above, the pancreas was carefully dissected away from the rest of the GI tract, placed on slides, and flat mounted avoiding multiple layers of tissue.
To determine relative β-cell area, 6–10 islet-containing images for each fish were analyzed. The images were obtained under a 20× DIC lens on a Zeiss AxioVert 200 microscope equipped with a CCD camera. The insulin-positive area and the area of the exocrine pancreas for each image was determined using ImageJ software.
Statistics
Differences among groups were analyzed by Student's t-test or by one-way ANOVA, followed by Fisher post-hoc test (SPSS, Chicago, IL). Data are shown as means±standard error (SE). Statistical significance was accepted when p<0.05.
Results
Generation of transgenic zebrafish with constitutive expression of CDK4R24C in β-cells
We obtained two independent Tg(−1.2ins:CDK4R24C; LR)VU508 lines. Although the intensity of lens tagRFP differs in the two lines, their β-cell phenotypes are indistinguishable. The body length of Tg(ins:CDK4R24C; LR)VU508 fish was not different from wild-type siblings at all stages measured (data not shown), indicating that the transgene does not impact growth. To ascertain that CDK4R24C was properly expressed, we crossed Tg(−1.2ins:CDK4R24C; LR)VU508 to Tg(−1.2ins:H2BmCherry)VU506 fish that express a nuclear localized mCherry protein only in β-cells18 and performed whole-mount immunostaining. Robust expression of CDK4R24C protein was detected in β-cells of transgenic fish at 6 dpf, but not in wild-type controls (Fig. 1B–D and data not shown).
CDK4R24C expression increases β-cell number in larvae
To determine the consequence of CDK4R24C expression on β-cell number, the number of β-cells was determined in Tg(−1.2ins:CDK4R24C; LR)VU508; Tg(−1.2ins:H2BmCherry)VU506 fish, and Tg(−1.2ins:H2BmCherry)VU506 siblings at various stages. Similar to other reports,18,19 we found 33.2±1.0 (mean±SE) β-cells in the principal islet of the control group at 6 dpf (Fig. 2A and E). Whereas in Tg(−1.2ins:CDK4R24C; LR)VU508; Tg(−1.2ins:H2BmCherry)VU506 fish, the number of β-cells increased to 41.3±0.9 (Fig. 2B and E). No significant difference in pancreatic α-cells was found between the two groups (data not shown). An increased number of β-cells was found in Tg(−1.2ins:CDK4R24C; LR)VU508; Tg(−1.2ins:H2BmCherry)VU506 fish as early as 3 dpf (32.4±1.1 vs. 28.2±0.7, p<0.05). In each of the subsequent 11 days, roughly one extra β-cell was added in Tg(−1.2ins:CDK4R24C; LR)VU508 larvae compared to control siblings. This results in Tg(−1.2ins:CDK4R24C; LR)VU508; Tg(−1.2ins:H2BmCherry)VU506 fish having 10 more β-cells than Tg(−1.2ins:H2BmCherry)VU506 fish (66.4±3.2 vs. 56.3±1.7, p<0.01) at 14 dpf. At some point after 14 dpf, substantially more β-cells were produced in Tg(−1.2ins:CDK4R24C; LR)VU508 fish. As a result, at 23 dpf there are 40% more β-cells in Tg(−1.2ins:CDK4R24C; LR)VU508 than control (192.6±18.8 vs. 141.1±9.6, p<0.05) (Fig. 2C–E). Taken together, these data indicate that expression of CDK4R24C causes supernumerary β-cells in zebrafish.
FIG. 2.
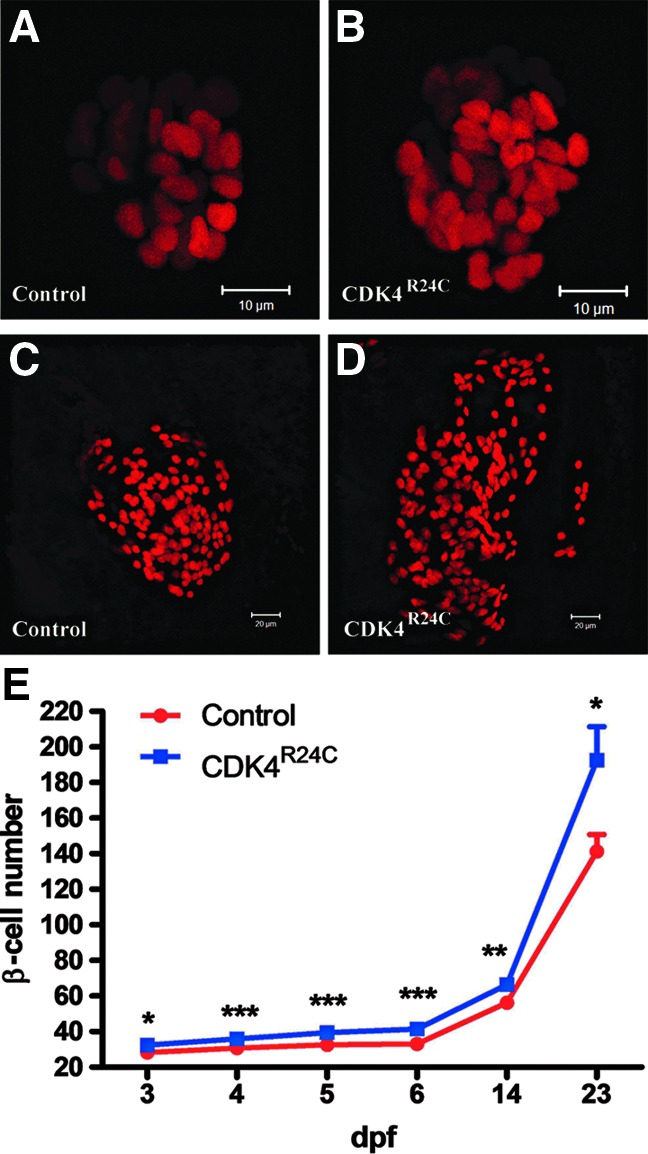
β-Cell specific overexpression of CDK4R24C increases β-cell number. Images of β-cells Tg(−1.2ins:H2BmCherry)VU506 (A,C), and double-transgenic Tg(−1.2ins:CDK4R24C)VU508;Tg(−1.2ins:H2BmCherry)VU506 (B,D) fish at 6 dpf (A,B) and 23 dpf (C,D). Scale bars, 10 μm. (E) β-Cell number in Tg(−1.2ins:H2BmCherry)VU506 and Tg(−1.2ins:CDK4R24C; LR)VU508; Tg(−1.2ins:H2BmCherry)VU506 at 3, 4, 5, 6, 14, and 23 dpf. Expression of CDK4R24C resulted in an increase in the number of β-cells throughout this developmental period. In each group, n=7∼18. The values shown are means±S.E., ***p<0.001, **p<0.01, *p<0.05.
CDK4R24C expression enhances β-cell replication in larvae
To determine whether the supernumerary β-cells in Tg(−1.2ins:CDK4R24C; LR)VU508 fish is due to replication, we labeled replicating cells for 24 h by pericardial EdU injection in 5 dpf larvae. In the control group, no EdU labeled β-cells were observed, consistent with previous data18,20,22 (Fig. 3A). However, we found 2.0±0.5 EdU positive β-cells in Tg(−1.2ins:CDK4R24C; LR)VU508 fish (Fig. 3B and F). These cells presented predominantly as doublets, consistent with β-cell replication. With long-term EdU labeling from 2 dpf to 6dpf, Tg(−1.2ins:CDK4R24C; LR)VU508 larvae had 11.2±0.8 EdU-positive β-cells, while control siblings had only 1.9±0.6 (Fig. 3G–I), indicating that all the extra β-cells in Tg(−1.2ins:CDK4R24C; LR)VU508 fish arose through replication.
FIG. 3.

Increased proliferation in Tg(−1.2ins:CDK4R24C; LR) fish. Twenty-four-hour cell proliferation analysis using EdU labeling in 6-dpf unfed Tg(−1.2ins:H2BmCherry)VU506 (A), Tg(−1.2ins:CDK4R24C; LR)VU508; Tg(−1.2ins:H2BmCherry)VU506 (B), or egg yolk fed Tg(−1.2ins:H2BmCherry)VU506 (C), and Tg(−1.2ins:CDK4R24C; LR)VU508; Tg(−1.2ins:H2BmCherry)VU506 (D) fish. In Tg(−1.2ins:H2BmCherry)VU506 larvae β-cells (red) rarely incorporated EdU, while Tg(−1.2ins:CDK4R24C; LR) VU508; Tg(−1.2ins:H2BmCherry)VU506 have multiple EdU positive β-cells. The images are confocal projections and scale bars indicate 10 μm. (E) Quantification of the β-cell number in 6-dpf Tg(−1.2ins:H2BmCherry) and Tg(−1.2ins:CDK4R24C; LR)VU508; Tg(−1.2ins:H2BmCherry)VU506, in unfed and egg yolk fed conditions. (F) Quantification of 24 h EdU labeled β-cells, in each group, n=8∼9. (G and H) EdU was incubated from 2-dpf to 6-dpf in unfed Tg(−1.2ins:H2BmCherry)VU506 (G) and Tg(−1.2ins:CDK4R24C; LR)VU508; Tg(−1.2ins:H2BmCherry)VU506 (H), the images are confocal slices and scale bars indicate 10 μm. (I) Quantification of 2-dpf to 6-dpf EdU labeled β-cells, in both group, n=10. All the values shown are means±S.E., ***p<0.001, *p<0.05.
To determine whether the transgenic fish with extra β-cells have an altered compensatory response to increased insulin demand, we challenged Tg(−1.2ins:CDK4R24C; LR)VU508 larvae in an overnutrition paradigm that stimulates compensatory β-cell expansion.18 Interestingly, although culturing in 5% egg yolk increased β-cell number in Tg(−1.2ins:CDK4R24C; LR)VU508 fish, (43.6±0.9 vs. 40.6±1.2, p=0.015) (Fig. 3E), the response is dampened compared to WT larvae, with only 3 additional β cells compared to the 7 β-cells added in WT larvae. The increase is likely through neogenesis, since there was not a significant increase of EdU-positive β-cells (2.6±0.6 vs. 2.0.±0.5, p=0.43) (Fig. 3C, D, and F).
CDK4R24C expression improves glucose tolerance
To determine if functional β-cells are continuously produced in Tg(−1.2ins:CDK4R24C; LR)VU508 fish, we determined β-cell content and glucose tolerance in 1-year-old fish. To determine β-cell content, the pancreas of Tg(−1.2ins:CDK4R24C; LR)VU508 fish and wild-type siblings was dissected and stained with an insulin antibody. The insulin-positive areas in Tg(−1.2ins:CDK4R24C; LR)VU508 pancreas was significantly greater than in the control siblings (Fig. 4A and B). The ratio of islet area to exocrine pancreas was ∼2 times greater in the Tg(−1.2ins:CDK4R24C; LR)VU508 fish than in their wild-type siblings (Fig. 4C). To investigate glucose clearance, blood glucose concentrations were examined at 0, 30, and 180 min after IP glucose injection of 0.5 mg glucose/g body weight. As shown in Figure 4D, glucose levels peaked at 30 min and declined to nearly pre-glucose administration levels at 180 min. When compared to control siblings, Tg(−1.2ins:CDK4R24C; LR)VU508 fish had lower blood glucose following glucose injection, although there was no difference in fasting blood glucose. These data suggest that adult Tg(−1.2ins:CDK4R24C; LR)VU508 fish have an increased β-cell mass, and this contributes to improved glucose tolerance.
FIG. 4.

Adult Tg(−1.2ins:CDK4R24C)VU508 fish have increased β-cell mass and improved glucose tolerance. (A, B) Images of insulin positive islets in adult Tg(−1.2ins:CDK4R24C; LR)VU508 (B) and nontransgenic siblings (A), scale bars indicate 20 μm. (C) β-Cell area relative to total pancreas area, 10 control and 8 Tg(−1.2ins:CDK4R24C; LR)VU508 fish were analyzed, **p<0.001. (D) glucose clearance in Tg(−1.2ins:CDK4R24C)VU508 and nontransgenic siblings. Glucose was injected intraperiotneally at 0.5 mg glucose/g body weight and blood glucose levels determined 30 and 180 min after injection. 10–14 fish per time point for each group.
Discussion
Postnatal plasticity of β-cell mass in mammals results mainly from self-replication.44 Yet the molecular mechanisms underlying β-cell replication is not fully understood. In contrast to perinatal mammals, larval zebrafish β-cells rarely replicate, even under conditions that promote β-cell mass expansion. Understanding the mechanism of this replication refractory period should also shed light on regulation of β-cell replication.
CDK4 plays a key role in cell cycle re-entry by integrating extracellular mitogenic and antimitogenic signals. CDK4 is normally inhibited by the INK4 family of CDK inhibitors. This inhibition is relieved by D-type cyclins that can be induced by mitogenic signals.28 To test if insufficient CDK4 activity underlies the replication resistance of larval β-cells, we generated transgenic fish to express CDK4R24C, a mutant that is insensitive to the INK4 family of CDK inhibitors,32,35,45 ectopically in the β-cells. As in mice, targeted expression of CDK4R24C result in increased β-cell content and improved glucose tolerance in adults (Fig. 4).35 Nevertheless, the 2-fold expansion of β-cells compared to wild-type siblings is not as large as seen in the CDK4R24C transgenic mice35, possibly due to intrinsic differences in the two animal models. In larvae, targeted expression of CDK4R24C resulted in one dividing β-cells (or 2%–4%) each day in the first 2 weeks of life. Much more β-cell replication occurred between 14 to 23 dpf. To our knowledge, this is the first report of genetically induced β-cell replication in zebrafish.
Since CDK4R24C still requires cyclin Ds for activity, the increased β-cell self-replication in the transgenic fish suggests sufficient cyclin D is present in β-cells to allow for replication. High levels of INK4 inhibitors relative to D-type cyclins may prevent these cells from self-replicating. More direct analyses will be required to dissect the precise mechanisms and contributions of cyclins and CKIs.
The impetus for generating the transgenic zebrafish model was to understand the lack of β-cell replication in larval zebrafish. Expression of CDK4R24C in zebrafish does lead to replication in larval β-cells (Figs. 2 and 3). Furthermore, these β-cells seem to be functional, since a less robust expansion was induced by overnutrition in the CDK4R24C fish than control siblings and adult transgenic fish are more glucose tolerant (Figs. 3E and 4). However, the fraction of replicating β-cells (2%–4%) is still very small compared to perinatal rodents. In addition, no significant increase of replication in induced by overnutrition in the CDK4R24C fish. The data suggest that insufficient CDK4 activity plays a minor role in limiting self-replication of larval β-cells and other unidentified factors are more predominant players. The adenosine pathway is unlikely to be involved, since its activation only increases β-cell replication after ablation.22 Identification of the factors that control β-cell replication in zebrafish larvae may shed light on the diminished self-replication of β-cells in adult mammals.
Acknowledgments
CDK4R24C construct (Weinberg laboratory, MIT) was obtained from Addgene (Cambridge, MA). This work was supported by the Vanderbilt Diabetes Research and Training Centers and DK088686 (WC).
We utilized the core(s) of the Vanderbilt Diabetes Research and Training Center funded by grant DK02593 from the National Institute of Diabetes and Digestive and Kidney Disease and confocal imaging was performed in the VUMC Cell Imaging Shared Resource (supported by NIH grants CA68485,DK20593, DK58404, HD15052, DK59637 and EY08126).
The authors also thank Dr. Patrick Page-McCaw (Vanderbilt University) for critical reading of the manuscript.
Disclosure Statement
No competing financial interests exist.
References
- 1.Oliver-Krasinski JM. Stoffers DA. On the origin of the beta cell. Genes Dev. 2008;22:1998–2021. doi: 10.1101/gad.1670808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Matveyenko AV. Butler PC. Relationship between beta-cell mass and diabetes onset. Diabetes Obes Metab. 2008;10:23–31. doi: 10.1111/j.1463-1326.2008.00939.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bonner-Weir S. Weir GC. New sources of pancreatic beta-cells. Nat Biotechnol. 2005;23:857–861. doi: 10.1038/nbt1115. [DOI] [PubMed] [Google Scholar]
- 4.Borowiak M. Melton DA. How to make beta cells? Curr Opin Cell Biol. 2009;21:727–732. doi: 10.1016/j.ceb.2009.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhou Q. Brown J. Kanarek A. Rajagopal J. Melton DA. In vivo reprogramming of adult pancreatic exocrine cells to beta-cells. Nature. 2008;455:627–632. doi: 10.1038/nature07314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xu X. D'Hoker J. Stange G, et al. Beta cells can be generated from endogenous progenitors in injured adult mouse pancreas. Cell. 2008;132:197–207. doi: 10.1016/j.cell.2007.12.015. [DOI] [PubMed] [Google Scholar]
- 7.Brennand KJ. Harvard University; 2007. Beta Cell Replication and Differentiation. [Google Scholar]
- 8.Georgia S. Bhushan A. p27 Regulates the transition of beta-cells from quiescence to proliferation. Diabetes. 2006;55:2950–2956. doi: 10.2337/db06-0249. [DOI] [PubMed] [Google Scholar]
- 9.Teta M. Rankin MM. Long SY. Stein GM. Kushner JA. Growth and regeneration of adult beta cells does not involve specialized progenitors. Dev Cell. 2007;12:817–826. doi: 10.1016/j.devcel.2007.04.011. [DOI] [PubMed] [Google Scholar]
- 10.Scaglia L. Cahill CJ. Finegood DT. Bonner-Weir S. Apoptosis participates in the remodeling of the endocrine pancreas in the neonatal rat. Endocrinology. 1997;138:1736–1741. doi: 10.1210/endo.138.4.5069. [DOI] [PubMed] [Google Scholar]
- 11.Kohler CU. Kreuter A. Rozynkowski MC, et al. Validation of different replication markers for the detection of beta-cell proliferation in human pancreatic tissue. Regul Pept. 2010;162:115–121. doi: 10.1016/j.regpep.2009.12.021. [DOI] [PubMed] [Google Scholar]
- 12.Teta M. Long SY. Wartschow LM. Rankin MM. Kushner JA. Very slow turnover of beta-cells in aged adult mice. Diabetes. 2005;54:2557–2567. doi: 10.2337/diabetes.54.9.2557. [DOI] [PubMed] [Google Scholar]
- 13.Finegood DT. Scaglia L. Bonner-Weir S. Dynamics of beta-cell mass in the growing rat pancreas. Estimation with a simple mathematical model. Diabetes. 1995;44:249–256. doi: 10.2337/diab.44.3.249. [DOI] [PubMed] [Google Scholar]
- 14.Chen H. Gu X. Su IH, et al. Polycomb protein Ezh2 regulates pancreatic beta-cell Ink4a/Arf expression and regeneration in diabetes mellitus. Genes Dev. 2009;23:975–985. doi: 10.1101/gad.1742509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dhawan S. Tschen SI. Bhushan A. Bmi-1 regulates the Ink4a/Arf locus to control pancreatic beta-cell proliferation. Genes Dev. 2009;23:906–911. doi: 10.1101/gad.1742609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sachdeva MM. Stoffers DA. Minireview: Meeting the demand for insulin: Molecular mechanisms of adaptive postnatal beta-cell mass expansion. Mol Endocrinol. 2009;23:747–758. doi: 10.1210/me.2008-0400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rhodes CJ. Type 2 diabetes—A matter of beta-cell life and death? Science. 2005;307:380–384. doi: 10.1126/science.1104345. [DOI] [PubMed] [Google Scholar]
- 18.Maddison LA. Chen W. Nutrient excess stimulates beta-cell neogenesis in zebrafish. Diabetes. 2012;61:2517–2524. doi: 10.2337/db11-1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hesselson D. Anderson RM. Beinat M. Stainier DY. Distinct populations of quiescent and proliferative pancreatic beta-cells identified by HOTcre mediated labeling. Proc Natl Acad Sci USA. 2009;106:14896–14901. doi: 10.1073/pnas.0906348106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moro E. Gnugge L. Braghetta P. Bortolussi M. Argenton F. Analysis of beta cell proliferation dynamics in zebrafish. Dev Biol. 2009;332:299–308. doi: 10.1016/j.ydbio.2009.05.576. [DOI] [PubMed] [Google Scholar]
- 21.Gnugge L. Meyer D. Driever W. Pancreas development in zebrafish. Methods Cell Biol. 2004;76:531–551. doi: 10.1016/s0091-679x(04)76024-0. [DOI] [PubMed] [Google Scholar]
- 22.Andersson O. Adams BA. Yoo D, et al. Adenosine signaling promotes regeneration of pancreatic beta cells in vivo. Cell Metab. 2012;15:885–894. doi: 10.1016/j.cmet.2012.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hesselson D. Anderson RM. Stainier DY. Suppression of Ptf1a activity induces acinar-to-endocrine conversion. Curr Biol. 2011;21:712–717. doi: 10.1016/j.cub.2011.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tiso N. Moro E. Argenton F. Zebrafish pancreas development. Mol Cell Endocrinol. 2009;312:24–30. doi: 10.1016/j.mce.2009.04.018. [DOI] [PubMed] [Google Scholar]
- 25.Tehrani Z. Lin S. Endocrine pancreas development in zebrafish. Cell Cycle. 2011;10:3466–3472. doi: 10.4161/cc.10.20.17764. [DOI] [PubMed] [Google Scholar]
- 26.Kimmel RA. Meyer D. Molecular regulation of pancreas development in zebrafish. Methods Cell Biol. 2010;100:261–280. doi: 10.1016/B978-0-12-384892-5.00010-4. [DOI] [PubMed] [Google Scholar]
- 27.Yee NS. Yusuff S. Pack M. Zebrafish pdx1 morphant displays defects in pancreas development and digestive organ chirality, and potentially identifies a multipotent pancreas progenitor cell. Genesis. 2001;30:137–140. doi: 10.1002/gene.1049. [DOI] [PubMed] [Google Scholar]
- 28.Sherr CJ. D-type cyclins. Trends Biochem Sci. 1995;20:187–190. doi: 10.1016/s0968-0004(00)89005-2. [DOI] [PubMed] [Google Scholar]
- 29.Zuo L. Weger J. Yang Q, et al. Germline mutations in the p16INK4a binding domain of CDK4 in familial melanoma. Nat Genet. 1996;12:97–99. doi: 10.1038/ng0196-97. [DOI] [PubMed] [Google Scholar]
- 30.Wolfel T. Hauer M. Schneider J, et al. A p16INK4a-insensitive CDK4 mutant targeted by cytolytic T lymphocytes in a human melanoma. Science. 1995;269:1281–1284. doi: 10.1126/science.7652577. [DOI] [PubMed] [Google Scholar]
- 31.Mettus RV. Rane SG. Characterization of the abnormal pancreatic development, reduced growth and infertility in Cdk4 mutant mice. Oncogene. 2003;22:8413–8421. doi: 10.1038/sj.onc.1206888. [DOI] [PubMed] [Google Scholar]
- 32.Rane SG. Dubus P. Mettus RV, et al. Loss of Cdk4 expression causes insulin-deficient diabetes and Cdk4 activation results in beta-islet cell hyperplasia. Nat Genet. 1999;22:44–52. doi: 10.1038/8751. [DOI] [PubMed] [Google Scholar]
- 33.Kim SY. Rane SG. The Cdk4-E2f1 pathway regulates early pancreas development by targeting Pdx1+ progenitors and Ngn3+ endocrine precursors. Development. 2011;138:1903–1912. doi: 10.1242/dev.061481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Annicotte JS. Blanchet E. Chavey C, et al. The CDK4-pRB-E2F1 pathway controls insulin secretion. Nat Cell Biol. 2009;11:1017–1023. doi: 10.1038/ncb1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hino S. Yamaoka T. Yamashita Y. Yamada T. Hata J. Itakura M. In vivo proliferation of differentiated pancreatic islet beta cells in transgenic mice expressing mutated cyclin-dependent kinase 4. Diabetologia. 2004;47:1819–1830. doi: 10.1007/s00125-004-1522-4. [DOI] [PubMed] [Google Scholar]
- 36.Kimmel CB. Ballard WW. Kimmel SR. Ullmann B. Schilling TF. Stages of embryonic development of the zebrafish. Dev Dyn. 1995;203:253–310. doi: 10.1002/aja.1002030302. [DOI] [PubMed] [Google Scholar]
- 37.Carten JD. Bradford MK. Farber SA. Visualizing digestive organ morphology and function using differential fatty acid metabolism in live zebrafish. Dev Biol. 2011;360:276–285. doi: 10.1016/j.ydbio.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hahn WC. Dessain SK. Brooks MW, et al. Enumeration of the simian virus 40 early region elements necessary for human cell transformation. Mol Cell Biol. 2002;22:2111–2123. doi: 10.1128/MCB.22.7.2111-2123.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kwan KM. Fujimoto E. Grabher C, et al. The Tol2kit: A multisite gateway-based construction kit for Tol2 transposon transgenesis constructs. Dev Dyn. 2007;236:3088–3099. doi: 10.1002/dvdy.21343. [DOI] [PubMed] [Google Scholar]
- 40.Urasaki A. Morvan G. Kawakami K. Functional dissection of the Tol2 transposable element identified the minimal cis-sequence and a highly repetitive sequence in the subterminal region essential for transposition. Genetics. 2006;174:639–649. doi: 10.1534/genetics.106.060244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kiyokawa H. Richon VM. Rifkind RA. Marks PA. Suppression of cyclin-dependent kinase 4 during induced differentiation of erythroleukemia cells. Mol Cell Biol. 1994;14:7195–203. doi: 10.1128/mcb.14.11.7195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kinkel MD. Eames SC. Philipson LH. Prince VE. Intraperitoneal injection into adult zebrafish. J Vis Exp. 2010:42. doi: 10.3791/2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Eames SC. Philipson LH. Prince VE. Kinkel MD. Blood sugar measurement in zebrafish reveals dynamics of glucose homeostasis. Zebrafish. 2010;7:205–213. doi: 10.1089/zeb.2009.0640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dor Y. Brown J. Martinez OI. Melton DA. Adult pancreatic beta-cells are formed by self-duplication rather than stem-cell differentiation. Nature. 2004;429:41–46. doi: 10.1038/nature02520. [DOI] [PubMed] [Google Scholar]
- 45.Kendall SD. Linardic CM. Adam SJ. Counter CM. A network of genetic events sufficient to convert normal human cells to a tumorigenic state. Cancer Res. 2005;65:9824–9828. doi: 10.1158/0008-5472.CAN-05-1543. [DOI] [PubMed] [Google Scholar]