

Abstract
Nutritional symbionts of insects include some of the most bizarre genomes studied to date, with extremely reduced size, biased base composition, and limited metabolic abilities. A monophyletic group of aphids within the subfamily Cerataphidinae have lost the bacterial symbiont common to all other Aphididae (Buchnera aphidicola), which have been replaced by a eukaryotic one, the yeast-like symbiont (YLS). As symbionts are expected to experience reduced effective population size (Ne) and largely clonal life cycles, we used this system as a model to test the hypothesis that chronically high levels of genetic drift will result in an increase in size of a eukaryotic genome. We sequenced the genome of the YLS of the aphid Cerataphis brasiliensis and observed elevated rates of protein sequence evolution and intron proliferation in YLS orthologs relative to those of its closest-sequenced relative, consistent with predictions. A moderate amount of repetitive DNA was found along with evidence of directed mutation to prevent proliferation of repetitive elements. Despite increased intron numbers, the overall genome structure appears not to have undergone massive expansion and is around 25 Mb in size. Compared with Buchnera, the YLS appears to have a much broader metabolic repertoire, though many gene families have been reduced in the YLS relative to related fungi. The patterns observed in the YLS genome suggest that its symbiotic lifestyle is permissive to intron proliferation and accelerated sequence evolution, though other factors appear to limit its overall genome expansion.
Keywords: yeast-like symbiont, Cerataphidinae, effective population size, introns, repeat-induced point mutation
Introduction
Small effective population size (Ne) reduces the efficacy of purifying selection and increases genetic drift, resulting in the increased fixation of deleterious mutations (Kimura and Ohta 1971; Ohta 1972, 1973). The effects of small effective population size have been elucidated through comparative genome sequencing and experimental evolution of bacteria and eukaryotes (Moran 1996; Lynch 2006; McCutcheon and Moran 2012). In both domains, reduced selection results in increased rates of sequence evolution compared with organisms with larger population sizes (Moran 1996; Lynch 2006; Kuo and Ochman 2009). The effect of these deleterious mutations is thought to have different consequences for eukaryotic and bacterial genome architecture.
Because of their stronger deletional bias, bacteria with reduced Ne undergo genome reduction (Mira et al. 2001; Kuo et al. 2009; Kuo and Ochman 2009). Genomes of certain symbionts of insects have shown how extreme this can be: the smallest sequenced cellular genomes are all obligate intracellular symbionts of insects (McCutcheon and Moran 2007; Nakabachi 2007; McCutcheon et al. 2009a; Lopez-Madrigal et al. 2011; McCutcheon and von Dohlen 2011). This habitat subjects the symbionts to severe population bottlenecks during vertical transmission and prevents exchange or acquisition of genetic material, leading to the advancement of Muller’s ratchet (Moran 1996; Mira and Moran 2002). Current models of bacterial genome evolution suggest that an initial burst of transposable element activity leads to inactivation of nonessential genes and large deletions, followed by erosion of the pseudogenes resulting in genome reduction (Mira and Moran 2002; Silva et al. 2001, 2003; Moran and Plague 2004).
Alternatively, eukaryotic genomes are hypothesized to expand when experiencing small Ne, primarily through the gains of mobile genetic elements and intronic sequences (Lynch and Conery 2003; Lynch 2006). Experimental evidence has shown that intronic sequences and mobile genetic elements can proliferate in populations with reduced Ne, and estimates of population size correlate inversely with genome size in eukaryotes (Lynch and Conery 2003; Gao and Lynch 2009; Li et al. 2009). However, in eukaryotic genomes with limited transposon or mobile genetic element activity, a deletional bias can still be observed (Kuo and Ochman 2009) and the genome size of many fungi fall within a range where proliferation of introns or mobile genetic elements may not occur (Lynch 2006). Recently, Kelkar and Ochman (2011) analyzed sequenced fungal genomes in search of a correlation between genome size and signatures of drift. Their results indicate that the pattern of increased drift leading to genome expansion in the phylum is consistent with that observed in eukaryotes, but lineage-specific genes and patterns of evolution distort the general trend when examined at different taxonomic levels. Specifically, certain lineages contained large fractions of novel, lineage-specific genes, which were less likely to exhibit signatures of genetic drift.
In the genomes of the obligate nutritional symbionts sequenced to date, the extent of genome erosion due to drift has resulted in a variety of interesting metabolic consequences. In the aphid symbiont Buchnera aphidicola, approximately 10% of the genome encodes amino acid biosynthesis genes, yet the bacterium is incapable of synthesizing most nonessential amino acids, cell wall components, or membrane lipids (Shigenobu et al. 2000). In several systems, loss of biosynthesis genes in a symbiont has been solved by division of essential nutrient production between multiple symbionts (Wu et al. 2006; McCutcheon et al. 2009b; Lamelas et al. 2011). In the most extreme cases, individual metabolic pathways have been divided between multiple symbionts, such as phenylalanine synthesis in the symbionts of mealybugs (McCutcheon and von Dohlen 2011). Although it is presumed that a eukaryotic symbiont could be capable of replacing the metabolic capabilities of a bacterial symbiont, the specific effects of drift on a eukaryotic symbiont’s metabolic potential are unclear.
To examine the relationship between reduced Ne and genome dynamics and to examine the effect of drift on the metabolic potential of a eukaryotic symbiont, we sequenced the genome of the yeast-like symbiont (YLS) of the aphid Cerataphis brasiliensis. Within the aphid subfamily Cerataphidinae, a monophyletic lineage of aphids has lost the obligate bacterial symbiont Buchnera aphidicola, found in all other members of the Aphididae (Buchner 1965; Fukatsu et al. 1994). In this clade of Cerataphidinae, Buchnera has been supplanted by a fungal symbiont (Fukatsu and Ishikawa 1992). The YLS, a member of the Cordycepitaceae (Suh et al. 2001) is transmitted maternally, as is Buchnera, though the fungus resides both intra- and extracellularly and is not found in the specialized cells Buchnera inhabits (Buchner 1965; Fukatsu and Ishikawa 1992; Suh et al. 2001; Braendle et al. 2003).
We used next-generation sequencing to interrogate the genome of the YLS. Comparative genomic methods were employed to examine whether the YLS genome exhibited signs of increased genetic drift and whether the genome of the symbiont appears to have expanded as predicted by the mutation–hazard model of Lynch and Conery (2003). The metabolic potential of the YLS was also investigated and compared with the metabolism of other insect symbionts.
Materials and Methods
Insect Cultures
Cerataphis brasiliensis aphids were collected from a population in Coral Gables, FL, at the Fairchild Tropical Botanic Garden from Cryosophila palms. A lab population was established at the University of Arizona from a single wingless female reared on Washingtonia robusta and W. filafera palm seedlings. Aphids were reared at 25 °C and 85% relative humidity on a 16:8 light-to-dark cycle.
DNA Extraction
Cerataphis brasiliensis (300–500 mg) was collected from lab cultures, washed twice in 75% ethanol, and then rinsed in sterile de-ionized water to remove the waxy secretions produced by adults and any external fungi. Aphids were then crushed in a 1.5 ml pestle tube with a plastic pestle in the presence of acid-washed 100 µm glass beads. The homogenate was suspended in 1 ml of buffer A (35 mM Tris pH 7.5, 25 mM KCl, 10 mM NaCl, and 250 mM sucrose). Aphid homogenate was filtered progressively through 100 and 20 µm nylon mesh. The resulting filtrate was pelleted then resuspended in DNAse I buffer (Fermentas) and treated with DNase I to remove aphid DNA. Fungal cells were pelleted by centrifugation and disrupted with Driselase (5 mg/ml) and Kitalase (6 mg/ml) in 0.7 M NaCl for 25 min at 30 °C. Isolation buffer (50 mM Tris pH 7.5, 100 mM ethylenediaminetetraacetic (EDTA) acid, 0.5% sodium dodecyl sulphate, 0.3 M NaOAc pH 8) and proteinase K (30 mg/ml) were used to lyse protoplasts. Crude DNA was precipitated by addition of 7.5 M NH4OAc and 100% isopropanol. DNA was then washed with phenol:chloroform:isoamyl alcohol. The resulting DNA was precipitated with 3 M NaOAc and 100% ethanol and stored in Tris-EDTA buffer prior to use.
Sequencing
Genomic DNA was sequenced at the Yale Center for Genome Analysis at Yale University. YLS genomic DNA was used to construct a paired-end library with an average 380 bp insert according to the Illumina Paired-End DNA Sample Preparation Kit. The library was sequenced on one lane of an Illumina HiSeq 1000 with 76 bp reads.
Quality Filtering and Assembly
Paired-end sequences were sorted by quality scores with an in-house perl script. Only sequences with 76 bases with PHRED-equivalence scores of 20 or higher and no ambiguous bases on both reads were kept. 115,878,346 paired-ends passed the filter. An additional 25,311,828 reads were kept in which the left or right read failed the quality filter but the paired read passed. Paired and single reads were randomly separated into three sets. Each set was independently assembled using Velvet (Zerbino and Birney 2008) with kmer = 43. The three assemblies were merged with minimus2, generating 34,063 contigs greater than 500 bp in length with an average coverage of 39× and an N50 length of 4,370 bp.
Contigs were also used as queries against the Genbank nonredundant (nr) protein database using BLASTx. Dinucleotide frequency analysis was performed using scripts provided by Vincent Denef and visualized using the ESOM program from Databionics (Ultsch and Moerchen 2005). Contigs were separated into two distinct bins based on the dinucleotide frequency analysis. All contigs with hits to fungal sequences were in a one bin (bin 0), which contained no contigs with strong hits to aphid sequences. Reads were then mapped back to the remaining contigs using BWA (Li and Durbin 2009) to determine coverage. GC content, ESOM bin, and coverage statistics were all determined for contigs with hits to fungi and insects. Contigs with top hits to fungal sequences had higher GC content (56%) than contigs with top hits to aphid sequences (22% GC) and higher coverage (mean coverage 35× for fungal contigs and 5× for aphid). Contigs with GC content over 45%, coverage over 10× and in the ESOM bin 0 were kept for further analysis. Reads mapping to these contigs were used to perform another assembly using Velvet, and was also visualized using Tablet (Milne et al. 2010) to assess assembly errors and possible allelic variants that were not separated in the assembly.
Annotation
Novel repetitive regions were identified with the program RepeatScout (Price et al. 2005), the output of which was combined with fungal repeats downloaded from RepBase version 17.1 (Jurka et al. 2005) to create a custom library of repetitive elements for the genome. Repetitive elements were masked using RepeatMasker version 3.3.0 (Smit et al. 2010). These elements were also used to assess the presence of repeat-induced point (RIP) mutations using the software RIP-cal (Hane and Oliver 2008). To assess the age of repetitive elements in the YLS genome, all elements with a copy number more than 4 were collected and aligned using MAFFT. These alignments were used to calculate average pairwise genetic distance within a repeat group using the program Dnadist and the F84 model in the PHYLIP package (Felsenstein 2005).
Masked fungal contigs over 10 kb were used as a training set for the self-training portion of GeneMark-es version 2.0 (Ter-Hovhannisyan et al. 2008). Genemark-e was then implemented to identify coding regions and introns. Transfer RNAs (tRNAs) were identified using tRNA-scan (Lowe and Eddy 1997), while metabolic pathways and information were mapped using the KEGG Automatic Annotation Server (KAAS, (Moriya et al. 2007). All open reading frames (ORFs) were queried against the NCBI nonredundant protein database using BLASTX. ORFs were also searched against the Pfam database version 26.0 (Punta et al. 2012) using Pfam-scan and against the pathogen–interaction database (PHI-base; Winnenburg et al. 2008), then families were identified in the Pfam database. Significant differences in gene family membership were determined using Café (De Bie et al. 2006).
Phylogenetics
To determine the closest fully sequenced relative of the YLS, gene sequences for Rpb1, Rpb2, and Ef1 were identified in YLS contigs based on KEGG annotation and BLAST hits. These YLS genes were used as queries against the sequenced genomes of the order Hypocreales (fig. 1). Gene sequences were downloaded from NCBI and aligned using MAFFT. Aligned sequences were trimmed to remove all gaps using Mesquite version 2.74 (http://www.mesequiteproject.org, last accessed December 1, 2011). A tree was constructed using PhyML (Guindon et al. 2010), using 100 bootstrap replicates and rooted with sequences from Aspergillus niger. Trees were visualized in FigTree.
Fig. 1.—

Cerataphis brasiliensis and the YLS. (A) Phylogenetic relationship of the YLS to sequenced fungal genomes based on a three-gene amino acid phylogeny. Phylogeny based on maximum likelihood tree of concatenated rpb1, rpb2, and ef1α amino acid sequence with 100 bootstraps. Thickened lines represent branches with maximum likelihood support >0.9 and bootstrap support >85. The closest sequenced relatives of the YLS are the entomopathogenic fungi Cordyceps militaris, Metarhizium anisopliae, and M. acridum, highlighted in blue. (B) YLS cells isolated from C. brasiliensis under phase-contrast microscopy. Scale bar = 20 µm. (C) C. brasiliensis feeding on leaves of W. filifera.
Evolutionary Rates Analysis and Deletional Bias
Comparison of evolutionary rates was performed by identifying all shared orthologs of the YLS, Metarhizium anisopliae (Gao et al. 2011) and Nectria haematococca (Coleman et al. 2009) by collecting three-way reciprocal best hits. The orthologs were then aligned in MUSCLE (Edgar 2004), and codeml (Yang 2007) was implemented to measure branch lengths on all trees using a restricted tree topology with M. anisopliae and the YLS being an ingroup to N. haematococca. The analysis was repeated with the genome sequences of Cordyceps militaris (Zheng et al. 2011) and Fusarium oxysporum (Ma et al. 2010).
The rate of nonsynonymous to synonymous substitutions (dN/dS) was estimated in codeml of the PAML package between the YLS, M. anisopliae and N. haematococca using “model = 2” and allowing rates to vary across branches (NSsites = 2). Only comparisons where all values of dS were below saturation (dS < 2) were considered. Radical to conservative amino acid substitution rates were calculated with HON-NEW (Zhang 2000).
Attempts were made to assess whether mutational patterns result in deletional bias in the YLS using previously established methods (Kuo and Ochman 2009). Homologous introns were collected from orthologous genes with single introns that were located at a similar position in all three orthologs (all intron start sites within 24 bp of each other). Orthologous introns were aligned using Muscle, and then manually inspected for well-aligned sequences. Similarly, intergenic spacers residing between pairs of syntenic orthologs were aligned and inspected. Because of the high level of divergence between the YLS and M. anisopliae, no useful alignments could be obtained for either orthologous introns or intergenic spacers, and further analysis of deletional biases was not possible.
Synteny Analysis, Intron Size, and Frequency
Gene coordinates for the YLS, M. anisopliae and N. haematococca were determined from gene feature files. The nucleotide sequence for each orthologous gene was obtained and aligned to the amino acid sequence using the program Genewise (Birney et al. 2004) revealing the intronic features of each ortholog. Only YLS genes for which the peptide sequence was at least 98% of the length of the N. haematococca sequence were considered for further analysis.
Results
Genome Sequencing
General features of the YLS draft genome are shown in table 1. A total of 25.4 Mb of sequence in 1,182 contigs with an N50 of 12,424 bp was binned as fungal according to coverage, GC content, and BLAST hits to fungi. This figure is potentially an underestimate of the YLS genome size, as contigs lacking ORFs were excluded and repetitive regions may have assembled together. Six contigs have terminal fungal telomeric repeat “TTAGGG/CCCTAAn,” indicating that the YLS has at least three chromosomes, though this is almost certainly an underestimate. Examination of the assembly and subsequent read mapping and a self-self BLAST search provided no evidence of polyploidy, which is consistent with the haploid state of most fungi in their “yeast-like” state though many bacterial symbionts, including Buchnera, display remarkable levels of polyploidy (Komaki and Ishikawa 1999; Vogel and Moran 2011). Ab initio annotation revealed 6,960 ORFs greater than 92 amino acids, giving a gene density of 274 genes per Mb, which is similar to the density observed in M. anisopliae and M. acridium (271 and 258 genes per Mb, respectively). Within the YLS genome sequence, we found 105 tRNA genes representing all 64 codons. The completeness of the set of essential genes (i.e., for complete ribosomal proteins and DNA/RNA polymerases) suggests that our sequencing captured nearly the entire protein-coding region of the YLS genome. The absence of high-coverage contigs encoding repetitive elements and the completeness of the gene set suggest that the genome is unlikely to be drastically larger than 25 Mb. In comparison with other sequenced fungal genomes in the Sordariomyceta, the YLS genome is noticeably smaller, though it maintains a similar coding density (fig. 2).
Table 1.
General Features of the Draft Genome Sequence of the YLS of Cerataphis brasiliensis
| Sizea (Mb) | 25.42 |
| No. of contigs | 1,182 |
| N50 | 12,424 |
| No. of ORFs | 6,960 |
| % Repetitive seqeuncea | 4.7 |
| % GC | 54 |
| tRNA operons | 106 |
aOnly contigs remaining after filtering by GC content, coverage, and BLAST hit to nonarthropod.
Fig. 2.—
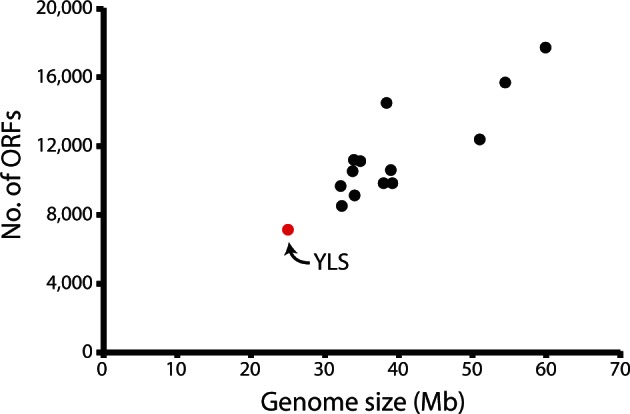
Size comparison of sequenced genomes of Sordariomyceta shown in figure 1. ORF numbers and genome size are those reported in NCBI database or the genome sequence publication. YLS ORF numbers were determined by de novo annotation as described in the Materials and Methods. Genome size for the YLS was estimated by summing the length of all contigs that were determined to be fungal as described in the Materials and Methods.
Phylogenetic Placement
Analysis of the aligned Rpb1, Rpb2, and Ef1 amino acid sequences of YLS and other species of Hypocreales supported placement of YLS within the Cordycepitaceae (fig. 1), as previously proposed on the basis of ribosomal RNA sequences (Suh et al. 2001). The entomopathogenic fungi Cor. militaris, M. anisopliae, and M. acridum are the closest sequenced relatives of the YLS, with F. oxysporum and N. haematococca more distantly related.
Evolutionary Rates
Bacterial symbionts are noteworthy for their accelerated rates of molecular evolution, often exhibiting rates several times higher than those of their free-living relatives (Moran 1996; Herbeck et al. 2003). The YLS symbiont of C. brasiliensis exhibits higher rates of evolution than do related free-living fungi, though the acceleration is not as extreme as that seen in some bacterial symbionts (McCutcheon and Moran 2012). At the amino acid level, orthologs shared between the YLS, M. anisopliae and N. haematococca are evolving on average 2.47 times faster than in M. anisopliae (table 2). This ratio is significantly higher than 1, which is the expected ratio if both genomes were evolving at the same rate (z statistic2,396 = 23.27, P < 0.0001). We also compared orthologs of the YLS with Cor. militaris and F. oxysporum, and again detected a significant increase in the YLS sequences (1.37, z statistic2,396 = 20.42, P < 0.0001), though this increase was smaller than the increase observed in the previous comparison. The lower increase observed in the YLS:Cor. militaris comparison may be related to reduced effective population size due to the limited host range of Cor. militaris (Zheng et al. 2011). These rates are consistent with amino acid substitution rates estimated for Buchnera (Moran 1996; Itoh et al. 2002), suggesting that the symbiotic environment has led to a similar increase in the substitution rate in the YLS.
Table 2.
Relative Rates Test of Orthologs of the YLS Compared to Free-Living Fungi
| Taxon A | Taxon B | Taxon C | N | Average Kab | Average Kac | Average Kbc | Kac–Kbc | Koa–Kob | Koa/Kob |
|---|---|---|---|---|---|---|---|---|---|
| YLS | M. anisopliae | N. haematacocca | 2,561 | 0.237 (0.241–0.234) | 0.266 (0.269–0.263) | 0.195 (0.198–0.192) | 0.071 (0.074–0.068) | 0.0709 (0.073–0.068) | 2.471* (2.596–2.347) |
| YLS | C. militaris | F. oxysporum | 2,397 | 0.297 (0.302–0.291) | 0.270 (0.275–0.264) | 0.246 (0.251–0.240) | 0.024 (0.027–0.021) | 0.024 (0.027–0.021) | 1.379** (1.416–1.343) |
Note.—Branch lengths were estimated for the amino acid sequence of each ortholog using codeml in the PAML package. Numbers in parentheses represent upper and lower 95% confidence intervals, bold number is average value for each comparison.
*Significantly different from ratio of 1 with similar standard deviation (z statistic2,396 = 23.27, P < 0.0001).
**Significantly different from ratio of 1 with similar standard deviation (z statistic2,396 = 20.42, P < 0.0001).
To further measure the evolutionary rates of the YLS, dN/dS was estimated for 452 YLS orthologs in which dS was not saturated. Relative to M. anisopliae, the YLS orthologs exhibited a significantly higher average dN/dS ratio (dN/dSYLS-Nh: 0.163, dN/dSMa-Nh: 0.118; t ratio450 = −5.20, P < 0.0001). Signatures of positive selection on YLS orthologs were not detected using this test: no YLS ortholog exhibited a dN/dS ratio above 1. Positive selection likely occurred in some YLS genes, though we currently lack the data necessary to detect it (i.e., polymorphism data or a more closely related fungal genome sequence).
Reduced selection has been shown in symbionts to elevate the rate of radical amino acid substitutions (dR) relative to conservative ones (dC), as the later are presumed to be more deleterious (Wernegreen 2011). Examination of deleterious mutations in YLS orthologs reveals a small but significantly elevated rate of radical substitution (dR/dCYLS-Nh: 0.545, dR/dCMa-Nh: 5.32; t ratio2,120 = −5.6, P < 0.001). The elevated rate of amino acid substitution, higher rate of dN/dS and dR/dC are consistent with an increased fixation of slightly deleterious mutations as expected with accelerated evolution due to higher genetic drift.
Intron Frequency and Size
In experimental evolution studies of Daphnia, small population size and strong drift have been shown to lead to intron gains (Li et al. 2009). We assessed intron frequency and size in the genomes of the YLS, M. anisopliae and N. haematococca to determine whether drift has led to an increase in the number of genes with introns, the number of introns per gene or the average length of introns (fig. 3). We compared intron number and size among the sequenced genomes of the YLS, M. anisopliae, and N. haematococca. The genes examined in the YLS had significantly more introns per gene (1.79 introns/gene) than the orthologs in either M. anisopliae (1.60 introns/gene, t ratio1,659 = −9.55, P < 0.0001) or N. haematococca (1.75 introns/gene, t ratio1,659 = −2.16, P < 0.0001). M. anisopliae had significantly fewer introns per ortholog than N. haematococca, suggesting that both gain of introns in the YLS orthologs as well as loss of introns in M. anisopliae orthologs contribute to the difference between the two.
Fig. 3.—
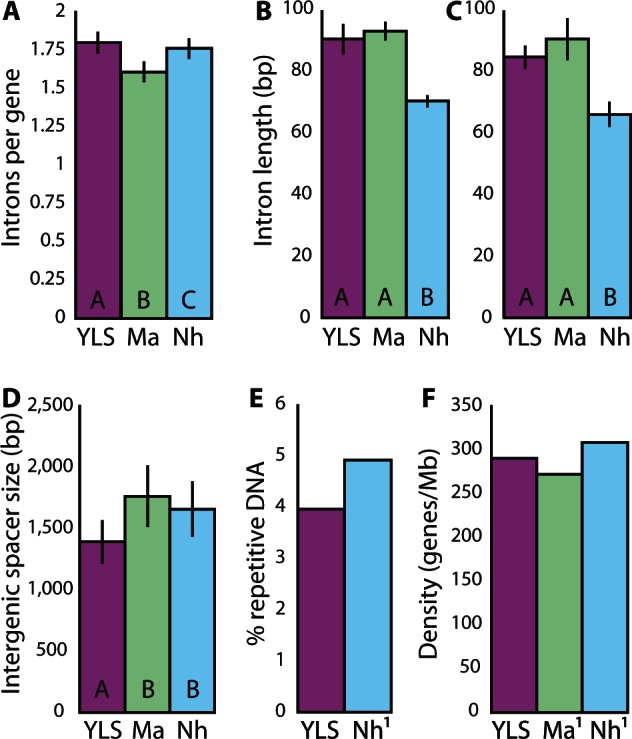
Comparison of genome features between the YLS, Metarhizium anisopliae (Ma) and Nectria haematococca (Nh). Letters within bars indicate significant differences at the levels noted below. Error bars in all cases represent the 95% confidence interval. (A) Average number of introns in orthologs of the three genomes. Number of introns was compared for each ortholog pairwise between the genomes. The YLS had significantly more introns per gene (1.79) than orthologous genes in either Ma (1.60 introns/gene; t ratio1659 = −9.55, P < 0.0001) or Nh (1.75 introns/gene; t ratio1659 = −2.16, P < 0.0001). Introns were only compared for YLS genes that were at least 98% as long as their Nh ortholog. A Wilcoxon rank-sum test indicated that the average difference in intron number between orthologs of the YLS and the other genomes was greater than 0, indicating a net gain of introns in the YLS (YLS:Ma P value < 0.0001; YLS:Nh P value = 0.033). (B) Average intron length in intron-containing orthologs of the YLS, Ma, and Nh. Only orthologs with at least one intron were considered. There was no significant difference between the length of introns in the YLS (90.5 bp) and Ma (93.2 bp; t ratio1255 = 1.02, P value = 0.30) though there was a significant increase in intron length relative to Nh (ratio1255 = −7.89, P value < 0.0001). (C) Average intron length in orthologs containing exactly 1 intron in all genomes. The average intron size of Ma was nearly significantly larger than that of the YLS (t ratio355 = 1.77, P value = 0.077). The YLS and Ma both had significantly larger introns than Nh (t ratio355 = −10.94, −15.91, respectively; P values < 0.0001). (D) Intergenic spacer size for pairs of syntenic orthologs in the three genomes. Intergenic spacers were significantly larger in the genomes of Ma and Nh relative to the YLS (YLS:Ma t ratio105 = 4.56, P value < 0.0001; YLS:Nh t ratio105 = 2.81, P value = 0.005). (E) Percent repetitive DNA in the genomes of YLS and Nh. Data for Ma was not comparable. (F) Gene density within the three genomes. 1Values are those reported in Gao et al. (2011) and Coleman et al. (2009) for Ma and Nh, respectively.
Another potential effect of reduced selection is the expansion of intron size. To compare intron sizes, the average length of all introns within an ortholog was determined for all orthologs with at least one intron. Compared with orthologs in M. anisopliae, there was no significant difference in intron size in the YLS (fig. 3). M. anisopliae orthologs had an average intron size of 93.2 bp, whereas the YLS had an average intron length of 90.5 bp (t ratio1,252 = 1.02, P = 0.31). The introns of the YLS and M. anisopliae were both significantly longer than the introns of N. haematococca (70.4 bp, t ratio1,252 = −7.89, P < 0.0001). Because of varying number of introns per ortholog, the average intron size includes comparison of nonhomologous introns. To reduce the effects of comparing nonhomologous introns, we also compared intron size between orthologs with only a single intron in each ortholog. YLS introns are not significantly different in size from their orthologous introns in M. anisopliae (84.7 and 90 bp, respectively, t ratio356 = 1.77, P = 0.077).
Intergenic spacers are also a potential point of genome expansion due to reduced efficacy of selection. Although recombination has disrupted all large regions of synteny between the three genomes, some regions of microsynteny persist. We measured the intergenic space between syntenic pairs of genes among the three genomes. The average intergenic spacer between YLS genes was 1,385 bp, compared to 1,758 bp in M. anisopliae and 1,653 bp in N. haematococca (fig. 3). The spacers in the two comparison genomes were significantly larger than the spacers in the YLS genome (YLS:Ma t ratio105 = 4.57, P < 0.0001; YLS:Nh t ratio105 = 2.81, P = 0.006).
Mobile and Repetitive Elements
Repetitive and mobile elements are predicted to proliferate in the early stages of bacterial symbiont evolution due to relaxed selection to maintain superfluous genes (Moran and Plague 2004). Within the contigs that were binned as fungal, 1.12 Mb of repetitive sequence were found in 8,108 sequences by RepeatMasker (Smit et al. 2010) representing 4.7% of the draft genome (fig. 3 and table 1). This is similar to the genome-wide amount of repetitive DNA in N. haematococca, though the distribution of repetitive sequence is highly heterogeneous (Coleman et al. 2009). Most families of repeats had fewer than 10 members (fig. 4A). All but 18 of these sequences were unclassified repeats found using RepeatScout. Of the 18 classified repetitive regions, 5 were long interspersed nuclear elements (LINES), 6 were long terminal repeats (LTRs), and 7 were DNA elements. Within the annotated ORFs, BLAST searches using the 18 identified transposons as queries revealed a total of 55 copies of these elements. Because of grouping of similar repetitive elements during assembly, it is possible that we have underestimated the total amount of repetitive DNA in the YLS genome as many repetitive regions may have assembled together. However, the average coverage of these regions is not elevated with respect to other fungal contigs.
Fig. 4.—

(A) Distribution of repetitive element abundance within each repetitive element family. (B) Average genetic distance of repeat elements in the YLS genome. The average genetic distance for each family of repeat element determined by Repeat Scout was measured using DNAdist in the PHYLIP package. There was no relationship between the abundance of a family (number of members) and the average genetic distance within a family (r2 = 0.0012, P = 0.47).
To examine the age of the repetitive sequences in the YLS genome, we measured the average pairwise genetic distance within each of the 407 families with more than 4 copies. The distribution of average pairwise distances within a repeat family shows a mean of 0.098 substitutions/site (fig. 4B). Although some elements have duplicated recently and exhibit low divergence, most of duplications of repetitive elements in the YLS genome have larger pairwise distances and are not recent.
RIP mutation is a mechanism by which fungi inactivate repetitive DNA through directed CpN → TpN mutations (Selker 1990). Given the lack of extensive repetitive element proliferation, RIP activity may be occurring in the YLS. We used an alignment-based consensus sequence as the “model” element for each group and compared all similar repeats with the model using RIPCAL (Hane and Oliver 2008). We found a large proportion of repetitive elements exhibited signatures of RIP, indicated by an excess of CpN → TpN transitions. Of the 10 most abundant repeat classes in the genome, all had a significant excess of CpN → TpN transitions relative to all other transitions (fig. 5). Transposons were less common in the YLS genome than were other types of repetitive sequence. However, of the 10 most abundant transposons present in the genome, all exhibited an abundance of CpN → TpN transitions relative to the expected frequency of these mutations, though due to small sample size only four of these transposons displayed significantly elevated rates. This suggests that RIP is likely a factor in limiting repetitive element replication.
Fig. 5.—
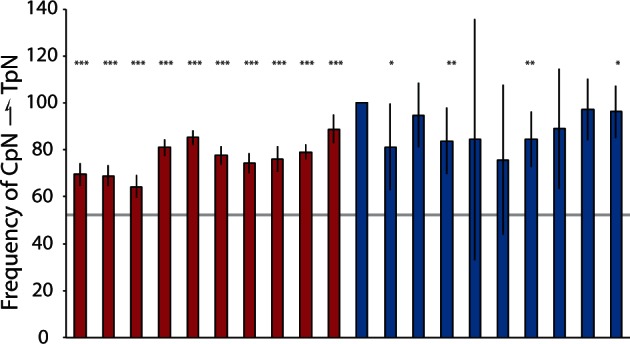
Evidenceof RIP mutation in repetitive elements within the YLS genome. Proportion of CpN → TpN transitions relative to all transitions for each of the 10 most abundant repetitive elements (red bars) and transposable elements (blue bars). Gray horizontal line represents expected frequency of CpN → TpN without RIP (54%). Asterisks above bars indicate classes of repetitive elements that exhibit significant elevation in CpN → TpN transitions relative to the expected frequency (Wilcoxon ranked-sign test; ***P < 0.0001, **P < 0.001, *P < 0.05). Error bars represent 95% confidence interval.
Gene Family Contraction in the YLS
Relative to the outgroup, N. haematococca, 54 protein families exhibited significant decreases in the number of members encoded by the YLS genome relative to M. anisopliae, and no families were significantly expanded in the YLS genome (figs. 6 and 7). Transporters are a major category for which there is significant contraction in the YLS, including those involved in movement of amino acids, sugars, and ATP-binding cassette (ABC) transporters. Many gene families implicated in synthesis and degradation of a wide variety of compounds are reduced in the YLS. These include three families of glycoside hydrolases, two mono-oxygenase families, the amidase family, and a crotonase family. These losses suggest that the YLS has a more limited metabolic potential than do related fungi (fig. 5).
Fig. 6.—

Pfam gene family size in the YLS, Metarhizium anisopliae and Nectria haematococca that have significantly reduced membership in the YLS (Verbiti P < 0.01). No family had higher membership in the YLS.
Fig. 7.—

Pathogenicity gene families in the YLS, Metarhizium anisopliae and Nectria haematococca with significantly higher or lower representation in the YLS genome (Verbiti P < 0.01). Families were identified by searching against the PHI-base database of pathogen–host interacting loci for fungi.
There has also been a significant contraction in many genes involved in pathogenicity and virulence. Twenty families of experimentally confirmed pathogenicity factors have contracted in the YLS (fig. 6). Many of these families appear to be involved in transport, though most are poorly characterized. Interestingly, fungal-specific pathogenicity transcription factors are reduced in the YLS. Similarly, most transcription factors have been lost in the numerous lineages of obligate bacterial symbionts of insects.
The YLS has lost many genes that are implicated in detoxification of secondary metabolites, including loss of several cytochrome p450 and glutathione-S transferase genes that are found in M. anisopliae and N. haematococca, though it still maintains a single member of each of these families (data not shown). Members of these families are involved in overcoming host defenses, providing a plausible reason for their reduction in the YLS. Similarly, the YLS has undergone a significant reduction in members of a family of tannanases, which are involved in metabolism of recalcitrant carbon compounds.
Metabolic Potential
Bacterial symbionts often have minimal metabolic networks, and are often missing pathways or genes essential for life outside of the host environment. In Buchnera, genes necessary for the biosynthesis of nonessential amino acids, phospholipids, the tricarboxylic acid cycle (TCA) cycle, and other key cellular processes have been lost (Shigenobu et al. 2000). The YLS appears to have a much more complete metabolic potential relative to Buchnera. Based on its genome, the YLS is inferred to be able to metabolize a variety of carbon sources, including glucose, sucrose, fructose, maltose, galactose, lactose, mannose, mannitol, as well as complex carbohydrates like starch, cellobiose, chitin, and glucans. The complete pathways for glucose oxidation to CO2: glycolysis, the TCA cycle, and oxidative phosphorylation are encoded in the genome. The YLS also encodes enzymes needed to anaerobically ferment glucose to ethanol via pyruvate decarboxylase and alcohol dehydrogenase. This ability to utilize diverse inputs and energy generation pathways stands in stark contrast to the metabolic capabilities of Buchnera, which lacks a functional TCA cycle, relying on its host for inputs for oxidative phosphorylation.
The YLS genome encodes an extensive set of genes for the generation of lipids and cell wall components. Genes for fatty acid precursors as well as lipids and phospholipids necessary for cell membranes are all found in the genome. The YLS also has the genetic capacity to synthesize glucans and chitin for cell wall construction, and electron microscopy has illustrated the fungus’s thick cell wall (Fukatsu et al. 1994). The relatively robust cell wall of the YLS renders it highly resistant to both physical and chemical disruption (this study). Unlike Buchnera, which is enclosed in a host-derived membrane within a host cell, the YLS is found both intra- and extracellularly and likely requires a more hardy cell wall and membrane.
Aphids rely on their obligate symbionts to synthesize essential amino acids, which are depauperate in the insects’ phloem sap diet (Douglas and Prosser 1992; Shigenobu et al. 2000). Ten percent of Buchnera’s genome is dedicated to genes for biosynthesis of essential amino acids, yet the bacterium lacks genes for almost all nonessential amino acids. The YLS encodes a more complete amino acid metabolic potential, as well as more diverse nitrogen metabolism abilities. Like most fungi, the YLS can synthesize all protein amino acids, including essential amino acids and nonessential amino acids. The YLS can reduce nitrate and nitrite to ammonia, which can then be incorporated into amino acids through the glutamine synthase/glutamate synthetase (GOGAT) cycle, as well as direct incorporation of ammonia into glycine. Buchnera is unable to incorporate ammonia directly into glutamine, relying instead on the host to perform this function (Hansen and Moran 2011). Unlike Buchnera, which lacks specific amino acid transporters, the YLS retains both general amino acid permeases and specific amino acid transporters for methionine, arginine, and asparagine. In addition to amino acids, the YLS can also make a variety of polyamines including spermidine, spermine, and putrescine. Intriguingly, the YLS has lost all members of the gene family nmrA, which silences the gene areA, which represses the utilization of nonglutamine nitrogen (Marzluf 1997).
Secondary Metabolites
Members of the Hypocreales are notable for their production of diverse toxins and secondary compounds. The YLS encodes multiple genes that are likely involved in production of secondary metabolites, including at least 10 polyketide synthase genes and at least 1 nonribosomal peptide synthase (NRPS). Further interrogation of the YLS genome may reveal additional evidence of genes involved in secondary metabolite production. The YLS also encodes a cytochrome P450 gene that may be involved in synthesis or detoxification of secondary metabolites.
Discussion
Obligate symbionts of insects experience severe bottlenecks during transmission between generations. In bacterial symbionts, this has led to the most extremely reduced cellular genomes studied to date. Experimental evolution studies and genomic analyses have suggested that eukaryotic genomes tend to expand when subject to reduced selective pressure. We examined the genome of the obligate fungal symbiont of C. brasiliensis to examine the consequences of small effective population size on the genome of a fungus.
The obligate bacterial symbionts of insects are notable for conserved synteny and lack of gene acquisition over millions of years of evolution (Tamas et al. 2002; van Ham et al. 2003; Sabree et al. 2010). Their strict maternal transmission imposes a clonal population structure, preventing homologous recombination with related strains, and their sequestered habitat potentially shields them from exogenous DNA. Moreover, these symbionts have lost the genes considered necessary to perform homologous recombination (McCutcheon and Moran 2012), which contributes to their accelerated rates of protein sequence evolution due to the action of Muller’s ratchet (Muller 1964). Strict maternal transmission, reinforced by loss of recombinational machinery, genes necessary for meiotic division, or genes involved in mating type recognition could impose Muller’s ratchet on the YLS as well. Although the YLS is maternally transmitted (Buchner 1965), the possibility of occasional horizontal or paternal transmission has not been eliminated; such events could present the opportunity for sexual recombination of distinct strains. However, phylogenies based on morphological characteristics of symbiont type are suggestive of long-term vertical transmission and co-evolution (Fukatsu et al. 1994).
Unlike the obligate bacterial symbionts, the YLS appears to have fully functional recombinational machinery, including the full suite of genes necessary for meiotic division. The presence of the mating type locus also suggests that the YLS is capable of sexual reproduction. The presence of RIP in the repetitive elements within the YLS genome also indicates occasional sexual reproduction (Selker 1990). The YLS could exhibit parasexuality, the fusion of unrelated hyphae, which is known to occur in Metarhizium sp. (Gao et al. 2011); this would also require opportunities for distinct strains to encounter one another within a host. Despite the presence of these genes, there has been no observation of the sexual phase of the YLS, and maternal transmission and a sequestered habitat may greatly restrict opportunities for genetic exchange.
The YLS’s genome reveals a diverse suite of metabolic abilities unlike the streamlined metabolism of the obligate bacterial symbionts of insects, though it has lost many genes found in related fungi. The symbiont appears to be capable of utilizing a number of carbon compounds and producing energy aerobically and anaerobically. Perhaps due to its extracellular habitat, the YLS has been forced to retain a full suite of cell membrane and wall biosynthesis genes.
Like Buchnera, the YLS encodes the full biosynthesis pathways for essential amino acids, though it can also produce the nonessential amino acids, which Buchnera mostly receives from the host. The ability of the YLS to produce essential amino acids supports the hypothesis that the YLS has replaced Buchnera’s functional role in these aphids. In a previous study, it was shown that the YLS of two planthoppers and of C. fransseni are closely related and encode putatively functional uricase genes, whereas related symbionts in two other Cerataphidine genera possessed only inactivated copies (Hongoh and Ishikawa 2000). This gene was also found to be present and intact in the YLS genome from C. brasiliensis, suggesting that the YLS can upgrade stored waste nitrogen (in the form of uric acid) for use in the biosynthesis of amino acids, as has been demonstrated in the YLS of the brown rice planthopper, Nilaparvata lugens (Hongoh and Ishikawa 2000). Phloem nitrogen content can vary within and between plants and varies with developmental stage (Ziegler 1975; Sandstrom et al. 2000), though it is unclear how often total nitrogen is limiting to aphids (Mittler 1953). Most homopterans, including aphids, produce ammonia as their waste product, rather than uric acid as in most insects, and pea aphid bacteriocytes show high expression of genes for incorporation of ammonia into glutamate via the GOGAT cycle (Hansen and Moran 2011). Potentially, uric acid wastes of C. brasiliensis are utilized with the assistance of uricase produced by YLS; presence of uric acid in Cerataphis species has not been examined.
Hypocreales produce a vast array of secondary metabolites, including antibiotics, toxins, and other bioactive compounds. The YLS genome encodes a number of genes which likely function in secondary metabolite synthesis, including polyketide synthases and a nonribosomal peptide synthase, compounds that are expected to have a toxic effect on many organisms. Symbionts of aphids have been demonstrated to provide a number of beneficial functions to their hosts, including protection from parasitoid wasps and heat tolerance (Oliver et al. 2010). While it is unknown what, if any, secondary metabolites the YLS produces, defensive functions are another potential role of the symbiont in addition to nutrient provisioning.
The YLS genome encodes significantly fewer virulence genes in comparison to its pathogenic relatives. Symbionts have evolved repeatedly from pathogenic ancestors, perhaps best illustrated by the facultative symbionts Serratia symbiotica str. Tucson (Burke and Moran 2011) and Hamiltonella defensa (Degnan et al. 2009). While H. defensa retains a complement of pathogenicity genes involved in its protective function, S. symbiotica has lost many ancestral virulence factors, likely through relaxed selection and reduced efficacy of purifying selection due to drift. The reduction in virulence-associated genes in the YLS genome suggests that selection to maintain virulence genes has been relaxed, likely due to the symbiotic relationship with the aphid.
Among the pathogenicity genes retained by the YLS are genes encoding polyketide synthases and NRPSs. Although these may be superfluous genes not yet lost, they may be involved in YLS functions such as protection of the host against parasites or predators. Cerataphidinae species are among the few groups of aphids that live in moist tropical regions where they form persistent colonies that can readily be targeted by parasites and predators. Perhaps for this reason, species of Cerataphidinae are notable for unusually elaborate defenses against natural enemies, with different species and life stages exhibiting associations with defending ants, waxy secretions that may limit parasite invasion, elaborate thick-walled galls, and soldier individuals with horns that attack predators (Stern and Foster 1996). Polyketides and NRPS are known to be symbiont-produced compounds that function in the defense of some insects and marine invertebrates (Piel 2002; Kwan et al. 2012), and their production by the YLS may serve to defend the host. Defensive bacterial symbionts are common in some other aphid species (Oliver et al. 2010).
Amino acid substitution rates appear elevated in proteins of the YLS. These elevated rates suggest that the symbiont is accumulating deleterious mutations as a result of a reduced effective population size, likely due to population bottlenecks experienced during maternal transmission. Additionally, the YLS appears to be gaining introns faster than its sequenced relatives. Such gains are also likely due to reduced selective efficacy imposed by low Ne. Intron size does not differ significantly between the symbiont and its closest sequenced relative, M. anisopliae, though the introns of the YLS are significantly larger than those of the free-living N. haematococca. Interestingly, the YLS appears to be gene-dense, exhibiting similar coding density to M. anisopliae, though a slightly lower density than N. haematococca. Also contrary to expectations of genome expansion is the reduction in intergenic spacer size in the YLS relative to M. anisopliae and N. haematococca. However, due to the fragmentary nature of the draft genome, it is possible that the coding density of the YLS is lower than our current estimate.
The genome of the YLS of C. brasiliensis appears to fit the patterns of evolution recently suggested by Kelkar and Ochman (2011) for Pezizomycotina experiencing genetic drift. The proliferation of introns and accelerated evolutionary rates indicate that the symbiont is accumulating deleterious mutations consistent with a reduced effective population size (Lynch and Conery 2003; Lynch 2006), likely due to transmission bottlenecks. The increase in introns, elevated rates of amino acid substitution, and repetitive elements are consistent with the mutation hazard model of Lynch and Conery (2003). However, several aspects of the YLS genome do not support the hypothesis of rampant genome expansion observed in fungi such as Tuber melanosporum (Martin et al. 2010). The high gene density and small intergenic spacers suggest that it may reside in a range of Ne and genome size that allow for expansion of introns but limit the rampant proliferation of mobile genetic elements (Lynch and Conery 2003).
An additional factor may influence the lack of repetitive DNA and compact genome structure. RIP mutations, first characterized in Neurospora crasssa (Selker 1990), are a mechanism unique to fungi in which duplicated regions of DNA are silenced by mutation. Genomic studies have revealed signatures of RIP in the genome of F. gramarium (Cuomo et al. 2007) and N. haematococca (Coleman et al. 2009), providing a mechanism for their lack of expansion. We see evidence of RIP in the genome of the YLS, providing a mechanism by which the expansion of repetitive elements may have been squelched. Consistent with the evidence of RIP, many of the repetitive elements in the YLS genome do not appear to be active or recently duplicated, suggesting that expansion of these elements have been effectively repressed in the YLS.
Based on fossils of their primary host plant, Styrax sp., Huang et al. (2012) suggest an origin of the subfamily Cerataphidinae in the late Cretaceous (99–65 Ma) and diversification between that time and the Eocene (55–35 Ma). As not all members of the subfamily harbor the YLS, its establishment as an obligate symbiont is unlikely to be older than 99 Myr and may be much more recent (up to 35 Myr). The Buchnera–aphid symbiosis is much older, and is thought to originate at least 200 Ma (Moran et al. 1993). The relatively young association of YLS with hosts may not have permitted sufficient time to allow for genome expansion. The genome-wide reduction in gene family size and intergenic spacer size do suggest a mutational bias favoring deletions, though this could not be directly measured in this study. Further analysis and sequencing of the YLS of C. brasiliensis and other eukaryotic symbionts will shed additional light on the specific mechanisms of genome evolution and possible genome-wide deletion biases in symbiotic fungi.
Acknowledgments
The authors thank C. Wasmann for the invaluable assistance in preparing the sample; J. Lopez of the Fairchild Tropical Botanic Gardens and D. Hodel of the University of California Cooperative Extension Service for providing aphids; V. Denef and G. Burke for providing scripts; and J. Hackett, N. Whiteman, and H. Van Etten for contributing reagents and equipment. M. Barker graciously provided access to computing resources. T. McDonald photographed C. brasiliensis. This work was supported by the Center for Insect Science at the University of Arizona to K.J.V., National Science Foundation grant 0723472 to N.A.M., and Yale University.
Literature Cited
- Birney E, Clamp M, Durbin R. GeneWise and genomewise. Genome Res. 2004;14:988–995. doi: 10.1101/gr.1865504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braendle C, et al. Developmental origin and evolution of bacteriocytes in the aphid-Buchnera symbiosis. PLoS Biol. 2003;1:E21. doi: 10.1371/journal.pbio.0000021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchner P. Animal symbiosis with plant microorganisms. New York: Interscience Inc; 1965. [Google Scholar]
- Burke GR, Moran NA. Massive genomic decay in Serratia symbiotica, a recently evolved symbiont of aphids. Genome Biol Evol. 2011;3:195–208. doi: 10.1093/gbe/evr002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman JJ, et al. The genome of Nectria haematococca: contribution of supernumerary chromosomes to gene expansion. PLoS Genet. 2009;5:e1000618. doi: 10.1371/journal.pgen.1000618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuomo CA, et al. The Fusarium graminearum genome reveals a link between localized polymorphism and pathogen specialization. Science. 2007;317:1400–1402. doi: 10.1126/science.1143708. [DOI] [PubMed] [Google Scholar]
- De Bie T, Cristianini N, Demuth JP, Hahn MW. CAFE: a computational tool for the study of gene family evolution. Bioinformatics. 2006;22:1269–1271. doi: 10.1093/bioinformatics/btl097. [DOI] [PubMed] [Google Scholar]
- Degnan PH, et al. Hamiltonella defensa, genome evolution of protective bacterial endosymbiont from pathogenic ancestors. Proc Nat Acad Sci U S A. 2009;106:9063–9068. doi: 10.1073/pnas.0900194106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas AE, Prosser WA. Synthesis of the essential amino-acid tryptophan in the pea aphid (Acyrthosiphon pisum) symbiosis. J Insect Physiol. 1992;38:565–568. [Google Scholar]
- Edgar RC. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics. 2004;5:1–19. doi: 10.1186/1471-2105-5-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenstein J. 2005. PHYLIP (Phylogeny Inference Package) Version 3.6. [Distributed by the author]. Seattle (WA): Department of Genome Sciences, University of Washington. [Google Scholar]
- Fukatsu T, Aoki S, Kurosu U, Ishikawa H. Phylogeny of Cerataphidini aphids revealed by their symbiotic microorganisms and basic structure of their galls—implications for host-symbiont coevolution and evolution of sterile soldier castes. Zool Sci. 1994;11:613–623. [Google Scholar]
- Fukatsu T, Ishikawa H. A novel eukaryotic extracellular symbiont in an aphid, Astegopteryx styraci (Homoptera, Aphididae, Hormaphidinae) J Insect Physiol. 1992;38:765–773. [Google Scholar]
- Gao Q, et al. Genome sequencing and comparative transcriptomics of the model entomopathogenic fungi Metarhizium anisopliae and M. acridum. PLoS Genet. 2011;7:e1001264. doi: 10.1371/journal.pgen.1001264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X, Lynch M. Ubiquitous internal gene duplication and intron creation in eukaryotes. Proc Natl Acad Sci U S A. 2009;106:20818–20823. doi: 10.1073/pnas.0911093106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon S, et al. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. 2010;59:307–321. doi: 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]
- Hane JK, Oliver RP. RIPCAL: a tool for alignment-based analysis of repeat-induced point mutations in fungal genomic sequences. BMC Bioinformatics. 2008;9:478. doi: 10.1186/1471-2105-9-478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen AK, Moran NA. Aphid genome expression reveals host-symbiont cooperation in the production of amino acids. Proc Natl Acad Sci U S A. 2011;108:2849–2854. doi: 10.1073/pnas.1013465108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbeck JT, Funk DJ, Degnan PH, Wernegreen JJ. A conservative test of genetic drift in the endosymbiotic bacterium Buchnera: slightly deleterious mutations in the chaperonin groEL. Genetics. 2003;165:1651–1660. doi: 10.1093/genetics/165.4.1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hongoh Y, Ishikawa H. Evolutionary studies on uricases of fungal endosymbionts of aphids and planthoppers. J Mol Evol. 2000;51:265–277. doi: 10.1007/s002390010088. [DOI] [PubMed] [Google Scholar]
- Huang X-L, et al. Molecular phylogeny and divergence times of Hormaphidinae (Hemiptera: Aphididae) indicate late Cretaceous tribal diversification. Zool J Linn Soc. 2012;165:73–87. [Google Scholar]
- Itoh T, Martin W, Nei M. Acceleration of genomic evolution caused by enhanced mutation rate in endocellular symbionts. Proc Natl Acad Sci U S A. 2002;99:12944–12948. doi: 10.1073/pnas.192449699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurka J, et al. Repbase update, a database of eukaryotic repetitive elements. Cytogenet Genome Res. 2005;110:462–467. doi: 10.1159/000084979. [DOI] [PubMed] [Google Scholar]
- Kelkar YD, Ochman H. Causes and consequences of genome expansion in fungi. Genome Biol Evol. 2011;4:13–23. doi: 10.1093/gbe/evr124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura M, Ohta T. Theoretical aspects of population genetics. Princeton (NJ): Princeton University Press; 1971. [PubMed] [Google Scholar]
- Komaki K, Ishikawa H. Intracellular bacterial symbionts of aphids possess many genomes per bacterium. J Mol Evol. 1999;48:717–722. doi: 10.1007/pl00006516. [DOI] [PubMed] [Google Scholar]
- Kuo CH, Moran NA, Ochman H. The consequences of genetic drift for bacterial genome complexity. Genome Res. 2009;19:1450–1454. doi: 10.1101/gr.091785.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo CH, Ochman H. Deletional bias across the three domains of life. Genome Biol Evol. 2009;1:145–152. doi: 10.1093/gbe/evp016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwan JC, et al. Genome streamlining and chemical defense in a coral reef symbiosis. Proc Natl Acad Sci U S A. 2012;109:20655–20660. doi: 10.1073/pnas.1213820109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamelas A, et al. Serratia symbiotica from the Aphid Cinara cedri: a missing link from facultative to obligate insect endosymbiont. PLoS Genet. 2011;7:e1002357. doi: 10.1371/journal.pgen.1002357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li WL, et al. Extensive, recent intron gains in Daphnia populations. Science. 2009;326:1260–1262. doi: 10.1126/science.1179302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Madrigal S, Latorre A, Porcar M, Moya A, Gil R. Complete genome sequence of “Candidatus Tremblaya princeps” strain PCVAL, an intriguing translational machine below the living-cell status. J Bacteriol. 2011;129:5587–5588. doi: 10.1128/JB.05749-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–964. doi: 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M. The origins of eukaryotic gene structure. Mol Biol Evol. 2006;23:450–468. doi: 10.1093/molbev/msj050. [DOI] [PubMed] [Google Scholar]
- Lynch M, Conery JS. The origins of genome complexity. Science. 2003;302:1401–1404. doi: 10.1126/science.1089370. [DOI] [PubMed] [Google Scholar]
- Ma LJ, et al. Comparative genomics reveals mobile pathogenicity chromosomes in Fusarium. Nature. 2010;464:367–373. doi: 10.1038/nature08850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin F, et al. Perigord black truffle genome uncovers evolutionary origins and mechanisms of symbiosis. Nature. 2010;464:1033–1038. doi: 10.1038/nature08867. [DOI] [PubMed] [Google Scholar]
- Marzluf GA. Genetic regulation of nitrogen metabolism in the fungi. Microbiol Mol Biol Rev. 1997;61:17–32. doi: 10.1128/mmbr.61.1.17-32.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCutcheon JP, McDonald BR, Moran NA. Origin of an alternative genetic code in the extremely small and GC-rich genome of a bacterial symbiont. PLoS Genet. 2009a;5:e1000565. doi: 10.1371/journal.pgen.1000565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCutcheon JP, McDonald BR, Moran NA. Convergent evolution of metabolic roles in bacterial co-symbionts of insects. Proc Natl Acad Sci U S A. 2009b;106:15394–15399. doi: 10.1073/pnas.0906424106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCutcheon JP, Moran NA. Parallel genomic evolution and metabolic interdependence in an ancient symbiosis. Proc Natl Acad Sci U S A. 2007;104:19392–19397. doi: 10.1073/pnas.0708855104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCutcheon JP, Moran NA. Extreme genome reduction in symbiotic bacteria. Nat Rev Micro. 2012;10:13–26. doi: 10.1038/nrmicro2670. [DOI] [PubMed] [Google Scholar]
- McCutcheon JP, von Dohlen CD. An interdependent metabolic patchwork in the nested symbiosis of mealybugs. Curr Biol. 2011;21:1366–1372. doi: 10.1016/j.cub.2011.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milne I, et al. Tablet—next generation sequence assembly visualization. Bioinformatics. 2010;26:401–402. doi: 10.1093/bioinformatics/btp666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mira A, Moran NA. Estimating population size and transmission bottlenecks in maternally transmitted endosymbiotic bacteria. Microb Ecol. 2002;44:137–143. doi: 10.1007/s00248-002-0012-9. [DOI] [PubMed] [Google Scholar]
- Mira A, Ochman H, Moran NA. Deletional bias and the evolution of bacterial genomes. Trends Genet. 2001;17:589–596. doi: 10.1016/s0168-9525(01)02447-7. [DOI] [PubMed] [Google Scholar]
- Mittler TE. Amino-acids in phloem sap and their excretion by aphids. Nature. 1953;172:207. doi: 10.1038/172207b0. [DOI] [PubMed] [Google Scholar]
- Moran NA. Accelerated evolution and Muller’s rachet in endosymbiotic bacteria. Proc Natl Acad Sci U S A. 1996;93:2873–2878. doi: 10.1073/pnas.93.7.2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran NA, Munson MA, Baumann P, Ishikawa H. A molecular clock in endosymbiotic bacteria is calibrated using the insect hosts. Proc R Soc B. 1993;253:167–171. [Google Scholar]
- Moran NA, Plague GR. Genomic changes following host restriction in bacteria. Curr Opin Genet Dev. 2004;14:627–633. doi: 10.1016/j.gde.2004.09.003. [DOI] [PubMed] [Google Scholar]
- Moriya Y, Itoh M, Okuda S, Yoshizawa AC, Kanehisa M. KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007;35:W182–W185. doi: 10.1093/nar/gkm321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller HJ. The relation of recombination to mutational advance. Mut Res. 1964;1:2–9. doi: 10.1016/0027-5107(64)90047-8. [DOI] [PubMed] [Google Scholar]
- Nakabachi A. The 160-kilobase genome of the bacterial endosymbiont Carsonella. Science. 2007;315:1221–1221. doi: 10.1126/science.1134196. [DOI] [PubMed] [Google Scholar]
- Ohta T. Fixation probability of a mutant influenced by random fluctuation of selection intensity. Genetical Res. 1972;19:33–38. [PubMed] [Google Scholar]
- Ohta T. Slightly deleterious mutant substitutions in evolution. Nature. 1973;246:96–98. doi: 10.1038/246096a0. [DOI] [PubMed] [Google Scholar]
- Oliver KM, Degnan PH, Burke GR, Moran NA. Facultative symbionts in aphids and the horizontal transfer of ecologically important traits. Ann Rev Entomol. 2010;55:247–266. doi: 10.1146/annurev-ento-112408-085305. [DOI] [PubMed] [Google Scholar]
- Piel J. A polyketide synthase-peptide synthetase gene cluster from an uncultured bacterial symbiont of Paederus beetles. Proc Natl Acad Sci U S A. 2002;99:14002–14007. doi: 10.1073/pnas.222481399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price AL, Jones NC, Pevzner PA. De novo identification of repeat families in large genomes. Bioinformatics. 2005;21(1 Suppl):i351–i358. doi: 10.1093/bioinformatics/bti1018. [DOI] [PubMed] [Google Scholar]
- Punta M, et al. The Pfam protein families database. Nucleic Acids Res. 2012;40:D290–D301. doi: 10.1093/nar/gkr1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabree ZL, Degnan PH, Moran NA. Chromosome stability and gene loss in cockroach endosymbionts. Appl Environ Microbiol. 2010;76:4076–4079. doi: 10.1128/AEM.00291-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandstrom J, Telang A, Moran NA. Nutritional enhancement of host plants by aphids—a comparison of three aphid species on grasses. J Insect Physiol. 2000;46:33–40. doi: 10.1016/s0022-1910(99)00098-0. [DOI] [PubMed] [Google Scholar]
- Selker EU. Premeiotic instability of repeated sequences in Neurospora crassa. Ann Rev Genet. 1990;24:579–613. doi: 10.1146/annurev.ge.24.120190.003051. [DOI] [PubMed] [Google Scholar]
- Shigenobu S, Watanabe H, Hattori M, Sakaki Y, Ishikawa H. Genome sequence of the endocellular bacterial symbiont of aphids Buchnera sp. APS. Nature. 2000;407:81–86. doi: 10.1038/35024074. [DOI] [PubMed] [Google Scholar]
- Silva FJ, Latorre A, Moya A. Genome size reduction through multiple events of gene disintegration in Buchnera APS. Trends Genet. 2001;17:615–618. doi: 10.1016/s0168-9525(01)02483-0. [DOI] [PubMed] [Google Scholar]
- Silva FJ, Latorre A, Moya A. Why are the genomes of endosymbiotic bacteria so stable? Trends Genet. 2003;19:176–180. doi: 10.1016/S0168-9525(03)00041-6. [DOI] [PubMed] [Google Scholar]
- Smit A, Hubley R, Green P. RepeatMasker Open-3.0. 2010. Available from: http://www.repeatmasker.org, last accessed February 15, 2012.
- Stern DL, Foster WA. The evolution of soldiers in aphids. Biol Rev Camb Philos Soc. 1996;71:27–79. doi: 10.1111/j.1469-185x.1996.tb00741.x. [DOI] [PubMed] [Google Scholar]
- Suh SO, Noda H, Blackwell M. Insect symbiosis: derivation of yeast-like endosymbionts within an entomopathogenic filamentous lineage. Mol Biol Evol. 2001;18:995–1000. doi: 10.1093/oxfordjournals.molbev.a003901. [DOI] [PubMed] [Google Scholar]
- Tamas I, et al. 50 million years of genomic stasis in endosymbiotic bacteria. Science. 2002;296:2376–2379. doi: 10.1126/science.1071278. [DOI] [PubMed] [Google Scholar]
- Ter-Hovhannisyan V, Lomsadze A, Chernoff YO, Borodovsky M. Gene prediction in novel fungal genomes using an ab initio algorithm with unsupervised training. Genome Res. 2008;18:1979–1990. doi: 10.1101/gr.081612.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ultsch A, Moerchen F. ESOM-Maps: tools for clustering, visualization, and classification with emergent SOM. 2005;Vol. 46 Technical Report of the Mathematics and Computer Science Department. Marburg (Germany): University of Marburg. p. 1–7. [Google Scholar]
- Vogel KJ, Moran NA. Effect of host genotype on symbiont titre in the Aphid-Buchnera symbiosis. Insects. 2011;2:423–434. doi: 10.3390/insects2030423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Ham RC, et al. Reductive genome evolution in Buchnera aphidicola. Proc Natl Acad Sci U S A. 2003;100:581–586. doi: 10.1073/pnas.0235981100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wernegreen JJ. Reduced selective constraint in endosymbionts: elevation in radical amino acid replacements occurs genome-wide. PLoS One. 2011;6:e28905. doi: 10.1371/journal.pone.0028905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winnenburg R, et al. PHI-base update: additions to the pathogen host interaction database. Nucleic Acids Res. 2008;36:D572–D576. doi: 10.1093/nar/gkm858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu D, et al. Metabolic complementarity and genomics of the dual bacterial symbiosis of sharpshooters. PLoS Biol. 2006;4:1079–1092. doi: 10.1371/journal.pbio.0040188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24:1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18:821–829. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J. Rates of conservative and radical nonsynonymous nucleotide substitutions in mammalian nuclear genes. J Mol Evol. 2000;50:56–68. doi: 10.1007/s002399910007. [DOI] [PubMed] [Google Scholar]
- Zheng P, et al. Genome sequence of the insect pathogenic fungus Cordyceps militaris, a valued traditional Chinese medicine. Genome Biol. 2011;12:R116. doi: 10.1186/gb-2011-12-11-r116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegler H. Nature of substances in phloem. In: Pirson A, Zimmermann MH, editors. Encyclopedia of plant physiology. New York: Springer-Verlag; 1975. [Google Scholar]