

Abstract
Prokaryotic genomes are small and compact. Either this feature is caused by neutral evolution or by natural selection favoring small genomes—genome streamlining. Three separate prior lines of evidence argue against streamlining for most prokaryotes. We find that the same three lines of evidence argue for streamlining in the genomes of thermophile bacteria. Specifically, with increasing habitat temperature and decreasing genome size, the proportion of genomic DNA in intergenic regions decreases. Furthermore, with increasing habitat temperature, generation time decreases. Genome-wide selective constraints do not decrease as in the reduced genomes of host-associated species. Reduced habitat variability is not a likely explanation for the smaller genomes of thermophiles. Genome size may be an indirect target of selection due to its association with cell volume. We use metabolic modeling to demonstrate that known changes in cell structure and physiology at high temperature can provide a selective advantage to reduce cell volume at high temperatures.
Keywords: streamlining, genome evolution, thermophilic bacteria
Introduction
Prokaryotic genomes are compact and contain little intergenic DNA compared with eukaryotes. Their compactness is often believed to be driven by genome streamlining, that is, by natural selection favoring a small genome (Doolittle and Sapienza 1980; Orgel and Crick 1980; Dufresne et al. 2005; Giovannoni et al. 2005; Ranea et al. 2005). Streamlining has sometimes been used to denote genome reduction caused by random genetic drift (Lynch 2006), but we refer to it here only if selection favors a small genome. Such streamlining might keep cell division times short, and thus ensure fast reproduction. It might also keep energy consumption for the synthesis of nucleotide precursors low. Although these arguments for the importance of streamlining would apply to many eukaryotes as well, the population genetic conditions for streamlining are more favorable in prokaryotes. Specifically, prokaryotes have larger population sizes than eukaryotes. In larger populations, selection—including selection for small genome sizes—is more powerful (Hartl and Clark 1997; Lynch 2007).
Although streamlining is an attractive concept, there are only few examples of it, all of which involve marine bacteria (Dufresne et al. 2005; Giovannoni et al. 2005; Yooseph et al. 2010) (all references to bacteria throughout the article refer to the domain Eubacteria). Giovannoni et al. (2005) showed that the Pelagibacter ubique genome—the smallest known genome of a free-living organism at the time—contains the smallest intergenic regions. Dufresne et al. (2005) showed that genome reduction in two Prochlorococcus species is associated with loss of several DNA-repair genes, leading to mutational bias and increased rate of evolution, similar to what is observed in some endosymbionts and pathogens. Yooseph et al. (2010) showed that the most abundant picoplankton species are characterized by small genomes and cells, and hypothesized that small cells are advantageous for decreasing predation. Several comparative genomics analyses suggest that examples like these may be the exception rather than the rule (Mira et al. 2001; Touchon and Rocha 2007; Koonin and Wolf 2008; Kuo et al. 2009; Vieira-Silva and Rocha 2010). Specifically, these studies found three lines of evidence that argue against widespread streamlining in prokaryotes and in favor of genetic drift as the predominant force behind compact prokaryotic genomes.
First, if streamlining occurred, noncoding regions should become preferentially reduced in size compared with protein-coding regions, because at least parts of these regions are more likely to be dispensable (Mira et al. 2001; Kuo et al. 2009). Their greater dispensability is suggested by patterns of molecular evolution, such as that more insertions and deletions can be tolerated in intergenic regions (Moran et al. 2009). However, the proportion of noncoding DNA in previously analyzed prokaryotic genomes is not correlated with genome size (Mira et al. 2001; Kuo et al. 2009). Second, generation time (cell division rate), a prime candidate for a quantity to be subject to selection, shows no relationship with genome size (Mira et al. 2001; Touchon and Rocha 2007; Vieira-Silva and Rocha 2010). Third, if streamlining occurred, the strength of selection to remove nonessential regions should be the highest in small genomes. The strength of selection can be estimated via the nonsynonymous/synonymous substitution rate ratio (dN/dS) in protein-coding genes. The smaller this ratio, the slower is the average rate of protein evolution, and the greater are genome-wide evolutionary constraints. In contrast to what would be expected for streamlining—if selection on protein-coding genes correlates with selection on genome streamlining—comparative studies show that larger genomes, not smaller genomes, are under stronger selective constraints (Koonin and Wolf 2008; Kuo et al. 2009).
Extant bacterial genomes are the end-products of a genome size evolution process that is difficult to study in time, because their extinct ancestors and their genome sizes are unknown. To date, this process has been studied only for one group of organisms, obligate parasites and (endo)symbionts, where a strong reduction in genome size has occurred, and was caused by genetic drift (Mira et al. 2001; Daubin and Moran 2004; Kuo et al. 2009). The reasons lie in these organisms’ biology. Many of them live in a relatively unvarying environment provided by their host. The host also provides metabolites or gene products essential to their life, such that many genes in their genomes have become superfluous (Moran and Wernegreen 2000). Moreover, these organisms also have small population sizes (Mira and Moran 2002), where selection is weaker than in large populations. Under these conditions, large genomic regions can be removed through DNA deletions that are effectively neutral (Mira et al. 2001; Moran et al. 2009).
Previous genome-wide analyses of prokaryotes evolutionarily adapted to high temperature have revealed several genomic footprints of thermophilic adaptation. First, the G + C content of helical regions in rRNA secondary structures increases with growth temperature (Galtier and Lobry 1997). Second, thermophiles tend to contain proteins specifically required for life at high temperatures (Forterre 2002; Makarova et al. 2003). Third, the frequency of specific amino acids and nucleotides correlate with growth temperature (Zeldovich et al. 2007; Vieira-Silva and Rocha 2010). Here, we propose an additional genomic signature of thermophilic adaptation: small genome size. We also show that the three lines of evidence we discussed earlier speak in favor of streamlining for thermophiles. This evidence suggests that small genome size, or an unknown factor highly correlated with it, is advantageous in adaptation to higher temperature. Thus, streamlining may not occur in all prokaryotes, but it occurs in some.
Materials and Methods
Collecting Species Information
We obtained the genome sequences and annotation of 1,553 prokaryotes from the National Center for Biotechnology Information (NCBI; ftp://ftp.ncbi.nlm.nih.gov/genomes/genomeprj/). A classification of growth temperature range (psychrophilic, mesophilic, thermophilic, and hyperthermophilic) exists for 1,283 of these species. Habitat classification (host-associated, specialized, aquatic, multiple, and terrestrial) exists for 1,225 species (ftp://ftp.ncbi.nlm.nih.gov/genomes/genomeprj/). We analyzed those 1,155 species further for which both a growth temperature range and a habitat classification is available. A specific (numerical) growth temperature is available for 518 species (453 bacteria and 65 archaea) and can be obtained from ftp://ftp.ncbi.nlm.nih.gov/genomes/genomeprj/. In cases where this temperature is given in the form of a range, we used the mean value of this range. A specific growth temperature and habitat classification is available for 494 species.
Testing for Phylogenetic Dependency
For the phylogenetic analysis, we used data from Vieira-Silva and Rocha (2010). These data consist of a curated list of growth-temperature for 214 species as well as the corresponding 16S rRNA multiple sequence alignment and phylogenetic tree. We matched the list of species to the NCBI data and excluded eukaryotes and species with host-associated habitats. To the remaining 115 species (94 bacteria and 21 archaea), we applied coevol (Lartillot and Poujol 2011), a Bayesian Markov Chain Monte Carlo sampling software for comparative analyses, to test for correlation between growth temperature and genome size. The program takes as an input a multiple sequence alignment, a matrix of continuous characters, and a phylogenetic tree. It then jointly estimates key evolutionary parameters (e.g., evolutionary rate, dN/dS ratio) and the correlations between the characters corrected for phylogenetic dependencies (Lartillot and Poujol 2011).
Estimation of dN/dS Ratios
We first identified pairs of closely related taxa in the following way: We identified the 16S rRNA processing (RimM) protein in 88 thermophile and 182 nonthermophile genomes. We aligned protein sequences of each group using MAFFT (Katoh et al. 2002) and constructed corresponding phylogenetic trees with the neighbor-joining method (Saitou and Nei 1987). We trimmed trees to include only pairs of closely related species with RimM divergence of 1–15%, resulting in 9 and 31 phylogenetically independent pairs of thermophiles and nonthermophiles, respectively. We used BlastClust (http://www.ncbi.nlm.nih.gov/IEB/ToolBox/C_DOC/lxr/source/doc/blast/blastclust.html) to identify 32 clusters of single-copy orthologous genes that are present in the genomes of all pairs (species pairs and nucleotide sequences of all orthologous genes are listed in supplementary file S1, Supplementary Material online). Further, we excluded species pairs in which less than 10 of the genes had nucleotide identity between 75% and 95% (sui for dN/dS analysis), resulting in 8 and 16 pairs of thermophiles and nonthermophiles, respectively. Finally, we estimated dN/dS ratios using Goldman and Yang’s Maximum-Likelihood method (Goldman and Yang 1994), implemented in the Matlab Bioinformatics toolbox. An analysis without the exclusion of species pairs and genes on basis of nucleotide identity resulted in qualitatively similar results (not shown).
Analysis of Protein Length and Protein Family Size
We used Pfam version 23 (Finn et al. 2010). This Pfam release contains 13,672 families. We included only families with sequences between 50 and 500 amino acids (11,771). We used the software HMMER (Eddy 1998) to annotate protein families in the thermophile and nonthermophile proteomes, employing the gathering cut-off criterion that minimizes false positives. In this way, we identified 6,654 single-domain families whose domains are covered over at least 90% by the hmm model. Of them, 19 families are present in all the studied genomes (table 3). For each protein family within each genome, we calculated the average protein length and the number of proteins per protein family. We then calculated the average protein length and the average number of proteins per protein family for the 19 families over all genomes.
Table 3.
Mean Protein Family Size Per Genome and Protein Length within 19 Common Protein Families
| S. No. | Family ID | Mean Family Size Per Genome |
Mean Protein Length |
Description | ||
|---|---|---|---|---|---|---|
| Thermophiles | Nonthermophiles | Thermophiles | Nonthermophiles | |||
| 1 | PF00005.22 | 28.4 | 46.4 | 295.6 | 304.0 | ABC transporter |
| 2 | PF00106.20 | 7.5 | 19.6 | 254.7 | 262.3 | Short chain dehydrogenase |
| 3 | PF00155.16 | 6.1 | 8.6 | 385.6 | 392.8 | Aminotransferase class I and II |
| 4 | PF00156.22 | 3.8 | 4.2 | 190.2 | 190.8 | Phosphoribosyl transferase domain |
| 5 | PF00238.14 | 1.0 | 1.0 | 122.2 | 122.3 | Ribosomal protein L14p/L23e |
| 6 | PF00266.14 | 3.6 | 4.0 | 391.8 | 404.3 | Aminotransferase class V |
| 7 | PF00275.15 | 2.4 | 2.1 | 426.1 | 430.6 | EPSP synthase |
| 8 | PF00416.17 | 1.0 | 1.0 | 123.9 | 121.6 | Ribosomal protein S13/S18 |
| 9 | PF00696.23 | 2.7 | 3.1 | 281.3 | 301.7 | Amino acid kinase family |
| 10 | PF00829.16 | 1.0 | 1.0 | 105.4 | 111.1 | Ribosomal prokaryotic L21 protein |
| 11 | PF00831.18 | 1.0 | 1.0 | 69.5 | 68.5 | Ribosomal L29 protein |
| 12 | PF01255.14 | 1.1 | 1.2 | 250.0 | 253.4 | Putative undecaprenyl diphosphate synthase |
| 13 | PF01327.16 | 1.2 | 1.7 | 172.7 | 175.9 | Polypeptide deformylase |
| 14 | PF01709.15 | 1.0 | 1.1 | 248.6 | 244.8 | Transcriptional regulator |
| 15 | PF01795.14 | 1.0 | 1.0 | 305.7 | 321.4 | MraW methylase family |
| 16 | PF02096.15 | 1.1 | 1.2 | 346.9 | 431.5 | 60 kDa inner membrane protein |
| 17 | PF06071.8 | 1.0 | 1.0 | 362.3 | 363.8 | Protein of unknown function (DUF933) |
| 18 | PF07690.11 | 7.3 | 21.7 | 443.5 | 446.8 | Major facilitator superfamily |
| 19 | PF07992.9 | 4.6 | 6.1 | 403.8 | 410.2 | Pyridine nucleotide-disulphide oxidoreductase |
Results
Figure 1a shows the distributions of genome sizes among prokaryotes with different growth temperature ranges (psychrophilic, mesophilic, thermophilic, and hyperthermophilic). We found that species living in warmer temperatures tend to have smaller genomes. The differences in genome size between mesophiles, thermophiles, and hyperthermophiles are significant (Wilcoxon rank-sum test, P < 1.9 × 10−5 and P < 7.9 × 10−3 for mesophiles–thermophiles and thermophiles–hyperthermophiles, respectively), but not between psychrophiles and mesophiles (Wilcoxon rank-sum test, P = 0.082). To the best of our knowledge, no such association has been described before.
Fig. 1.—
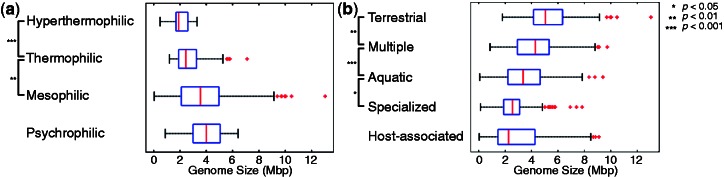
(a) Distribution of genome sizes among prokaryotes with different growth temperature ranges. The differences in genome size between mesophiles, thermophiles, and hyperthermophiles are significant (Wilcoxon rank-sum test, P < 1.9 × 10−5 and P < 7.9 × 10−3 for mesophiles–thermophiles and thermophiles–hyperthermophiles, respectively), but not between psychrophiles and mesophiles (Wilcoxon rank-sum test, P = 0.082). (b) Distribution of genome sizes among different habitats. Habitats are ordered according to environmental variability from unvarying (host-associated) to the most variable environment (terrestrial). The distributions of genome sizes differ between habitats (Wilcoxon rank-sum test, P < 0.018, P < 0.0005, P < 0.0028, for specialized-aquatic, aquatic-multiple, and multiple-terrestrial, respectively), with the exception of host-associated habitats (Wilcoxon rank-sum test, P = 0.67, for comparison between host-associated and specialized). The red vertical marks are the medians, the edges of the box are the 25th and 75th percentiles, the whiskers extend to the most extreme data points not considered outliers (99% of all data if the data are normally distributed) and outliers are plotted individually as red crosses.
Why are genome size and growth temperature negatively associated? One possibility is that both are associated with a third, confounding factor. A prominent candidate is the extent to which the environment varies. It is easy to see why environmental variability could be associated with genome size (Parter et al. 2007; Rodrigues and Wagner 2009). For example, free living organisms in which the availability of different nutrients varies greatly need to have metabolic enzymes to metabolize each nutrient. Such organisms would need to have larger genomes, to accommodate all the genes that encode these enzymes. In a relevant study of 117 bacterial species, Parter et al. (2007) have shown that bacteria living in more variable environments tend to have larger metabolic networks with more enzymatic reactions. Conversely, organisms that live in environments with low variability, such as parasites or symbionts that live in close association with a host organism—which provides an unchanging environment—tend to have smaller genomes (Moran and Wernegreen 2000; Mira et al. 2001). Perhaps, we reasoned, organisms in high temperature habitats simply experience less environmental variability.
Data on environmental variability are difficult to come by, especially if needed for many organisms. We here used a classification of environments available from the NCBI (Wheeler et al. 2008) ftp://ftp.ncbi.nlm.nih.gov/genomes/genomeprj/). We follow the definition of habitat variability from (Parter et al. 2007) to order habitats according to increasing variability as host-associated, specialized, aquatic, multiple, and terrestrial. Using this classification of habitats, we found that the distributions of genome sizes indeed differ between habitats (Wilcoxon rank-sum test, P < 0.018, P < 0.0005, P < 0.0028, for specialized-aquatic, aquatic-multiple, and multiple-terrestrial habitats, respectively), with the exception of host-associated habitats (Wilcoxon rank-sum test, P = 0.67, for comparison between host-associated and specialized). Genome size decreases in less variable habitats (fig. 1b).
We next asked which of the two factors, habitat temperature or variability, affects genome size more strongly? To answer this question, we performed a two-way analysis of variance (ANOVA) with growth temperature and habitat as the independent factors. We found that the effect of growth temperature is significant (P = 0.0002), whereas there is no significant effect of habitat, and no significant interaction (P = 0.52, P = 0.60, respectively). In a next analysis, we used 494 species for which data on growth temperature and habitat is available (NCBI), to examine the association between genome size and temperature within each habitat type (fig. 2a and table 1). Within each habitat type, temperature is negatively correlated with genome size, the only exception being host-associated organisms (table 1). The correlations within habitat types support the ANOVA result and suggest a direct effect of growth temperature on genome size. Subsequently, we asked whether the association between genome size and growth temperature differs between bacteria and archaea. We found that the association is much stronger in bacteria than in archaea, especially when host-associated species are excluded (fig. 2b and table 1).
Fig. 2.—

(a) Growth temperature and genome size of species from different habitat types. (b) Growth temperature and genome size of species from different kingdoms. See table 1 for statistical analysis.
Table 1.
Statistical Association between Growth Temperature and Genome Size
| Number of Species | Spearman's ρ | P | |
|---|---|---|---|
| Habitat | |||
| Host associated | 173 | −0.14 | 0.063 |
| Specialized | 83 | −0.56 | 3.75 × 10−8 |
| Aquatic | 65 | −0.66 | 2.29 × 10−9 |
| Multiple | 145 | −0.45 | 1.29 × 10−8 |
| Terrestrial | 28 | −0.63 | 3.50 × 10−4 |
| Kingdom | |||
| Bacteria | 453 (260) | −0.43 (−0.56) | 4.86 × 10−22 (4.06 × 10−23) |
| Archaea | 65 (61) | −0.36 (−0.36) | 0.0034 (0.0048) |
| All | 518 (321) | −0.48 (−0.64) | 4.46 × 10−31 (6.78 × 10−38) |
Note.—Numbers in parentheses indicate values after excluding host-associated species.
The phylogenetic relationship between species is a potential source of error in analyses like ours, because the species share an evolutionary history and are thus not independent (Felsenstein 2008). We therefore tested whether the association between genome size and growth temperature holds when the phylogenetic dependencies between the species are controlled for. To this end, we used an approach proposed by (Lartillot and Poujol 2011) and implemented in the software coevol to test for correlation between growth temperature and genome size within bacteria and archaea. The approach corrects for spurious associations due to shared evolutionary history and accounts for potential uncertainties in the phylogenetic relationships of species. In this analysis, we used a 16S rRNA phylogenetic tree delineating the phylogenetic relationships among 214 species for which curated information on growth temperatures is available (Vieira-Silva and Rocha 2010). We excluded eukaryotes and species with host-associations from this analysis, which left us with 115 species (94 bacteria and 21 archaea). We found a significant negative correlation between genome size and temperature in bacteria (posterior probability of 0.04), but not in archaea (posterior probability of 0.58). Hence, the correlation between genome size and temperature in archaea may be due to shared evolutionary history. We therefore focus in the remainder of our analyses solely on bacteria.
Percentage of Intergenic DNA Correlates with Genome Size and Growth Temperature
If selection acts to decrease the size of a genome, the size reduction should preferentially affect the regions of a genome that are least constrained (Mira et al. 2001; Kuo et al. 2009). These regions are the intergenic regions (Moran et al. 2009). Bacterial genomes in general do not meet this criterion, which is one major earlier line of evidence against streamlining in bacteria (Mira et al. 2001; Kuo et al. 2009). To ask whether this criterion is met for our study organisms, we examined the percentage of a genome’s DNA contained in intergenic regions (denoted as %IG) separately for thermophilic and nonthermophilic bacteria. Specifically, we calculated %IG for thermophilic and hyperthermophilic bacteria together—we refer to these groups as thermophiles for brevity—and for nonthermophilic bacteria (mesophiles and psychrophiles). Table 2 shows that thermophiles have lower %IG than nonthermophiles, a difference that is statistically significant (P = 0.0003, Wilcoxon rank-sum test). We also note that the %IG of thermophiles living at the highest temperatures (above 65 °C) is especially low (table 2).
Table 2.
Percentage of Intergenic Regions
| Number of Genomes | % Intergenic Regions, Mean (SD) | |
|---|---|---|
| Nonthermophiles | 192 | 13.3 (3.2) |
| Thermophiles | 48 | 10.8 (4.0) |
| Thermophiles above 65 °C | 22 | 8.8 (3.4) |
We next compared %IG and genome size (fig. 3a) and found a strong positive correlation in thermophiles (Spearman’s ρ = 0.63, P < 2.5 × 10−6). That is, those genomes of thermophiles that are small also contain a smaller percentage of their DNA in noncoding regions. In contrast, we found no such correlation between %IG and genome size in nonthermophiles (P = 0.58, fig. 3a). Intriguingly, the percentage of intergenic DNA is also negatively correlated with growth temperature in thermophiles (Spearman’s ρ = −0.54, P < 7.6 × 10−5, fig. 3b). In other words, those thermophiles growing at higher temperatures are affected to a greater extent by streamlining. Such an association is absent for mesophiles (Spearman’s ρ = 0.12, P = 0.09, fig. 3b), suggesting that streamlining occurs at the higher growth temperatures that are characteristic of thermophiles, but not at the lower growth temperatures characteristic of mesophiles. As expected, we found similar trends as with %IG when we examined the gene density (i.e., the number of genes in a genome divided by genome size) in thermophiles and nonthermophiles (data not shown).
Fig. 3.—

The percentage of a genome occupied by intergenic regions (%IG, vertical axes) in nonthermophilic bacteria (blue) and thermophilic bacteria (red) is plotted against genome size (a) and growth temperature (b) on the horizontal axes. (a) %IG and genome size are positively correlated in thermophiles (Spearman’s ρ = 0.63, P < 2.5 × 10−6) but not in nonthermophiles (P = 0.58). (b) %IG and temperature are negatively correlated in thermophiles (Spearman’s ρ = −0.54, P < 7.6 × 10−5) but not in nonthermophiles (Spearman’s ρ = 0.12, P = 0.09).
Generation Time
The compactness of prokaryotic genomes has often been attributed to selection for short generation times (reviewed in Lynch 2006). However, so far there has been no evidence supporting this view (Mira et al. 2001; Touchon and Rocha 2007; Vieira-Silva and Rocha 2010). For example, Vieira-Silva and Rocha (2010) have examined the generation times of 214 prokaryotes and found no correlation with genome size. We re-analyzed the data from Vieira-Silva and Rocha (2010), excluding host-associated species, and differentiated between thermophilic and nonthermophilic bacteria (fig. 4). In agreement with Vieira-Silva and Rocha (2010), we found no significant correlation between generation time and genome size, either in thermophiles (ρ = 0.56, P = 0.096) or in nonthermophiles (ρ = −0.01, P = 0.92, fig. 4a). However, we noted a conspicuous positive association for thermophiles (fig. 4a), whose nonsignificance could be due to the small sample size of 10 species. Highly intriguing is moreover that thermophiles that live at higher temperatures also divide significantly faster (Spearman’s ρ = −0.91, P < 2.1 × 10−4, fig. 4b), an association that is absent for nonthermophiles (P = 0.8). Such an association is expected if increasing temperature favors shorter generation times.
Fig. 4.—

Generation time (vertical axes) in nonthermophilic bacteria (blue) and thermophilic bacteria (red) is plotted against genome size (a) and growth temperature (b) on the horizontal axes. Data are from Vieira-Silva and Rocha (2010). (a) The associations between generation time and genome size are not significant (Spearman’s ρ = 0.56, P = 0.096 and ρ = −0.01, P = 0.92, for thermophiles and nonthermophiles, respectively), but the nonsignificance in thermophiles could be due to the small sample size of 10 species. (b) Generation time and temperature are negatively correlated in thermophiles (Spearman’s ρ = −0.91, P < 2.1 × 10−4) but not in nonthermophiles (P = 0.8).
No Reduction in Selective Constraints on Proteins in Thermophile Genomes
Genome size reduction could be the result of drift for genomes that experience weaker selective constraints (Mira et al. 2001; Kuo et al. 2009). Thus, we determined the ratios of dN/dS (Goldman and Yang 1994), whose value increases with increasing selective constraints, in thermophiles and nonthermophiles. We identified 40 phylogenetically independent pairs of closely related taxa (9 thermophile pairs and 31 nonthermophile pairs). Within the genome of these pairs, we identified 32 groups of single-copy orthologous genes that are present in all genomes. We excluded species pairs from our analysis in which less than 10 gene pairs had a nucleotide identity between 75% and 95% (suitable for analysis of dN/dS), resulting in 8 and 16 pairs of thermophile and nonthermophile species, respectively. Comparison between average dN/dS ratios shows lower dN/dS values in thermophiles (average dN/dS = 0.039 and 0.048 for thermophiles and nonthermophiles, respectively), but the difference is not significant (p = 0.0922, Wilcoxon rank-sum test). We found no significant correlation between average dN/dS and genome size, either in thermophiles (P = 0.58) or in nonthermophiles (P = 0.39, fig. 5a). Similarly, we found no significant correlation between average dN/dS and temperature, either in thermophiles (P = 0.11) or in nonthermophiles (P = 0.11, fig. 5b), but future analysis with larger samples might reveal a negative association in thermophiles. A previous study compared 17,957 pairs of orthologous genes from 22 pairs of closely related species and reported lower dN/dS values in both bacterial and archaeal thermophiles compared with mesophiles (Friedman et al. 2004). Although our analysis did not show an equivalent significant decrease in dN/dS ratios (possibly because Friedman et al. used different genes from their species pairs), it shows that selective constraints are not weaker in thermophiles (as they are in obligate parasites and endosymbionts). Thus, genome size reduction is unlikely to be the result of drift.
Fig. 5.—

Average dN/dS ratios (vertical axes) in nonthermophilic bacteria (blue) and thermophilic bacteria (red) are plotted against genome size (a) and growth temperature (b) of phylogenetically independent species-pairs on the horizontal axes. (a) The associations between dN/dS ratios and genome size are not significant (P = 0.58 and P = 0.39, for thermophiles and nonthermophiles, respectively). (b) The associations between dN/dS ratios and temperature are not significant (P = 0.11 and P = 0.11, for thermophiles and nonthermophiles, respectively).
Distinct Characteristics of Protein Families in Thermophile Genomes
Thermophile genomes contain unique protein families (Makarova et al. 2003). To further examine the influence of protein families on size reduction of thermophile genomes, we compiled a set of 19 single-domain protein families that are shared by all thermophile and nonthermophile genomes. For each protein family within each genome, we calculated the average protein length and the number of proteins per protein family. We then calculated the average protein length and the average number of proteins per protein family for the 19 families of each genome (figs. 6 and 7, and table 3). In agreement with previous studies (Thompson and Eisenberg 1999; Chakravarty and Varadarajan 2000), we found that proteins in thermophile genomes are shorter than their homologous counterparts in nonthermophile genomes (P < 6.7 × 10−7, Wilcoxon rank-sum test). In addition, protein families in thermophile genomes contain fewer proteins then protein families in nonthermophile genomes (P < 8.6 × 10−13, Wilcoxon rank-sum test), as expected by the reduction of gene number in thermophile genomes. All association presented in figures 6 and 7 (between genome size and protein length, between genome size and family size, between temperature and protein length, and between temperature and family size) are significant (P < 0.05).
Fig. 6.—

Average protein length across 19 common protein families (vertical axes) in nonthermophilic bacteria (blue) and thermophilic bacteria (red) is plotted against genome size (a) and growth temperature (b) on the horizontal axes. (a) The associations between average protein length and genome size are significant (Spearman’s ρ = 0.34, P < 0.015 and ρ = 0.53, P < 2.7 × 10−16, for thermophiles and nonthermophiles, respectively). (b) The associations between average protein length and temperature are significant (Spearman’s ρ = −0.32, P < 0.025 and ρ = −0.25, P < 2.7 × 10−4, for thermophiles and nonthermophiles, respectively).
Fig. 7.—

Average protein family size per genome for 19 common protein families (vertical axes) in nonthermophilic bacteria (blue) and thermophilic bacteria (red) is plotted against genome size (a) and growth temperature (b) on the horizontal axes. (a) The associations between average family size and genome size are significant (Spearman’s ρ = 0.88, P < 3.3 × 10−17 and ρ = 0.81, P < 5.5 × 10−50, for thermophiles and nonthermophiles, respectively). (b) The associations between average protein length and temperature are significant (Spearman’s ρ = −0.55, P < 3.9 × 10−5 and ρ = −0.23, P < 8.6 × 10−4, for thermophiles and nonthermophiles, respectively).
No Selection against Proteins Unable to Adapt to High Temperature
Finally, we examined two hypotheses that might explain why thermophile genomes have small size. The first hypothesis is that genome size reduction occurs because selection preferentially eliminates genes that encode proteins with low thermodynamic stability from a genome. This hypothesis is motivated by the observation that organisms adapted to high temperature have thermodynamically more stable proteins (Jaenicke 2000; Kumar and Nussinov 2001). We reasoned that some proteins may not be able to evolve higher stability, and thus would become nonfunctional (or even toxic) at high temperatures. The encoding genes might thus be preferentially eliminated. Unlike previous studies that compared homologous proteins in mesophiles and thermophiles (Jaenicke 2000; Kumar and Nussinov 2001), we compared the thermodynamic stability of proteins that were lost during thermal adaptation with that of proteins that remained in the genome (supplementary material, Supplementary Material online). However, our analysis (supplementary material, Supplementary Material online) did not provide any support for this hypothesis. A second hypothesis, again prompted by previous observations (Burra et al. 2010), is that selection may preferentially eliminate genes encoding proteins with disordered tertiary structures. Again, these data do not support this hypothesis (supplementary material, Supplementary Material online). Thus, instead of selection against specific types of proteins, selection may have operated genome-wide to reduce the size of thermophilic genomes, independently of the stability or disorder of individual gene products.
Discussion
We showed that bacteria that live at higher temperatures tend to have smaller genomes (figs. 1a and 2). The correlation is striking: all species that live at temperatures above 60 °C have genomes smaller than 4 Mb, and all species with genomes larger than 6 Mb, live in temperatures below 45 °C (fig. 2). We excluded the possibility that this correlation is the result of phylogenetic dependencies between species. Our observations thus suggest that adaptation to high temperature involves a reduction in genome size.
To find out whether this reduction is caused by random genetic drift or genome streamlining, that is, natural selection, we turned to three major lines of evidence that can speak to either hypothesis (Mira et al. 2001; Touchon and Rocha 2007; Koonin and Wolf 2008; Kuo et al. 2009; Vieira-Silva and Rocha 2010).
First, if drift is responsible for genome size reductions, the fraction of intergenic DNA as a proportion to total genomic DNA should remain constant with decreasing genome size. This has been observed in previous studies that did not focus on thermophiles (Mira et al. 2001; Kuo et al. 2009). In contrast, we found that the proportion of intergenic DNA correlates strongly with genome size and growth temperature for thermophiles: Smaller thermophile genomes have significantly less intergenic DNA; thermophiles growing at higher growth temperatures also have less intergenic DNA (fig. 3). Thus, this line of evidence argues against drift and for streamlining. We note that previous evidence on the size reduction of coding DNA is also in line with the streamlining hypothesis. Specifically, proteins in thermophilic bacteria are shorter than their orthologs in mesophiles (fig. 6), presumably because structure-destabilizing loops get lost in the proteins of thermophiles (Thompson and Eisenberg 1999; Chakravarty and Varadarajan 2000).
The second line of evidence regards generation time, an important fitness component. If drift is behind genome size reductions, generation time should be independent of genome size. This is indeed generally the case. However, among thermophiles, we find that organisms with smaller genomes have shorter generations. The association is not significant, possibly because of a small number of species available for this analysis (fig. 3a). However, because generation time and temperature are also strongly and significantly correlated (fig. 3b), a larger sample might reveal a significant relationship between genome size and generation time.
The third line of evidence relates to selective constraints, indicated by average dN/dS values over all protein-coding regions that a genome experiences. If drift is responsible for genome size reduction, small genomes should experience weaker constraints than large genomes (Mira et al. 2001; Kuo et al. 2009). Conversely, if selection is at work, one would expect to find smaller genomes to be more constrained, that is, to show lower average dN/dS ratios. Previous studies (Koonin and Wolf 2008; Kuo et al. 2009; Novichkov et al. 2009) have shown that larger genomes are more constrained. Thermophiles, in contrast, show a different pattern. A comparison of 17,957 pairs of orthologous genes from 22 pairs of closely related species revealed that in both bacteria and archaea thermophiles are more constrained than mesophiles (Friedman et al. 2004). Using a more restricted set of orthologous genes, we show that proteins in thermophile genomes are similarly constrained as their orthologs in nonthermophile genomes (fig. 5). Thermophiles also have reduced mutation rates, perhaps in response to the increased fitness cost of mutations at high temperatures (Mackwan et al. 2008; Drake 2009). Together, these observations indicate that the small genomes of thermophiles are not the product of neutral evolution, as in obligate parasites and (endo)symbionts (Mira et al. 2001; Daubin and Moran 2004; Kuo et al. 2009).
Previous studies have argued for genome streamlining with limited evidence from several species (Dufresne et al. 2005; Giovannoni et al. 2005). In contrast, we present three lines of evidence from a large assemblage of bacteria—thermophiles—in favor of streamlining. Whether genome size itself or some other quantity related to it is the direct target of selection is unknown. We next discuss three candidate targets and propose a fourth such target. First, selection for fast replication is an unlikely target, because the energetic cost of DNA replication is relatively low (below 2% of the entire energy usage) (Wagner 2005; Lynch 2006). Second, organisms adapted to high temperature have proteins that are thermodynamically more stable (Jaenicke 2000; Kumar and Nussinov 2001). Thus, selection may preferentially eliminate dispensable low-stability proteins. Third and relatedly, thermophile proteins are less disordered (Burra et al. 2010). Hence, selection may preferentially eliminate disordered proteins. However, our analysis (supplementary material, Supplementary Material online) did not provide any support for any of these hypotheses.
A fourth potential target is cell size itself. Smaller cells tend to have smaller genomes in both prokaryotes and unicellular eukaryotes (Shuter et al. 1983; Lynch 2007), for reasons that are not fully understood (but see Dill et al. 2011). This correlation suggests that cell and genome size are functionally related, such that an evolutionary change in one would induce an evolutionary change in the other. Previous studies have hypothesized that reduction in cell size could be advantageous, either to optimize surface-to-volume ratio for uptake of scarce nutrients (Dufresne et al. 2005; Giovannoni et al. 2005; Moya et al. 2009), or to decrease predation by bacterivores or bacteriophages (Yooseph et al. 2010). Similarly, we speculate that bacteria adapted to high temperature could be subject to selection favoring smaller cell size, which also requires smaller genome size—regardless of gene content. The amount by which natural selection could reduce cell size would be limited by the ability to minimize genome size. For example, in P. ubique, the smallest free-living microorganism, the genome occupies a substantial fraction (30%) of the cell volume (Giovannoni et al. 2005). If our hypothesis is true, small genomes would be a by-product of selection for small cells.
Although we cannot provide conclusive evidence for this hypothesis, we investigated what advantages smaller cells could have at high temperature in addition to those mentioned earlier (Dufresne et al. 2005; Giovannoni et al. 2005; Moya et al. 2009; Yooseph et al. 2010). Growth at high temperature has two important effects on cells. First, it requires cells to increase the lipid content and change the lipid composition of cell membranes (Nordstrom and Laakso 1992). Part of this change is required to reduce proton leakage at high temperatures, and thus to maintain the amount of energy available to cells through proton gradients (Mitchell 1966). Second, high temperature increases the amount cells need to expend on nongrowth-associated maintenance (Coultate and Sundaram 1975; Kuhn et al. 1980; McKay et al. 1982; Pennock and Tempest 1988; Sonnleitner 1983).
In the supplementary material, Supplementary Material online, we used a genome-scale metabolic model of the thermophile Thermotoga maritima to show that these effects can reduce biomass synthesis—and thus maximally achievable generation times—substantially. For example, a mere doubling of nongrowth-associated maintenance requirements—much higher increases have been reported experimentally (McKay et al. 1982; Pennock and Tempest 1988)—may reduce biomass growth by more than 60% (supplementary fig. S2, Supplementary Material online). Thus, evolutionary adaptations that lower these effects of high temperature could have strong fitness benefits. Reduced cell size is one such adaptation because cell size has a direct impact on both nongrowth-associated maintenance requirements and lipid requirements. In support of this idea, the cell size of planktonic bacteria was shown to decrease with increasing temperatures in controlled chemostat incubations, as well as in their natural habitat (Chrzanowski et al. 1988). Improved energy usage of small cells at high temperature could explain the experimental observation that bacteria evolved at high temperature did not have reduced fitness in the original temperature, suggesting that there is not necessarily a tradeoff between growth at different temperatures (Elena and Lenski 2003).
In sum, our analysis showed that prokaryotic species adapted to high temperature have small genomes. This association does not appear to be a by-product of lower environmental variability at higher temperature, at least based on available data. It persists for bacteria when phylogenetic relationships among species are accounted for. Several lines of evidence argue that drift alone is not sufficient, and selection needs to be invoked to explain this correlation. Whether genome size is a direct or indirect target of selection is unknown. A candidate direct target is cell size—correlated with genome size—because metabolic analysis suggests that large cells may suffer significant fitness costs at high temperatures. Future work will show whether this explanation is correct. Genome size reduction in thermophiles is currently a unique candidate case of selection for streamlining in bacteria.
Supplementary Material
Supplementary file S1, tables S1–S4, and figures S1 and S2 are available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org/).
Literature Cited
- Burra PV, Kalmar L, Tompa P. Reduction in structural disorder and functional complexity in the thermal adaptation of prokaryotes. PLoS One. 2010;5:e12069. doi: 10.1371/journal.pone.0012069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakravarty S, Varadarajan R. Elucidation of determinants of protein stability through genome sequence analysis. FEBS Lett. 2000;470:65–69. doi: 10.1016/s0014-5793(00)01267-9. [DOI] [PubMed] [Google Scholar]
- Chrzanowski TH, Crotty RD, Hubbard GJ. Seasonal variation in cell volume of epilimnetic bacteria. Microb Ecol. 1988;16:155–163. doi: 10.1007/BF02018911. [DOI] [PubMed] [Google Scholar]
- Coultate TP, Sundaram TK. Energetics of Bacillus stearothermophilus growth: molar growth yield and temperature effects on growth efficiency. J Bacteriol. 1975;121:55–64. doi: 10.1128/jb.121.1.55-64.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daubin V, Moran NA. Comment on “The origins of genome complexity”. Science. 2004;306:978. doi: 10.1126/science.1100559. author reply 978. [DOI] [PubMed] [Google Scholar]
- Dill KA, Ghosh K, Schmit JD. Physical limits of cells and proteomes. Proc Natl Acad Sci U S A. 2011;108:17876–17882. doi: 10.1073/pnas.1114477108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doolittle WF, Sapienza C. Selfish genes, the phenotype paradigm and genome evolution. Nature. 1980;284: 601–603. doi: 10.1038/284601a0. [DOI] [PubMed] [Google Scholar]
- Drake JW. Avoiding dangerous missense: thermophiles display especially low mutation rates. PLoS Genet. 2009;5:e1000520. doi: 10.1371/journal.pgen.1000520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dufresne A, Garczarek L, Partensky F. Accelerated evolution associated with genome reduction in a free-living prokaryote. Genome Biol. 2005;6:R14. doi: 10.1186/gb-2005-6-2-r14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddy SR. Profile hidden Markov models. Bioinformatics. 1998;14:755–763. doi: 10.1093/bioinformatics/14.9.755. [DOI] [PubMed] [Google Scholar]
- Elena SF, Lenski RE. Evolution experiments with microorganisms: the dynamics and genetic bases of adaptation. Nat Rev Genet. 2003;4:457–469. doi: 10.1038/nrg1088. [DOI] [PubMed] [Google Scholar]
- Felsenstein J. Comparative methods with sampling error and within-species variation: contrasts revisited and revised. Am Nat. 2008;171:713–725. doi: 10.1086/587525. [DOI] [PubMed] [Google Scholar]
- Finn RD, et al. The Pfam protein families database. Nucleic Acids Res. 2010;38:D211–D222. doi: 10.1093/nar/gkp985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forterre P. A hot story from comparative genomics: reverse gyrase is the only hyperthermophile-specific protein. Trends Genet. 2002;18:236–237. doi: 10.1016/s0168-9525(02)02650-1. [DOI] [PubMed] [Google Scholar]
- Friedman R, Drake JW, Hughes AL. Genome-wide patterns of nucleotide substitution reveal stringent functional constraints on the protein sequences of thermophiles. Genetics. 2004;167:1507–1512. doi: 10.1534/genetics.104.026344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galtier N, Lobry JR. Relationships between genomic G + C content, RNA secondary structures, and optimal growth temperature in prokaryotes. J Mol Evol. 1997;44:632–636. doi: 10.1007/pl00006186. [DOI] [PubMed] [Google Scholar]
- Giovannoni SJ, et al. Genome streamlining in a cosmopolitan oceanic bacterium. Science. 2005;309:1242–1245. doi: 10.1126/science.1114057. [DOI] [PubMed] [Google Scholar]
- Goldman N, Yang Z. A codon-based model of nucleotide substitution for protein-coding DNA sequences. Mol Biol Evol. 1994;11:725–736. doi: 10.1093/oxfordjournals.molbev.a040153. [DOI] [PubMed] [Google Scholar]
- Hartl DL, Clark AG. Principles of population genetics. Sunderland (MA): Sinauer associates; 1997. [Google Scholar]
- Jaenicke R. Stability and stabilization of globular proteins in solution. J Biotechnol. 2000;79:193–203. doi: 10.1016/s0168-1656(00)00236-4. [DOI] [PubMed] [Google Scholar]
- Katoh K, Misawa K, Kuma K, Miyata T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002;30:3059–3066. doi: 10.1093/nar/gkf436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koonin EV, Wolf YI. Genomics of bacteria and archaea: the emerging dynamic view of the prokaryotic world. Nucleic Acids Res. 2008;36:6688–6719. doi: 10.1093/nar/gkn668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn HJ, Cometta S, Fiechter A. Effects of growth temperature on maximal specific growth rate, yield, maintenance, and death rate in glucose-limited continuous culture of the thermophilic Bacillus caldotenax. Appl Microbiol Biotechnol. 1980;10:303–315. [Google Scholar]
- Kumar S, Nussinov R. How do thermophilic proteins deal with heat? Cell Mol Life Sci. 2001;58:1216–1233. doi: 10.1007/PL00000935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo CH, Moran NA, Ochman H. The consequences of genetic drift for bacterial genome complexity. Genome Res. 2009;19:1450–1454. doi: 10.1101/gr.091785.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lartillot N, Poujol R. A phylogenetic model for investigating correlated evolution of substitution rates and continuous phenotypic characters. Mol Biol Evol. 2011;28:729–744. doi: 10.1093/molbev/msq244. [DOI] [PubMed] [Google Scholar]
- Lynch M. Streamlining and simplification of microbial genome architecture. Annu Rev Microbiol. 2006;60:327–349. doi: 10.1146/annurev.micro.60.080805.142300. [DOI] [PubMed] [Google Scholar]
- Lynch M. The origins of genome architecture. Sunderland (MA): Sinauer Associates; 2007. [Google Scholar]
- Mackwan RR, Carver GT, Kissling GE, Drake JW, Grogan DW. The rate and character of spontaneous mutation in Thermus thermophilus. Genetics. 2008;180:17–25. doi: 10.1534/genetics.108.089086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarova KS, Wolf YI, Koonin EV. Potential genomic determinants of hyperthermophily. Trends Genet. 2003;19:172–176. doi: 10.1016/S0168-9525(03)00047-7. [DOI] [PubMed] [Google Scholar]
- McKay A, Quilter J, Jones CW. Energy conservation in the extreme thermophile Thermus thermophilus HB8. Archives Microbiol. 1982;131:43–50. [Google Scholar]
- Mira A, Moran NA. Estimating population size and transmission bottlenecks in maternally transmitted endosymbiotic bacteria. Microb Ecol. 2002;44:137–143. doi: 10.1007/s00248-002-0012-9. [DOI] [PubMed] [Google Scholar]
- Mira A, Ochman H, Moran NA. Deletional bias and the evolution of bacterial genomes. Trends Genet. 2001;17: 589–596. doi: 10.1016/s0168-9525(01)02447-7. [DOI] [PubMed] [Google Scholar]
- Mitchell P. Chemiosmotic coupling in oxidative and photosynthetic phosphorylation. Biochim Biophys Acta. 1966;1807:1507–1538. doi: 10.1016/j.bbabio.2011.09.018. [DOI] [PubMed] [Google Scholar]
- Moran NA, McLaughlin HJ, Sorek R. The dynamics and time scale of ongoing genomic erosion in symbiotic bacteria. Science. 2009;323:379–382. doi: 10.1126/science.1167140. [DOI] [PubMed] [Google Scholar]
- Moran NA, Wernegreen JJ. Lifestyle evolution in symbiotic bacteria: insights from genomics. Trends Ecol Evol. 2000;15: 321–326. doi: 10.1016/s0169-5347(00)01902-9. [DOI] [PubMed] [Google Scholar]
- Moya A, et al. Toward minimal bacterial cells: evolution vs. design. FEMS Microbiol Rev. 2009;33:225–235. doi: 10.1111/j.1574-6976.2008.00151.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordstrom KM, Laakso SV. Effect of growth temperature on fatty acid composition of ten thermus strains. Appl Environ Microbiol. 1992;58:1656–1660. doi: 10.1128/aem.58.5.1656-1660.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novichkov PS, Wolf YI, Dubchak I, Koonin EV. Trends in prokaryotic evolution revealed by comparison of closely related bacterial and archaeal genomes. J Bacteriol. 2009;191:65–73. doi: 10.1128/JB.01237-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orgel LE, Crick FH. Selfish DNA: the ultimate parasite. Nature. 1980;284:604–607. doi: 10.1038/284604a0. [DOI] [PubMed] [Google Scholar]
- Parter M, Kashtan N, Alon U. Environmental variability and modularity of bacterial metabolic networks. BMC Evol Biol. 2007;7:169. doi: 10.1186/1471-2148-7-169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennock J, Tempest DW. Metabolic and energetic aspects of the growth of Bacillus stearothermophilus in glucose-limited and glucose-sufficient chemostat culture. Arch Microbiol. 1988;150:452–459. [Google Scholar]
- Ranea JA, Grant A, Thornton JM, Orengo CA. Microeconomic principles explain an optimal genome size in bacteria. Trends Genet. 2005;21:21–25. doi: 10.1016/j.tig.2004.11.014. [DOI] [PubMed] [Google Scholar]
- Rodrigues JFM, Wagner A. Evolutionary plasticity and innovations in complex metabolic reaction networks. PLoS Comput Biol. 2009;5:e1000613. doi: 10.1371/journal.pcbi.1000613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4:406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- Shuter BJ, Thomas JE, Taylor WD, Zimmerman AM. Phenotypic correlates of genomic DNA content in unicellular eukaryotes and other cells. Am Naturalist. 1983;122:26–44. [Google Scholar]
- Sonnleitner B. Biotechnology of thermophilic bacteria—growth, products, and application. In: Fiechter A, editor. Advances in biochemical engineering/biotechnology. Berlin (Germany): Springer; 1983. pp. 69–138. [Google Scholar]
- Thompson MJ, Eisenberg D. Transproteomic evidence of a loop-deletion mechanism for enhancing protein thermostability. J Mol Biol. 1999;290:595–604. doi: 10.1006/jmbi.1999.2889. [DOI] [PubMed] [Google Scholar]
- Touchon M, Rocha EP. Causes of insertion sequences abundance in prokaryotic genomes. Mol Biol Evol. 2007;24:969–981. doi: 10.1093/molbev/msm014. [DOI] [PubMed] [Google Scholar]
- Vieira-Silva S, Rocha EP. The systemic imprint of growth and its uses in ecological (meta)genomics. PLoS Genet. 2010;6:e1000808. doi: 10.1371/journal.pgen.1000808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner A. Energy constraints on the evolution of gene expression. Mol Biol Evol. 2005;22:1365–1374. doi: 10.1093/molbev/msi126. [DOI] [PubMed] [Google Scholar]
- Wheeler DL, et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2008;36:D13–D21. doi: 10.1093/nar/gkm1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yooseph S, et al. Genomic and functional adaptation in surface ocean planktonic prokaryotes. Nature. 2010;468:60–66. doi: 10.1038/nature09530. [DOI] [PubMed] [Google Scholar]
- Zeldovich KB, Berezovsky IN, Shakhnovich EI. Protein and DNA sequence determinants of thermophilic adaptation. PLoS Comput Biol. 2007;3:e5. doi: 10.1371/journal.pcbi.0030005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.