

Abstract
Arsenic is widespread in the environment and its presence is a result of natural or anthropogenic activities. Microbes have developed different mechanisms to deal with toxic compounds such as arsenic and this is to resist or metabolize the compound. Here, we present the first reference set of genomic, transcriptomic and proteomic data of an Alphaproteobacterium isolated from an arsenic-containing goldmine: Rhizobium sp. NT-26. Although phylogenetically related to the plant-associated bacteria, this organism has lost the major colonizing capabilities needed for symbiosis with legumes. In contrast, the genome of Rhizobium sp. NT-26 comprises a megaplasmid containing the various genes, which enable it to metabolize arsenite. Remarkably, although the genes required for arsenite oxidation and flagellar motility/biofilm formation are carried by the megaplasmid and the chromosome, respectively, a coordinate regulation of these two mechanisms was observed. Taken together, these processes illustrate the impact environmental pressure can have on the evolution of bacterial genomes, improving the fitness of bacterial strains by the acquisition of novel functions.
Keywords: arsenic metabolism, motility/biofilm, Rhizobium/Agrobacterium, transcriptomics/proteomics, phylogeny, rhizosphere
Introduction
To deal with high concentrations of toxic metals, micro-organisms have evolved various strategies, which enable them to detoxify their environment. These processes involve physicochemical reactions, for example, precipitation or solubilization, adsorption or desorption (Borch et al. 2010), and metabolic oxido-reduction reactions (Gadd 2010). In addition, most of the metallic elements found in the periodic table may play a crucial role in microbial physiology, for example, as components of metalloproteins, or as electron donors or acceptors in energy metabolism (Stolz 2011). Such a metabolism may have been important in the early stages of life, due to a high concentration of metals, including arsenic, in the primordial planet (reviewed in van Lis et al. 2012).
In recent years, the various “omics” methods, which include genome sequencing, comparative genomics, and transcriptome or proteome analysis, have allowed to address the physiology of organisms in a global way. Such approaches have therefore greatly improved the understanding of microbial metabolism (Bertin et al. 2008; Holmes et al. 2009; Wilkins et al. 2009), including the global functioning of ecosystems, as recently demonstrated for an arsenic-rich microbial community (Bertin et al. 2011). To date, the genomes of more than 20 arsenic-metabolizing strains have been sequenced. They originate from various environments, belong to unrelated taxonomic groups, and have different carbon and energy requirements (reviewed in Bertin et al. 2012; van Lis et al. 2013).
Regarding arsenic, which is mainly present in two oxidation states in aquatic environments, that is, arsenite [As(III)] and arsenate [As(V)], microorganisms have acquired various metabolic capacities. These include As(V) reduction, which is usually part of the resistance mechanism, but also functions involved in As(III) oxidation or methylation (reviewed in Stolz 2011). Unlike arsenite methyltransferase genes, which are not often found in bacterial genomes, genes encoding arsenite oxidase are widespread in Bacteria and Archaea (Heinrich-Salmeron et al. 2011 and reviewed in Osborne and Santini 2012). In Herminiimonas arsenicoxidans, the arsenite oxidase aioBA genes are located in an arsenic genomic island, which also contains genes involved in arsenic resistance and biosynthesis of a molybdenum cofactor of the Aio enzyme (Muller et al. 2007). Such a genetic organization has also been observed in Thiomonas arsenitoxydans (Arsène-Ploetze et al. 2010; Bertin et al. 2012) and the presence of aio genes on a plasmid has been reported in Nitrobacter hamburgensis and Thermus thermophilus str. HB8 (Bertin et al. 2012). These observations suggest that aioBA genes may be acquired by horizontal gene transfer.
The adaptative response to arsenic has been recently shown as occurring in two steps (Cleiss-Arnold et al. 2010). First, bacterial cells express various genes involved in defence mechanisms, for example, oxidative stress and arsenic efflux. Next, several metabolic activities are induced, including arsenite oxidation which, in heterotrophic bacteria like H. arsenicoxydans, may be principally considered as a detoxification mechanism (Muller et al. 2007). In contrast, in bacteria that can grow autotrophically such as T. arsenitoxydans (Arsène-Ploetze et al. 2010) arsenite oxidation is part of a bioenergetic mechanism involved in energy generation. Despite some similarities, the genome organization of these two bacteria and their arsenic response, including biofilm formation, have been shown to differ markedly (Marchal et al. 2010, 2011).
In their natural environment, bacteria usually grow in biofilms, which are structured microbial communities embedded in extracellular polymeric substances (EPS) composed of sugars, proteins, and DNA (Hall-Stoodley et al. 2004; McDougald et al. 2012). Even though biofilm formation can be a problem in the field of human health, it allows bacteria to survive and thrive in highly toxic environments, including those characterized by high concentrations of heavy metals or metalloids such as arsenic (Guibaud et al. 2006; Muller et al. 2007). Unlike H. arsenicoxydans (Marchal et al. 2010), T. arsenitoxydans has been shown to induce biofilm formation in the presence of As(III) (Marchal et al. 2011). In addition, after biofilm development, the induction of cell motility has led to accelerated cell dispersion, an important process in the colonization of alternative ecological niches.
To gain a better understanding of the genetic determinants involved in the metabolism of arsenic, we have investigated the response to As(III) in Rhizobium sp. NT-26, a motile, chemolithoautotrophic arsenite oxidizer isolated from a gold mine in Australia (Santini et al. 2000). This strain belongs to the Rhizobiaceae family of the Alphaproteobacteria, which includes many species living in association with plants, such as plant mutualists of the Rhizobium and Ensifer (formerly Sinorhizobium) genera (Martens et al. 2007) and plant pathogens or plant growth-promoting rhizobacteria of the Agrobacterium genus (Hao et al. 2011). The Rhizobium sp. NT-26 genome was sequenced and annotated, and its physiology was investigated using differential transcriptomics and proteomics, and random mutagenesis. Remarkably, the synthesis of flagella was shown to be controlled by arsenite, suggesting a possible coordinate regulation between clusters located on two genetic elements. Indeed, proteins involved in As(III) oxidation were shown to be encoded by genes present on a megaplasmid, whereas flagellar genes are located on the chromosome.
Materials and Methods
Bacterial Strains, Plasmid, and Growth Conditions
Rhizobium sp. NT-26 and its mutant strains were cultivated at 28 °C in minimal salts medium (MSM) containing 0.04% yeast extract (Santini et al. 2000) and supplemented with As(III) and agar when required. Escherichia coli S17.1 λpir was cultivated at 28 °C in Luria–Bertani (LB) (MP Biomedicals) medium supplemented with 20 mg/l kanamycin (Sigma) for the maintenance of the pTGN/mini-Tn5 gfp-km plasmid (Tang et al. 1999).
Random Mutagenesis and Screening
Using the suicide vector pTGN carrying the mini-Tn5 transposon, random mutagenesis was performed to construct a mutant library and to identify genes involved either in arsenite oxidation or in motility. Mobilization of the plasmid was performed using E. coli S17.1 λpir carrying plasmid pTGN as the donor and Rhizobium sp. NT-26 as the recipient. For conjugation, both strains in exponential phase, respectively, corresponding to an optical density (OD) of 0.6 and 0.135 at 600 nm, were superposed on LB plates at 28 °C for 24 h. As the Rhizobium sp. NT-26 strain used in this study is rifampicin resistant (Santini and vanden Hoven 2004), mutants were then selected on LB plates supplemented with 20 mg/l kanamycin and rifampicin.
Colonies from the library were screened for the loss of arsenite oxidation or the loss of motility. Briefly, mutants were individually inoculated into 96-well microtiter plates containing MSM with 0.04% yeast extract and 8 mM As(III) and incubated at 28 °C in 1.5% agar for 48 h or 0.3% agar for 24 h, respectively. The library was screened in the following two ways: 1) the silver nitrate method was used to detect arsenite oxidation (Lett et al. 2001; Muller et al. 2003) and 2) the diameter of the swarming ring was used to determine whether the cells were motile (Muller et al. 2007). Each phenotype was subsequently confirmed on Petri dishes in the corresponding medium. Mutants unable to oxidize arsenite were also tested for motility and vice versa.
To identify the disrupted gene in each mutant, the genomic region close to the mini-Tn5 insertion site was amplified by inverse polymerase chain reaction (PCR). Total DNA was extracted with the Wizard Genomic DNA purification kit according to the manufacturer’s instructions (Promega). One microgram of DNA was digested with 50 U of restriction enzymes that do not cut the transposon sequence (ClaI or PstI) in a 50 µl reaction volume at 37 °C for 2 h. After precipitation by ethanol and sodium acetate, digested DNA was ligated with 10 U of DNA ligase (Fermentas) in a volume of 20 µl overnight at 16 °C. PCR was carried out on 25 ng of this template in a 25 µl volume reaction with iProof DNA Polymerase (Bio-Rad) and Oend (ACTTGTGTATAAGAGTCAG) and Iend (AGATCTGATCAAGAGACAG) primers. The program used involved a denaturation step at 98 °C for 30 s, followed by 35 cycles of denaturation at 98 °C for 10 s, annealing at 52 °C for 30 s and elongation at 72 °C for 3 min, and a final elongation step at 72 °C for 10 min. Amplification products were checked on an agarose gel and sequenced with Oend by MilleGen (http://www.millegen.com/, last accessed April 30, 2013). The Blastn tool on the MaGe interface (Vallenet et al. 2006) was used to align the sequences with that of the Rhizobium sp. NT-26 genome allowing for identification of the disrupted gene. For each mutant, the precise insertion site and orientation of the mini-Tn5 was determined by PCR, combining the Oend and Iend primers with new specific primers (supplementary table S1, Supplementary Material online) designed around each probable insertion site.
Biofilm Quantification
Biofilm formation by Rhizobium sp. NT-26 wild-type and mutant strains grown in the presence or absence of arsenite was measured by the crystal violet method. Cultures were grown in MSM medium containing 0.04% yeast extract supplemented with and without 8 mM As(III) and incubated at 28 °C overnight with shaking (120 rpm). The cultures were then diluted with fresh medium to an OD of 0.1 at 600 nm. Each strain was tested with six replicates of 200 µl in two flat-bottomed polystyrene 96-well microtiter plates (Nunc). Cultures were incubated at 28 °C for 24 h and 48 h without agitation. Biofilm formation was quantified using crystal violet, as previously described (Hommais et al. 2002). Briefly, after removing the culture medium, wells were gently rinsed three times with 0.1 M phosphate-buffered saline (PBS). To fix biofilms, plates were dried at 55 °C for 25 min, then 200 µl of 0.1% [w/v] crystal violet solution (Merck) was added to the wells and the plates were incubated at 30 °C for 30 min. Free crystal violet was removed and wells were washed three times with PBS. Plates were dried at room temperature and the biofilm was subsequently dissolved in 200 µl of 95% [v/v] ethanol over 30 min. Finally, the absorbance was read at 595 nm with a microplate reader (Synergy HT).
Pulsed-Field Gel Electrophoresis
Plasmid profiles were determined by a modified Eckhardt agarose gel electrophoresis technique, as described previously (Hynes and McGregor 1990). Rhizobium sp. NT-26 was grown in LB until an OD of 0.5 at 600 nm was reached, and 150 µl of culture were used per well. Electrophoresis was carried out at 4 °C, 5 V for 30 min and 85 V for 7 h on a 0.7% agarose gel containing 1% [w/v] sodium dodecyl sulfate (SDS) (Ramírez-Bahena et al. 2012). Plasmid size was estimated by comparison with those from Agrobacterium tumefaciens C58 (Wood et al. 2001).
Plant Trapping Tests
Nodulation experiments were performed under gnotobiotic conditions. Seeds of Macroptilium atropurpureum, Vicia faba, Phaseolus vulgaris, and Pisum sativum were surface sterilized for 2 min in 95% ethyl alcohol and then three times for 3 min in 1% sodium hypochlorite, each time washed with sterile water. Germination was carried out at 28 °C in dark conditions on glass plates covered with sterile filter paper moistened with sterile water. Pots with a capacity of 1.5 l were filled with sterile vermiculite, and 200 ml of nutrient sterile solution (Rigaud and Puppo 1975) was added per pot. Two seedlings were sown in each pot and plants were inoculated with a suspension of 105 CFU/ml 1 week after their transfer to hydroponic growth. Rhizobium sp. NT-26 was grown on YMB medium (Mannitol 0.7%, Yeast extract 0.2%, KH2PO4 0.02%, MgSO4 0.02%) and 1 ml of inoculum was applied to each seedling. Plants were regularly observed for nodule formation, and nodulation was quantified after inoculation as described in Gremaud and Harper (1989).
Genome Sequencing
The complete genome sequence of Rhizobium sp. NT-26 was obtained by combining Sanger and 454 sequencing methods. Sanger reads were obtained from a 10 kb insert library constructed after mechanical shearing of the genomic DNA and cloning of the generated inserts into the plasmid pCNS, as described previously (Muller et al. 2007). Plasmid DNA was purified and end-sequenced (26,888 reads) by dye-terminator chemistry with ABI3730 sequencers (Applied Biosystems) leading approximately to a 4× coverage. Reads were assembled by Newbler with around 20× coverage of 454 GS FLX reads (Roche) and validated via the Consed interface. Finishing steps were performed using primer walking of clones, PCR and in vitro transposition technology with the Template Generation System II Kit (Finnzyme), corresponding to 252, 32 and 8,404 additional reads, respectively. Approximately 70× coverage of 36 bp Illumina reads were mapped in the polishing phase, using SOAP (http://soap.genomics.org.cn/, last accessed April 30, 2013), as previously described (Aury et al. 2008).
Comparative Analysis of 24 Rhizobiaceae Genomes
The 23 genomes of Rhizobiaceae publicly available at the time of experiments (supplementary table S2, Supplementary Material online) were retrieved from ENA database (http://www.ebi.ac.uk/ena/, last accessed April 30, 2013). A homologous gene family database was built under the HOGENOM procedure (Penel et al. 2009) based on these 23 genomes and the one of Rhizobium sp. NT-26. Homologous protein sequences were aligned using MUSCLE (v.3.8.31, default parameters) (Edgar 2004) and then retro-translated with the pal2nal program (v.14) (Suyama et al. 2006). Nucleic acid alignments were restricted to conserved blocks with Gblocks (v.0.91b, minimum 50% of sequences in conserved and flank positions and all gaps allowed, codon mode) (Castresana 2000) and gene trees were computed from these alignments with PhyML (v.3.0, GTR + G8 + I model of evolution, best of SPR and NNI moves, SH-like branch supports) (Guindon and Gascuel 2003). All alignments and phylogenetic trees are shown in supplementary methods S1 and S2, Supplementary Material online. Replicon mapping and gene content comparison were done with custom Python scripts.
Species Phylogenies
The “core” set contained 822 gene families present in every 24 strains in only one copy. The “ribosomal” set contained 51 gene families whose products were annotated as “ribosomal protein” or related terms in at least one genome and were present in at least 22 strains. Full alignments of both family sets, and third codon-removed version of core family set were concatenated and used for species tree construction with RaxML (version 7.2.8-ALPHA, GTRCAT model with 50 categories, branch supports from 200 and 1,000 rapid bootstrap trees for “core” and “ribosomal” alignments, respectively) (Stamatakis 2006) (species trees are stored in supplementary methods S3, Supplementary Material online).
Tree Pattern Matching
Phylogenetic trees of gene families were searched for particular phylogenetic patterns, that is, subtrees with specific arrangement of relative branching leaves representing taxa, with TPMS software (Bigot et al. 2012): “NT26outAgro”, that is Rhizobium sp. NT-26 as a direct outgroup of Agrobacterium genus; “NT26inAgro”, that is Rhizobium sp. NT-26 as an ingroup of Agrobacterium and sister group of A. tumefaciens. Both searches were made first without considering branch support and then matching only with >0.9 SH-like branch support at nodes of interest (supplementary methods S4, Supplementary Material online).
Aio Phylogenies
Homologs of AioA, AioB, AioR, AioS, and AioX were retrieved from the nr database at the NCBI (http://www.ncbi.nlm.nih.gov/, last accessed April 30, 2013) using the BlastP program (Altschul et al. 1997) with the protein sequences of Rhizobium sp. NT-26 as queries and default parameters except the “Max target sequences” parameter which was set to 1,000. For each Aio protein, the 500 homologs displaying the highest similarity with the sequence of Rhizobium sp. NT-26 were retrieved and aligned using MAFFT (version 6, default parameters) (Katoh and Toh 2008). The resulting alignments were trimmed using BMGE (default parameters) (Criscuolo and Gribaldo 2010). Preliminary phylogenies were inferred using the Neighbor-Joining method implemented in SeaView (Poisson evolutionary distance) (Gouy et al. 2010). The robustness of the resulting trees was estimated with the nonparametric bootstrap procedure implemented in SeaView (100 replicates of the original alignments). Based on the resulting trees, the closest relatives of Rhizobium sp. NT-26 sequences were identified and used for more detailed phylogenetic analyses. The corresponding sequences were realigned and the resulting alignments trimmed using the same procedure. Final phylogenetic analyses were performed using the maximum likelihood and Bayesian approaches implemented in PhyML (version 3) (Guindon et al. 2009) and MrBayes (version 3.2) (Ronquist et al. 2012), respectively. PhyML was run with the LG evolutionary model (Le and Gascuel 2008) and a gamma distribution with four categories of substitution rates (Γ4) and an estimated alpha parameter. The robustness of the maximum likelihood trees was estimated by the nonparametric procedure implemented in PhyML (100 replicates of the original alignments). MrBayes was run with a mixed substitution model and a Γ4 distribution. Four chains were run in parallel for 1,000,000 generations. The first 2,000 generations were discarded as “burnin.” The remaining trees were sampled every 100 generations to build the consensus tree.
Total RNA Extraction, Microarrays, and Data Analysis
A custom 15 K microarray with a probe length of 60 mer was manufactured by Agilent Technologies following the protocol used for H. arsenicoxydans (Weiss et al. 2009). Total RNA was extracted from Rhizobium sp. NT-26 strain grown heterotrophically in MSM containing 0.04% yeast extract in the absence and presence of 5.3 mM As(III) until late log phase (OD at 600 nm of 0.115 and 0.152, respectively) as described previously (Santini et al. 2007). RNA quality was checked using an Agilent Bioanalyzer. Ten micrograms of total RNA was reverse transcribed using the Fairplay III Microarray labeling kit (Agilent Technologies) and cDNA were indirectly labeled using Cy3 or Cy5 Mono reactive dyes (GE Healthcare). Labeled cDNA quality and quantity were determined by spectroscopy at 260, 280, 550, and 650 nm. The labeled Cy3 and Cy5 target quantities were adjusted to 250 pmol, mixed together and concentrated with Microcon YM-30 (Millipore). Hybridization was performed for 17 h at 65 °C. Three distinct biological RNA samples as well as dye swap experiments were performed for each culture condition. Arrays were scanned as described previously (Weiss et al. 2009). Data were acquired by Genepix Pro 6.0 (Axon Instrument) and statistically analyzed as described previously (Koechler et al. 2010). Genes having a BH adjusted P value lower than 0.05 were considered as differentially expressed between the two conditions and were retained for further study. Microarray data were deposited in ArrayExpress (E-MEXP-3021).
Preparation of Proteins Extracts and 2D Gel Electrophoresis
Experiments were performed with four protein extracts from four replicates for each growth condition. Strain NT-26 was grown heterotrophically in MSM containing 0.04% yeast extract in the absence or presence of 5.3 mM As(III). Exponential phase cultures were harvested by centrifugation at 6,000 × g for 10 min at 4 °C. Pellets were suspended in 400 µl of distilled water supplemented with 1 µl Benzonase Nuclease (Sigma) and 4 µl of Protease Inhibitor Mix (GE Healthcare). Cell suspensions were sonicated on ice with 10 pulses of 30 s at 28% of amplitude with 30 s intervals using a VC 750 sonicator (Bioblock Scientific). Cellular debris were removed by two centrifugations, the first at 6,000 × g for 5 min and the second at 16,000 × g for 90 min. Protein concentrations were measured using the Bradford method (Bradford 1976).
Differential accumulation of proteins was either monitored by Colloidal Brilliant Blue staining or DIGE (Marouga et al. 2005). For Colloidal Brilliant Blue staining experiments, 300 μg of protein extract were diluted to a final volume of 350 µl with rehydration buffer (8 M urea, 2% [w/v] CHAPS, 0.5% [v/v] IPG buffer pH 3-10, 40 mM DTT, and 0.01% [w/v] bromophenol blue). For DIGE experiments, 50 µg of protein was adjusted to pH 8.8 by adding 50 mM Tris–HCl final concentration, and either stained with 400 pmol of Cy3 or Cy5 (GE Healthcare). In addition, 25 µg of each 8 extracts (4 replicates for 2 conditions) were pooled and stained with 1,600 pmol of Cy2 to serve as an internal standard. For the staining, each CyDye DIGE fluor stock solution was diluted in high grade dimethylformamide to a final concentration of 400 pmol/µl. One microliter of the dilution was added to 50 µg of protein (Cy3 and Cy5) or 4 µl to 200 µg of the internal standard pool (Cy2) and kept in the dark and on ice for 30 min. The reaction was stopped by adding 1 µl of a 10 mM solution of lysine to 50 µg of protein (4 µl to 200 µg) and then 1 volume of 2× sample buffer (9 M urea, 3 M thiourea, 130 mM DTT, 4% [w/v] CHAPS, 2% [v/v] IPG buffer Pharmalyte 3-10) was added prior to incubation on ice for 10 min. One Cy3-labeled sample (condition 1) and one Cy5-labeled sample (condition 2) were mixed with one-fourth of the Cy2-labeled pool and rehydration buffer was added to reach a volume of 350 µl. Dye swap experiments were performed for each culture condition.
For protein separation, samples were first loaded onto an 18 cm pH 4–7 IPG strip. IEF was conducted using the Ettan IPGphor system (GE Healthcare), as previously described (Weiss et al. 2009). The strips were equilibrated in SDS equilibration buffer (30 mM Tris–HCl pH 8.8, 6 M urea, 34.5% [v/v] glycerol, 2% [w/v] SDS, 0.01% [w/v] bromophenol blue) supplemented with 1% [w/v] DTT for 15 min and then with 2.5% [w/v] iodoacetamide for 15 min. SDS polyacrylamide gel electrophoresis was subsequently performed using 11.5% SDS gels, using the Ettan DAltsix system (GE Healthcare) with the following steps: 1 h at 60 mA, 80 V, 4 W and 1 h at 240 mA, 500 V, 52 W. Gels were stained with Colloidal Brilliant Blue or digitized using a Typhoon Scanner (GE Healthcare).
Differential protein expression analysis was performed as previously described (Bryan et al. 2009; Weiss et al. 2009). Spots were selected and identified by MALDI-TOF and Nano LC-MS/MS, and data analysis were performed with Mascot (Matrix Science Ltd.) as described previously (Bryan et al. 2009) against a Rhizobium sp. NT-26 protein database. All identifications were incorporated into the “InPact” proteomic database developed previously (http://inpact.u-strasbg.fr/∼db/, last accessed April 30, 2013) (Bertin et al. 2008).
Transmission Electron Microscopy
Rhizobium sp. NT-26 or the aioR mutant were grown in MSM containing 0.04% yeast extract in the presence or absence of 8 mM As(III) for 24 h. A drop of culture was deposited onto Formvar-coated nickel grids and after cell decantation, the liquid excess was removed. Uranyl acetate 2% was added to negatively stain bacteria and flagella and these samples were dried. Grids were observed with a Hitachi H-600 transmission electron microscope (TEM) at 75 kV and photographed with a Hamamatsu ORCA-HR camera using the AMT software (Advanced Microscopy Techniques).
Results and Discussion
General Genome Features
Chromosome, Plasmids, and Genomic Plasticity
The Rhizobium sp. NT-26 genome includes a single 4.2 Mbp chromosome and two plasmids. The circular chromosome consists of 4,239,731 bp with 4,380 coding sequences, including 4,303 coding DNA sequences and 59 RNA genes, and representing 90.28% of the whole genome (fig. 1). Among these coding sequences (CDS), 34.40% are of unknown function.
Fig. 1.—

Circular representation of the Rhizobium sp. NT-26 genome. The chromosomal (A) and plasmidic (B) characteristics are 4,239-Mb long, 61.97% GC, 9 16S-23S-5S rRNA, 50 tRNA, and 4,294 CDSs; and 322-kb long, 60.19% GC, 0 16S-23S-5S rRNA, 0 tRNA, and 367 CDSs, respectively. From outside, circles display 1) the GC percent deviation in a 1,000 bp window (GC window – mean GC); 2) and 3) predicted CDSs transcribed in the clockwise and counterclockwise direction, respectively; red and blue colors correspond to validated annotations, orange to automatic annotation and purple to primary automatic annotation; 4) GC skew (G + C/G − C) in a 1,000 bp window; 5) rRNA are shown in blue, tRNA in green, miscRNA in orange, transposable elements in pink, and pseudogenes in gray. The regions with the genes coding for proteins involved in motility, reduction of arsenate or oxidation of arsenite are highlighted in black, blue, or yellow, respectively. The figure does not represent the p2 plasmid and the scale between the two genetic determinants is not respected (https://www.genoscope.cns.fr/agc/microscope/home/index.php, last accessed April 30, 2013).
The mean G + C content of the chromosome is 61.97% but its distribution is not homogenous (fig. 1), and the Rhizobium sp. NT-26 chromosome exhibits 65 regions of genomic plasticity (RGP, supplementary table S3, Supplementary Material online) (Vallenet et al. 2009) in comparison with that of A. tumefaciens 5A. The G + C content of these regions, their size (5–207 kb) and the codon adaptation index lower than the average are characteristic of genomic islands (GEI) (Juhas et al. 2009). Moreover, transposable elements and tRNA encoding genes are present in several of these regions, which further support the lateral transfer of these potential genomic islands (Daubin et al. 2003). Such genetic events are known to promote bacterial adaptation under environmental stresses by the acquisition of various capacities through horizontal gene transfer, an important mechanism of microbial genome evolution (Juhas et al. 2009). In agreement with this, more than 15 loci coding for metabolic functions that may improve the fitness of the strain to its environment were found among the 65 RGP identified in the genome of Rhizobium sp. NT-26. These include amino acids and carbon sources transport, inorganic carbon fixation, nitrogen metabolism, and sulfur oxidation (supplementary table S3, Supplementary Material online).
The genomes of bacteria in the Agrobacterium and Rhizobium genera are known to include several extrachromosomal replicons, which encode various functions required for the adaptation to specific niches (López-Guerrero et al. 2012). The Rhizobium sp. NT-26 genome comprises two plasmids, including a megaplasmid of 322,264 bp containing 367 coding sequences (CDS) (fig. 1). The presence and the size of the megaplasmid were confirmed experimentally by a modified Eckhardt gel electrophoresis method (supplementary fig. S1, Supplementary Material online). The second plasmid is 15,430 bp and more than half of its CDS encode proteins with unknown functions.
The large plasmid harbors four different replication systems, in particular three repABC operons. The first one is duplicated. These two repABC operons are related to repABC of Dinoroseobacter (>90% identity) of the Rhodobacteraceae, a family of the Rhodobacterales. The third repABC operon is related to repABC of Rhizobiales. The fourth replication system is composed of a ParB/RepC replication system homolog of A. tumefaciens NCPPB925 plasmid origin of replication. Such a redundancy is not rare in Rhizobiales. For example, two replicons in R. etli CFN42, one in R. leguminosarum 3841 and one in Ruegeria sp. PR1b contain two repABC operons (Zhong et al. 2003; González et al. 2006; Young et al. 2006).
The smaller plasmid replication system is different from the canonical repABC replication system. It is constructed as the replication system described in pTAR of A. vitis (Gallie and Kado 1988), that is, the origin region carries a repA-like gene, a parA gene and a putative regulator locus coding for a putative segregation protein. Besides these replication transfer genes, the 15 kb plasmid harbors a toxin antitoxin system, which may explain its maintenance in Rhizobium sp. NT-26.
Plasmidic Adaptive Traits
In symbiotic bacteria, plasmids are known to play a role in their interaction with plants (López-Guerrero et al. 2012). In addition to multiple transposases and insertion sequences, the megaplasmid identified in Rhizobium sp. NT-26 encodes a putative type IV secretion system known to be involved in conjugal DNA transfer, including between bacteria and plants. Indeed, two complete tra clusters were found on the 322 kb plasmid: the first one is related to the type IV secretion system found in the Rhizobium/Agrobacterium genus, whereas the second one is related to the type IV secretion system of marine bacteria members of the Rhodobacteraceae family, that is, Oceanibulbus indolifex or Ruegeria sp. PR1b plasmid pSD25. However, canonical nodABC genes coding for proteins NodA (acetyl transferase), NodC (oligomerization of N-acetyl-glucosamine), and NodB (chitooligosaccharide deacatylase) that are required for the synthesis of the core structure of lipo-chitooligosaccharide (i.e., Nod factor) (Dénarié et al. 1996) were not identified in the genome of Rhizobium sp. NT-26. CDS displaying some similarities with other nod genes, that is, encoding enzymes that control specific substitutions on the chitooligosaccharide backbone, or the fix operon are present (supplementary table S4, Supplementary Material online), but these genes are also well conserved in nonsymbiotic prokaryotes. Nevertheless, it has been demonstrated that the nodulation of some Fabaceae by rhizobia occurs in the absence of the nodABC genes and lipo-chitooligosaccharidic Nod factors (Giraud et al. 2007). This indicates that other signaling strategies can trigger nodule organogenesis in some legumes. Nodulation assays on various Fabaceae plants were therefore performed as previously described (Gremaud and Harper 1989), but no nodules were observed at 3 weeks after inoculation or later at 4 weeks (data not shown).
Despite a lack of plant nodulation, root inoculation by Rhizobium sp. NT-26 suggested a potential phytobeneficial effect (fig. 2). Direct plant-growth promotion can be derived from phosphorus solubilization (Richardson et al. 2009), production of plant growth regulators (phytohormones) such as auxins, gibberellins, and cytokinins (Spaepen et al. 2009), NO production and/or by supplying biologically fixed nitrogen (Creus et al. 2005). Increasing the bioavailability of phosphate as micronutrient is mediated by bacterial phosphatase activity, and a phosphatase homolog, that is, NT26v4_0651, is present in the Rhizobium sp. NT-26 genome. Moreover, two main classes of dissimilatory nitrite reductase (Nir) involved in NO production exist among denitrifying bacteria: the heme-cytochrome cd1 type encoded by nirS genes and the copper-containing type encoded by nirK genes (Zumft 1997). A nirK homolog but no nirS homolog was identified in the Rhizobium sp. NT-26 genome. Finally, no other homolog of classical phytobeneficial functions was identified when analyzing the genome, for example, phytohormone synthesis such as auxin by ipdC/ppdC or acetoin-2,3-butanediol by budABC, or nitrogen fixation by nitrogenase nifHDK.
Fig. 2.—

Phytobeneficial effect of Rhizobium sp. NT-26 on Phaseolus vulgaris. Erlenmeyer flasks of P. vulgaris were inoculated with Rhizobium sp. NT-26 on the left, and with water on the right.
Indirect plant growth-promoting mechanisms used by plant-growth-promoting rhizobacteria (PGPR) include induced systemic resistance, antibiotic protection against pathogens, reduction of iron availability in the rhizosphere by sequestration with siderophores, synthesis of fungal cell wall-lysing or lytic enzymes, and competition for nutrients and colonization sites with pathogens (Dobbelaere and Okon 2007). Rhizobium sp. NT-26 contains loci coding for polyketide synthases (NT26v4_3331, NT26v4_3332, and NT26v4_3333), which are involved in nonribosomal synthesis of antibiotics, or coding for proteins involved in siderophore transport (NT26v4_2008, NT26v4_4195, and NT26v4_4199). Taken together, these observations suggest that Rhizobium sp. NT-26 does not exert any direct interaction with plants but it may have an indirect role in plant growth and protection by its metabolic activities in the rhizosphere.
Unlike H. arsenicoxydans (Muller et al. 2007) and T. arsenitoxydans (Arsène-Ploetze et al. 2010), which metabolize and provide resistance to arsenic using proteins encoded by chromosomally borne genes, proteins involved in arsenic resistance in Rhizobium sp. NT-26 are encoded by ars genes present on both the chromosome and the megaplasmid. The aio genes involved in arsenite oxidation are present only on the megaplasmid (fig. 1B), as shown in The. thermophilus str. HB8. The Rhizobium sp. NT-26 aio cluster also contains genes coding for phosphate transport and molybdenum cofactor biosynthesis, as previously observed in other arsenite-oxidizing bacteria (Arsène-Ploetze et al. 2010; Bertin et al. 2011). In addition, like the metallo-resistant strain Cupriavidus metallidurans (Janssen et al. 2010), the Rhizobium sp. NT-26 megaplasmid contains numerous genes involved in resistance to heavy metals such as chromium, cadmium, and mercury. These observations suggest a loss of most plasmid-encoded functions known to be involved in bacteria–plant interactions and an acquisition of multiple genes allowing the organism to grow in its natural habitat, a goldmine known to contain toxic metals and metalloids.
The gene cluster coding for arsenite oxidase contains 5 aio genes in Rhizobium sp. NT-26. The survey of the nr database revealed the existence of numerous homologs of AioA, AioB, AioR, AioS, and AioX. Preliminary phylogenetic analyses of AioA homologs showed that the sequence from Rhizobium sp. NT-26 belongs to a well-supported clade of proteobacterial sequences corresponding to the groups I and II, which were recently described (Heinrich-Salmeron et al. 2011). Subsequent phylogenetic analyses revealed that the Rhizobium sp. NT-26 AioA branched among alphaproteobacterial sequences (group I), within a strongly supported clade composed of sequences from various Agrobacterium species and Sinorhizobium sp. M14 (Rhizobiaceae), from Ochrobacterium tritici (Brucellaceae), and uncultured organisms (bootstrap value [BV] = 98% and posterior probability [PP] = 1.00, supplementary fig. S2A, Supplementary Material online). Phylogenetic analyses of other Aio proteins showed similar branching patterns (supplementary fig. S2B–E, Supplementary Material online). This suggests that the five aio genes have co-evolved, which is not entirely surprising given that they are functionally related and clustered together when present in a genome.
A careful examination of the taxonomic distribution within the subgroups (supplementary fig. S2, Supplementary Material online) revealed that only 2 Rhizobium/Agrobacterium complete genomes contain the aio genes although nearly 30 genome sequences are available at NCBI. In addition, the relationships among the sequences were not always in agreement within the phylogeny of species, for example, the grouping of a member of Brucellaceae within Rhizobiaceae. This strongly suggests that horizontal gene transfers may have played a role in the spread of aio genes among these species. The alternative hypothesis, that is the presence of aio genes in the common ancestor of the Rhizobium/Agrobacterium group followed by multiple independent gene losses during the diversification of this lineage, appears less likely and does not explain the discrepancies among the Aio phylogenies and the taxonomy. On the contrary, these inconsistencies may be easily explained by the initial acquisition of the aio genes by one member of the Rhizobium/Agrobacterium group followed by a few horizontal gene transfers to related species or strains. Such transfers may have been favored by the colocation of aio genes on genomes and their location on plasmids in a few strains (e.g., pSinA in Sinorhizobium sp. M14 and pAt5A for A. tumefaciens 5A).
Taxonomic Relationship of Rhizobium sp. NT-26 with Other Rhizobiaceae
Rhizobium sp. NT-26 strain has been previously assigned to the Rhizobium genus on the basis of 16S RNA sequence (Santini et al. 2000), but its phylogenetic relationship with other Rhizobiaceae remains quite unclear. Therefore, the sequence of the Rhizobium sp. NT-26 chromosome has been compared with the sequences present in the “Prokaryotic Genome DataBase” (PkGDB) (Vallenet et al. 2006) and RefSeq (NCBI Reference Sequences) data banks. The highest synteny conservation was observed with the A. tumefaciens C58 circular chromosome, which is 67.85% of the CDS in this genome share synteny with the chromosome of Rhizobium sp. NT-26 and the average size of the syntons is 8.4 CDS. This gene order conservation is higher than that observed with Rhizobium spp. strains (6.9–7.4), suggesting a closer evolutionary relationship of strain Rhizobium sp. NT-26 with the Agrobacterium lineage. To determine more precisely the taxonomic position of Rhizobium sp. NT-26 among Rhizobiaceae, we compared its genome with a set of 23 other sequenced genomes of this family in a phylogenetic framework. We computed maximum-likelihood (ML) trees based on the concatenated alignments of sequences of either all-homologous genes that are common to and unique in all strains (822 “core” genes) or genes of ribosomal proteins (51 “ribosomal” genes). Intriguingly, both data sets yielded phylogenies that agree on all major splits in the taxon, but not on the position of Rhizobium sp. NT-26. According to core genes, this strain branched as an “in-group” of the Agrobacterium subgroup, being the brother clade of A. tumefaciens after the split with A. vitis (fig. 3A). Instead, according to ribosomal genes, strain NT-26 branched as an “out-group” of the Agrobacterium clade that encompasses A. vitis and A. tumefaciens (fig. 3B). In both cases, the conflicting bipartitions are well supported, and removing third codon positions in the alignment of core genes, because of the possible saturation of the substitution signal in non-housekeeping genes, did not change the observed pattern (supplementary methods S3, Supplementary Material online). This suggests that among the core gene set, which include the majority of the ribosomal gene set, different genes have different histories, causing the average history (core phylogeny) to be different from that of a subset (ribosomal phylogeny). This may have been caused by horizontal gene transfer to and from A. tumefaciens, A. vitis, Rhizobium, or other more phylogenetically distant taxon that would blur the signal for vertical inheritance.
Fig. 3.—

Phylogeny of Rhizobium sp. NT-26 among Rhizobiaceae. ML phylogenies of 24 Rhizobiaceae including strain NT-26 were built from concatenated alignments of (A) 822 core genes and (B) 51 ribosomal genes. Branch supports are percentage of the 200 and 1,000 bootstrap trees having the bipartition, respectively.
We therefore computed individual phylogenies for all homologous families to determine what scenario each gene supported. On a set of 2,878 homologous gene family trees containing 3,537 Rhizobium sp. NT-26 genes, we searched for subtrees displaying the unambiguous patterns of either Rhizobium sp. NT-26 as a direct out-group of the Agrobacterium clade (“NT26outAgro”) or as an in-group of the Agrobacterium clade and a brother group of A. tumefaciens (“NT26inAgro”). “NT26inAgro” was prevalent with 338 genes (268 considering only high branch support) versus 255 (146) for “NT26outAgro,” and even though the amount of genes displaying such unambiguous patterns was relatively low, “NT26inAgro” was significantly more frequent (χ2 test, P value < 10−3). The location of those genes along the chromosome of Rhizobium sp. NT-26 showed no grouping with a particular pattern that could support a large-scale transfer event (Mann–Whitney–Wilcoxon test, P value >0.9; supplementary fig. S3, Supplementary Material online). The homogeneous dispersal of phylogenetic signatures rather suggests that numerous small-scale transfer events occurred, as observed in the case of frequent homologous recombination with partners of different taxa (Didelot et al. 2010). Alternatively, our observations may be the consequence of a poor resolution of phylogenies. This may be due to the frequent artifacts in the phylogenetic reconstruction of the relationship of strain NT-26 to Agrobacterium, such as those caused by the long branch leading to Rhizobium sp. NT-26. In the future, more phylogenetic information might be provided by sampling strains branching at the base of the Rhizobium/Agrobacterium group.
Most of Rhizobiaceae contain a secondary chromosome or megaplasmids, generally referred to as chromids (Harrison et al. 2010), that are members of a same family of large replicons derived from a plasmid (Slater et al. 2009). Although Rhizobium sp. NT-26 genome contains a megaplasmid with chromid characteristics (Harrison et al. 2010), this megaplasmid show very limited homology with chromids of this family (supplementary table S5, Supplementary Material online). The absence of a typical Rhizobiaceae secondary replicon makes the genome structure of Rhizobium sp. NT-26 unusual when compared with other members of the family. Comparison of the homologous gene location in Rhizobium sp. NT-26 and related organisms may help with the understanding of its evolution history. With this aim, the closest homolog of each of its genes present in several strains of Rhizobium and Agrobacterium were mapped along the Rhizobium sp. NT-26 chromosome (supplementary fig. S4, Supplementary Material online). It appeared that the vast majority of strain NT-26 chromosomal genes map to the principal chromosome in Rhizobium and A. vitis, suggesting that the Rhizobium sp. NT-26 lineage has completely lost the secondary chromosome of the Rhizobium/Agrobacterium ancestor. The history of intragenomic translocations have been documented in this taxon (Slater et al. 2009) and locating Rhizobium sp. NT-26 genes whose homologs have migrated at a specific divergence time would help to date the age of the divergence of the Rhizobium sp. NT-26 lineage. In this respect, its chromosome possesses a large chromosomal fragment (spanning from 2.55 to 3.40 Mb), which is specifically present on the secondary (linear) chromosome in A. tumefaciens (supplementary fig. 3, Supplementary Material online), further supporting a divergence of the Rhizobium sp. NT-26 lineage predating the A. tumefaciens speciation and synapomorphic translocation events. Similarly, strain NT-26 appears to have conserved the majority of the genes that are specifically borne by the secondary chromosome of R. rhizogenes (119 homologs in Rhizobium sp. NT-26 over 129 specific translocated genes). If strain NT-26 belonged to the Rhizobium lineage, the majority of these genes would probably have been lost with this whole chromid, although we cannot rule out potential translocations of those genes back to the main chromosome along with the chromid loss. Taken together, and even though our current data do not allow a more accurate classification, our observations support the inclusion of Rhizobium sp. NT-26 in the Agrobacterium subgroup. Nevertheless, according to the current nomenclature (Young et al. 2001), Rhizobium is still a valid genus name for strain NT-26.
Finally, although chromosomes are mainly dedicated to housekeeping functions, chromids carry genes involved in specific ecological functions, that is, legume symbiosis enabled by symbiotic plasmids in Rhizobium and Sinorhizobium (Harrison et al. 2010) and plant-related functions in A. tumefaciens C58 (Lassalle et al. 2011). All these functions relate to the interactions inside the rhizosphere and soil that represent the canonical habitat of Rhizobiaceae. The loss by Rhizobium sp. NT-26 of this large replicon housing rhizosphere-associated functions may be related to the drastic shift in environment the lineage has experienced. Indeed (discussed earlier), this loss coincides with the gain of genes enabling resistance to heavy metals, and arsenic and sulfur metabolisms, both traits with a potentially great adaptive value in sustaining life on an arsenopyrite (FeAsS)-containing rock.
Functional Approaches to Investigate Arsenic Metabolism and Resistance
Proteomic and Transcriptomic Profiling
Genomic tools have been used to study the bacterial response to arsenic mainly in arsenite-oxidizing Betaproteobacteria such as H. arsenicoxydans, a chemoorganotroph (Carapito et al. 2006; Muller et al. 2007; Weiss et al. 2009), and T. arsenitoxydans, a chemolithoautotroph (Bryan et al. 2009; Arsène-Ploetze et al. 2010). Arsenic metabolism was investigated in the chemolithoautotrophic Alphaprotebacterium Rhizobium sp. NT-26 by two complementary approaches: protein and RNA profiling using 2D gel electrophoresis and DNA microarrays, respectively. The comparisons of expression were done on proteins and RNA isolated from strain NT-26 grown heterotrophically with and without arsenite. The main results are summarized in table 1 and a complete list of the data is presented in supplementary table S6, Supplementary Material online.
Table 1.
Major Arsenic-Regulated Functional Categories Identified in Transcriptomics and Proteomics Experiments
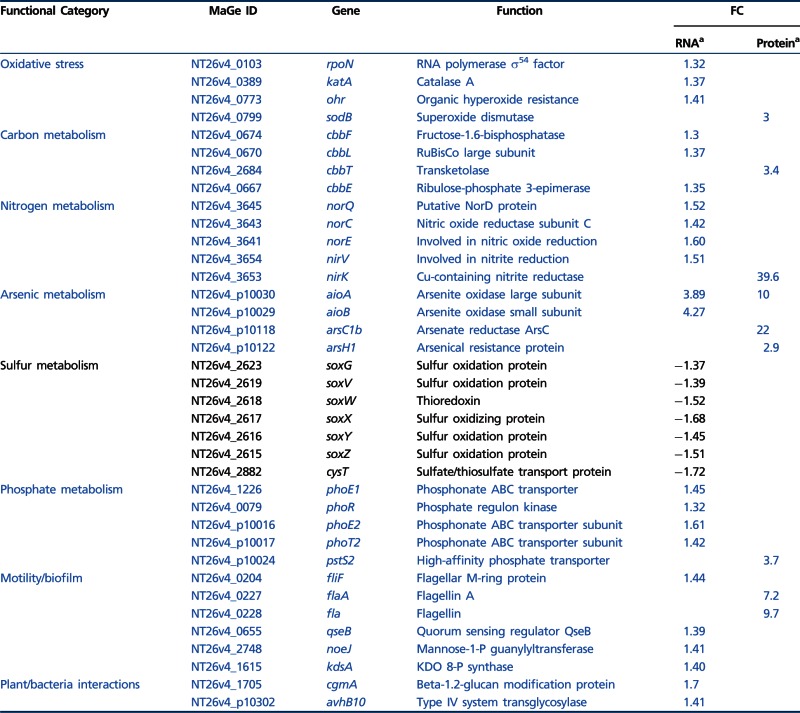 |
Note.—Induced and repressed functions are shown in blue and black, respectively. No value is indicated in the FC column if the gene is not statistically differentially expressed in transcriptomics or if the protein has not been identified in proteomics. Complete data are presented in supplementary table S6, Supplementary Material online.
aFold-change observed in transcriptomics and proteomics data, respectively.
The 2D gel proteomic profile of Rhizobium sp. NT-26 was quite similar to those previously obtained for H. arsenicoxydans (Carapito et al. 2006) and T. arsenitoxydans (Bryan et al. 2009), which are also neutrophilic bacteria. Sixty-three spots showed a significant difference in their accumulation pattern in strain NT-26 grown with and without As(III). Their analysis by mass spectrometry led to the identification of 141 proteins (supplementary table S6a, Supplementary Material online), including arsenite oxidase, which was identified for the first time on 2D gels. Like membrane proteins, such periplasmic proteins are often eliminated with cell debris before their solubilization during sample preparation. Proteins up- or downregulated with a fold-change ranging from +39.6 to −5.8 when Rhizobium sp. NT-26 was grown in the presence of As(III) had a molecular mass ranging from 15 to 109 kDa and a pI value from 4.2 to 7.9. Among them, 24% were involved in cell envelope and cellular processes, 12% in transport and binding proteins, 11% in information and regulation pathways, 42% in metabolism, 1% in transcription, and 10% were of unknown function.
The second approach used whole genome microarrays to perform a differential expression profiling experiment. Under As(III) stress, the transcript level of 199 genes, that is 4.5% of the whole genome, showed an increase of up to more than four times with a P value ≤0.05. At the same time, the expression of 416 genes, that is 9.5% of the whole genome, decreased by up to more than three times (supplementary table S6c, Supplementary Material online).
General Response to Arsenic Stress
Several proteins involved in arsenic resistance were shown to be accumulated on 2D gels when the organism was grown in the presence of As(III), for example, an ArsH1 NADPH-dependent FMN reductase and an ArsC1 arsenate reductase (table 1) with fold changes of 2.9 and 22, respectively. These two proteins are encoded by an ars operon located on the megaplasmid, which also contains genes coding for an ArsB efflux pump and an ArsR regulator. Moreover, a second operon located on the chromosome contains an arsA gene coding for an ATPase associated with an ArsB arsenite efflux pump. ArsA enables the strain to increase arsenic resistance by ATP-dependent extrusion of the metalloid out of the cell (Branco et al. 2008).
Microarray experiments showed that genes encoding the two arsenite oxidase subunits, that is, the small subunit, AioB, which contains the Rieske 2Fe-2S cluster and the catalytic subunit, AioA, which contains a molybdopterin guanine dinucleotide at the active site and a 3Fe–4S cluster (Santini and vanden Hoven 2004), were about 4-fold induced in the presence of arsenite (table 1). In contrast, the expression of two genes located downstream of the aioBA operon, that is, cytC encoding the periplasmic cytochrome c552, which can serve as an electron acceptor to the arsenite oxidase (Santini et al. 2007) and moeA1 encoding a molybdenum cofactor biosynthesis gene, was not significantly affected under arsenite stress (supplementary table S6c, Supplementary Material online). These observations are further supported by proteomic experiments showing that, among these proteins, AioA was found to be preferentially accumulated in the presence of As(III). All these results are consistent with previous data (Santini et al. 2007), which suggests that the expression of aioBA genes is induced by arsenite while genes located downstream are constitutively expressed even though they may have a role in arsenic metabolism.
Located upstream of aioBA are two regulatory genes, aioS and aioR, which encode a sensor histidine kinase and a response regulator, respectively (Sardiwal et al. 2010). Both proteins have been shown to be required for the transcriptional regulation of the aioBA genes (Koechler et al. 2010; Sardiwal et al. 2010). Moreover, it has been demonstrated that the expression of the aioBA genes requires the RpoN alternative sigma factor (σ54) in H. arsenicoxydans (Koechler et al. 2010). Similarly, a role for RpoN in arsenite oxidation has been recently highlighted in A. tumefaciens 5A (Kang et al. 2012). In this respect, a putative σ54-dependent promoter region has been detected upstream of the aioB gene in Rhizobium sp. NT-26 (Santini et al. 2007), suggesting that it is also involved in the expression of the aioBA operon in strain NT-26 (Sardiwal et al. 2010). This hypothesis is supported by our transcriptomic data, which revealed an induced expression of rpoN in Rhizobium sp. NT-26 when it was grown in the presence of As(III) (table 1), in contrast to the constitutive expression recently observed in A. tumefaciens 5A (Kang et al. 2012). Similarly, microarray and 2D-gel data showed a 2-fold increase in the expression of genes coding for general chaperones, that is, DnaK and GroEL, in the presence of arsenite (supplementary table S6a and c, Supplementary Material online), which is in agreement with the role played by proteins of the heat-shock family in As(III) oxidation in H. arsenicoxydans (Koechler et al. 2010).
Rhizobium sp. NT-26 also tolerates arsenate concentration greater than 0.5 M (Clarke A. and Santini J.M., unpublished data), suggesting the existence of an alternative mode of resistance. The first one is an Ars-type arsenic resistance system, components of which were found to be upregulated when the strain was grown with arsenite (discussed earlier) and the second is the presence of a specific phosphate transport system which is thought to limit arsenate entry into the cell (Weiss et al. 2009). Indeed, in Rhizobium sp. NT-26, the arsenic genomic island contains a pst operon in the vicinity of the aio operon. The pst operon encodes proteins implicated in the specific transport of phosphate into the cell to maintain a sufficient level of this ion despite the presence of arsenate, a structural analog of phosphate (Muller et al. 2007; Cleiss-Arnold et al. 2010). PstS2, a periplasmic protein involved in phosphate transport and encoded by this operon, had a 3.7-fold increase in expression when strain NT-26 was grown in the presence of As(III) (table 1). The pst operon is regulated by phoR, which encodes a membrane-associated protein kinase that phosphorylates PhoB in response to environmental signals. Indeed, microarray data showed that the expression of phoR was also upregulated in the presence of As(III) (table 1). Moreover, the PhoR protein may be involved in biofilm formation as phoB overexpression has been shown to increase biofilm formation in A. tumefaciens (Danhorn et al. 2004). This is supported by the presence in the vicinity of the pho chromosomal operon of a cluster of genes involved in EPS biosynthesis (NT26v4_1233–NT26v4_1263).
Arsenic is known to induce oxidative stress by generating free radicals (Bernstam and Nriagu 2000). An induction of genes involved in the resistance to such a stress has been previously observed in Pseudomonas aeruginosa and in H. arsenicoxydans under arsenite exposure (Parvatiyar et al. 2005; Weiss et al. 2009; Cleiss-Arnold et al. 2010). In Rhizobium sp. NT-26, an increase in katA mRNA, which encodes a catalase involved in the protection against oxidative stress by scavenging endogenously produced H2O2, was observed in microarray experiments (table 1). Similarly, the expression of ohr, which promotes bacterial resistance to hydroperoxide, was also up-regulated in Rhizobium NT-26 (table 1). Finally, results of the proteomic experiments showed a 3-fold increase in the SodB superoxide dismutase accumulation when strain NT-26 was grown in the presence of As(III). These observations further support the strong link, which exists in bacteria between arsenic response and protection against oxidative stress (Bertin et al. 2012).
Carbon, Nitrogen, and Energy Metabolism
Rhizobium sp. NT-26 is able to use various carbon or electron sources for growth. Indeed, multiple carbohydrates such as acetate, succinate, fumarate, lactate, glucose, fructose, xylose, and galactose are potential carbon sources for this bacterium (Santini et al. 2000). Alternatively, Rhizobium sp. NT-26 is able to grow chemolithoautotrophically in the presence of bicarbonate as a carbon source, oxygen as an electron acceptor and arsenite as an electron donor (Santini et al. 2000). Transcriptomics and proteomics experiments revealed that several genes and proteins involved in the fixation of CO2 via the Calvin cycle were upregulated with a fold change ranging from 1.3 to 3.4 when strain NT-26 was grown in the presence of As(III) (table 1). This is in agreement with the proteomics results obtained in T. arsenivorans, where an accumulation of the ribulose-1,5-biphosphate carboxylase/oxygenase large subunit and of the fructose-1,6-biphosphate has been observed when the organism was grown in the presence of As(III) (Bryan et al. 2009). Both strains may thus improve their capacity to fix CO2 when arsenite is present.
In addition, our microarrays data showed that the expression of nirV encoding a protein involved in nitrite reduction was induced. Moreover, 2D-gel data showed that NirK, which also participates in the reduction of nitrite to nitric oxide, was 39.6 times more accumulated when strain NT-26 was grown in the presence of arsenite. These experiments also showed an induction of several genes of the norEFCBQD nitric oxide reductase gene cluster when Rhizobium sp. NT-26 was grown in the presence of As(III) (table 1). These genes encode proteins that catalyze the reduction of nitric oxide to nitrous oxide, that is, norQ, norE and norC coding for a protein involved in nitric oxide reduction, a nitric oxide reductase activating protein, and the small subunit of the nitric oxide reductase, respectively. This suggests that the chemolithoautotrophic bacterium Rhizobium sp. NT-26 may fix CO2 and couple nitrite reduction with As(III) oxidation. However, as no growth was observed with arsenite on either nitrate or nitrite, arsenite oxidation using nitrite as electron acceptor in autotrophic conditions seems unable to support sufficient energy (ATP) generation to sustain growth.
Rhizobium sp. NT-26 has been shown to grow with hydrogen sulfide, elemental sulfur, and thiosulfate (Santini J.M., unpublished data). In agreement with these observations, a sox cluster implicated in the oxidation of thiosulfate is present in the Rhizobium sp. NT-26 genome. Nevertheless, many genes involved in sulfur metabolism, that is, soxGVWXYZ and cysT were downregulated by up to 2-fold when the strain was grown in the presence of arsenite (table 1). Our results therefore suggest that, even though Rhizobium sp. NT-26 may be able to grow by using sulfur as an electron donor, the strain represses sulfur oxidation when grown in the presence of As(III). One hypothesis may be that, in such a case, the strain expresses a repressor of the sox genes. Their products serve for the oxidation of thiosulfate to sulfate and the reaction intermediates, that is, sulfite, sulfide, and hydrogen sulfide, have been shown to inhibit arsenite oxidase activity (Lieutaud et al. 2010).
Physiological and Genetic Approaches: Flagellar Motility and Biofilm Formation
Flagellum Cascade Features
Rhizobium sp. NT-26 is motile by the means of two subterminal flagella (Santini et al. 2000). Genes involved in their biosynthesis are organized in a large chromosomal cluster of 55 genes showing a perfect synteny with those of S. meliloti. In this flagellar regulon, visN and visR form part of the master operon and encode the proteins forming the VisNR heterodimer that acts as a global transcriptional regulator. This master regulator activates the expression of genes located in the cascade that encode flagella, motor, and chemotaxis proteins (Sourjik et al. 2000). In Rhizobium sp. NT-26, microarray data showed that the expression of fliF, coding for the flagellum M-ring protein, was induced when the organism was grown in the presence of As(III) (table 1). Furthermore, proteomic data showed a 9.7- and 7.2-fold-increase in the accumulation of flagellin proteins Fla and FlaA, respectively (table 1). The expression of qseB was also induced when Rhizobium sp. NT-26 was grown in the presence of As(III). QseB has been shown to participate in the flagellum and motility bacterial regulatory network via a quorum-sensing mechanism. Indeed, in E. coli, qseBC expression enhances the transcription of flagellar genes in response to the autoinducer by a direct binding of QseB to the flhDC master operon promoter (Clarke and Sperandio 2005). Finally, microarray data showed the induction of genes possibly involved in the synthesis of an exopolysaccharide matrix in Rhizobium sp. NT-26, that is, noeJ, coding for a mannose-1-phosphate guanylyltransferase and kdsA, coding for a 2-dehydro-3-deoxyphosphooctonate aldolase. These observations suggest that As(III) has an impact on flagellum synthesis, that is to say motility, and biofilm formation in Rhizobium sp. NT-26, as observed in H. arsenicoxydans (Muller et al. 2007; Marchal et al. 2010). To test this hypothesis, swarming assays were performed on 0.3% agar plates. The presence of As(III) was shown to increase the swarming ring by up to 2-fold in the presence of 8 mM As(III) (fig. 4A). Remarkably, cell observation under a TEM revealed that flagellum biosynthesis occurred immediately in the presence of 8 mM As(III) while more than two days were needed to observe flagella in the absence of arsenite (fig. 4B and C), providing evidence that arsenite promotes motility in Rhizobium sp. NT-26. In addition, a two-fold reduction in biofilm formation was observed in the first 24 h of growth in the presence of As(III) (fig. 4D), which suggests a preferential development as motile planktonic cells rather than as unflagellated sessile cells as in H. arsenicoxydans (Marchal et al. 2010).
Fig. 4.—

Motility phenotype of Rhizobium sp. NT-26 grown at different concentration of arsenite. (A) Swarming diameter measured after 48 h in MSM containing 0.04% yeast extract and supplemented by different concentrations of As(III). Results are the mean values of three independent experiments. (B) and (C) TEM observations of Rhizobium sp. NT-26 at 24 h of culture, without or with 8 mM As(III), respectively. The scale bar corresponds to 500 nm and the pictures are representative of 10 pictures. (D) Biofilm formation by strain NT-26, without or with 8 mM As(III) visualized by the crystal violet method. Results are the mean values of 24 replicates.
Random Mutagenesis
The Rhizobium sp. NT-26 genome organization suggests that motility and arsenite oxidation depend on genes located on its chromosome and on its megaplasmid, respectively. With the aim to analyze the possible link between these physiological processes, a mutant library was constructed by random transposon mutagenesis (Tang et al. 1999). The motility of 6,000 kanamycin-resistant transposition derivatives was tested on semisolid medium, which led to the isolation of 22 motility-deficient mutants. The mutations that resulted in a loss of motility were identified by sequencing the mini-Tn5 transposon insertion sites. Fourteen mutations were shown to directly disrupt motility genes (table 2), and the proteins encoded by these genes are either structural or regulatory components of the flagellum cascade, that is, 5 Flg proteins (FlgE, FlgF, FlgG, FlgI, and FlgL), 5 Fli proteins (FliF, FliK, Flip, and FliR), 1 Flh protein (FlhA), and 1 Vis protein (VisR). No flagellin-defective mutant was obtained, which may be explained by the presence of four different flagellin-encoding genes on the chromosome.
Table 2.
Rhizobium sp. NT-26 Mutants Isolated on the Basis of a Loss of Motility or Arsenite Oxidation
| Mutant | MaGe IDa | Gene | Function | Gene Locationb | Insertionc |
|---|---|---|---|---|---|
| Motility | |||||
| 2B5 | NT26v4_0222 | flgI | Flagellar P-ring protein precursor | 221267–222391 | 258I-O |
| 2D6 | NT26v4_0245 | flhA | Flagellar biosynthesis protein | 243262–245349 | 38I-O |
| 4H7 | NT26v4_0204 | fliF | Flagellar M-ring protein | 206351–208027 | 477I-O |
| 5B7 | NT26v4_0226 | fliP | Flagellar biosynthetic protein | 224272–225009 | 109O-I |
| 6E4 | NT26v4_0220 | flgG | Flagellar basal-body rod protein | 219981–220769 | 182I-O |
| 10A11 | NT26v4_0248 | Putative FlgJ-like protein | 246658–247212 | ||
| 10G2 | NT26v4_2965 | Conserved hypothetical protein | 2880029–2881285 | 44O-I | |
| 16B6 | NT26v4_0245 | flhA | Flagellar biosynthesis protein | 243262–245349 | 55I-O |
| 16B7 | NT26v4_0240 | flgL | Flagellar hook-associated protein | 240428–241549 | 11I-O |
| 18D11 | NT26v4_0238 | flgE | Flagellar hook protein | 237398–238930 | 363I-O |
| 20E7 | NT26v4_0246 | fliR | Flagellar biosynthetic protein | 245378–246130 | 110O-I |
| 23E5 | NT26v4_0214 | flgF | Flagellar basal-body rod protein | 216054–216788 | 44O-I |
| 29G6 | NT26v4_0247 | Putative FliR/FliJ-like chaperone | 246137–246553 | 91I-O | |
| 35A8 | NT26v4_2314 | Putative two-component sensor histidine kinase | 2263060–2264472 | 113O-I | |
| 37C12 | NT26v4_2314 | Putative two-component sensor histidine kinase | 2263060–2264472 | 48O-I | |
| 37H1 | NT26v4_2267 | Putative ATP-dependent hydrolase protein | 2217585–2219546 | 337O-I | |
| 38B4 | NT26v4_0206 | visR | Master transcriptional regulator of flagellar regulon | 209070–209798 | 53O-I |
| 38G3 | NT26v4_0204 | fliF | Flagellar M-ring protein | 206351–208027 | 235I-O |
| 39G12 | NT26v4_2671 | Conserved protein of unknown function | 2586119–2587117 | 94O-I | |
| 40E5 | NT26v4_0234 | fliK | Flagellar hook-length regulator | 234324–235835 | 83I-O |
| 50E11 | NT26v4_0250 | Conserved integral membrane protein of unknown function | 247603–248142 | 92O-I | |
| 61C2 | NT26v4_3970 | Conserved exported protein of unknown function | 3918374–3918853 | 196I-O | |
| Arsenite oxidation | |||||
| 8G1 | NT26v4_p10026 | aioX | Putative periplasmic phosphite-binding-like protein precursor; PtxB-like protein | 23892–24806 | 275I-O |
| 11B3 | NT26v4_4048 | moeB | Putative molybdopterin biosynthesis protein MoeB | 3998958–3999725 | 245O-I |
| 24B7 | NT26v4_p10028 | aioR | Two-component response regulator | 26262–27584 | 65I-O |
| 37C3 | NT26v4_p10026 | aioX | Putative periplasmic phosphite-binding-like protein precursor; PtxB-like protein | 23892–24806 | 191I-O |
| 55H7 | NT26v4_p10029 | aioB | Arsenite oxidase small subunit | 27721–28248 | |
| 60E6 | NT26v4_p10030 | aioA | Arsenite oxidase large subunit | 28261–30798 | 818I-O |
aIdentification number of the gene in the MaGe interface.
bPosition of the corresponding gene on the chromosome or the plasmid.
cPosition of the codon immediately upstream of the transposon insertion site. Subscripts indicate the orientation of the insertion.
Similarly, six mutations resulting in a lack of arsenite oxidation as compared with the wild-type and motility mutants were obtained after screening 6,000 kanamycin-resistant clones with the silver nitrate method (Muller et al. 2007) (table 2). First, two mutations were identified in the arsenite oxidase genes, that is, aioA and aioB. One mutation was also shown to affect aioR, which encodes the regulatory protein of the AioRS two-component system. No oxidation of As(III) to As(V) was detected by HPLC-ICP-AES (Muller et al. 2007) in this mutant, as compared with complete As(III) oxidation determined in the motility mutant deficient in the flagellum master regulator VisR. A fourth mutation was located in the moeB gene involved in the synthesis of the molybdopterin cofactor required for arsenite oxidase activity. Finally, the inactivation of the aioX gene, which is located upstream of aioSR, also resulted in a loss of arsenite oxidation in Rhizobium sp. NT-26. In A. tumefaciens 5A, the periplasmic AioX has been recently shown to be involved in the regulation of As(III) oxidation (Liu et al. 2012).
To determine the link between As(III) oxidation and colonization properties in Rhizobium sp. NT-26, the ability of various mutants to move and to form a biofilm was evaluated in the presence of arsenite (fig. 5). Mutations in flagellar genes resulted in a loss of motility and in a decrease in biofilm formation. Indeed, all the mutants we tested were nonmotile (fig. 5A) and lost between 12% and 45% of their ability to form a biofilm when compared with the wild-type strain (fig. 5B). This observation demonstrates that, although Rhizobium sp. NT-26 has a preferential motile life style in the presence of arsenite, flagella have a role as adhesive appendages in the first steps of biofilm formation, which has been shown previously in other studies (Kirov et al. 2004; Nejidat et al. 2008). This result is in agreement with those obtained with H. arsenicoxydans, where mutations resulting in nonfunctional flagella led to a more rapid adhesion as compared with the wild-type. Finally, aioA and aioR mutants were less motile and formed 30% and 45%, respectively, less biofilm than the wild-type (fig. 5), further supporting the role of motility and flagella in biofilm formation.
Fig. 5.—

Percentage of motility and biofilm formation in various mutants as compared with Rhizobium sp. NT-26 wild-type strain. The left and the right panels show the results obtained in mutants affected in motility and As(III) oxidation, respectively. (A) % of swarming motility measured after 24 h. Results are the mean values calculated from the % of three independent experiments. In each experiment, mutant and wild-type strains were tested in triplicates. (B) % of biofilm formation visualized by crystal violet coloration. Results are the % calculated from the mean values of six replicates for each strain.
Regulation of Flagella Synthesis by AioR
The “omics” data showed that flagellar proteins and genes were upregulated when strain NT-26 was grown in the presence of arsenite (table 1). Remarkably, both aioA and aioR mutations resulted in a moderate reduction in motility (fig. 5A). This can be explained by the reduction in energy available to the cells as they are unable to metabolize arsenite (Santini et al. 2000). In addition, TEM observations of the aioR mutant, affected in the two-component signal transduction system, revealed the presence of flagella in the early log phase of growth even in the absence of As(III), which suggests that AioR may be involved in the repression of motility when no arsenite is present (fig. 6). AioR may thus interact, directly or indirectly, with components of the flagellar cascade.
Fig. 6.—

TEM observations of the aioR mutant. The aioR mutant was cultivated 24 h (A) in the absence of As(III) and (B) in the presence of 8 mM As(III). Pictures are representative of 10 photographs. The scale bar corresponds to 500 nm.
To identify possible AioR-binding sites in the Rhizobium sp. NT-26 genome, multiple sequence alignments of all aioBA regulatory sequences available in databases were performed with fuzznuc (Rice et al. 2000). This enabled us to suggest the possible existence of two AioR putative binding sites upstream of the aioBA transcriptional start site, that is, GT[CT]CGN(6)CG[GA]AC in the Rhizobiales strains and GTTNCN(6)GNAAC in the Burkholderiales strains. In contrast, strains lacking the two-component system aioSR operon did not harbor any of these putative AioR-binding sites, which further supports a role for these motifs in the regulation of aioBA operon expression by AioR. Although the GT[CT]CGN(6)CG[GA]AC putative binding site was found at 49 locations on the Rhizobium sp. NT-26 chromosome, it is only in the upstream region of the aioBA operon that this motif was associated with the -12/-24 σ54-dependent promoter sequence needed for the RpoN-dependent transcription initiation of arsenite oxidase genes (Koechler et al. 2010). Moreover, no clear connection could be observed between those putative binding sites and motility-related genes. Nevertheless, a search of the whole genome of strain NT-26 with a relaxed version of the pattern allowing any nucleotide at its degenerated positions, GTNCGN(6)CGNAC, yielded 39 more hits than with the canonical pattern. This low number of new hits suggests that the presence of this signature is not due to chance and this sequence may therefore have a potential regulatory role. None of the new hits was associated with a RpoN motif site although one hit was found within the coding sequence of the flagellar master regulatory gene visN. Therefore, although we cannot rule out an indirect effect of AioR by controlling another regulatory protein, we can hypothesize that the binding to this mildly degenerate motif of unphosphorylated AioR in the absence of arsenic would result in a visN repression and a delayed motility. Such a transcriptional repression via binding of the coding sequence of target genes has already been observed for other regulatory proteins and two-component system regulators, for example, OxyR (Zheng et al. 2001) and PrrA (Eraso et al. 2008). Taken together, our results demonstrate the importance of arsenite oxidation in the behavioral response of Rhizobium sp. NT-26, suggesting that arsenic metabolism enhances the ability of the organism to explore and colonize its environment.
Conclusion
This study extends our knowledge of the physiological response to arsenic in arsenite-oxidizing bacteria. Our results provide for the first time a reference set of genomic, transcriptomic, and proteomic data of an Alphaproteobacterium isolated from an arsenopyrite-containing goldmine, which allowed us to propose a model for the Rhizobium sp. NT-26 response to arsenite exposure (fig. 7). Although phylogenetically related to the plant-associated bacteria, strain sp. NT-26 has lost the major colonizing capabilities needed for symbiosis. Instead, this bacterium has acquired on a megaplasmid the various genes which allow it to metabolize arsenate. Remarkably, a link between flagellar motility/biofilm formation and arsenite oxidation was observed although the genes required for these physiological activities are carried by different genetic determinants, that is, the chromosome and the megaplasmid, respectively. This suggests the existence of a mechanism, probably indirect and which remains to be characterized at a molecular level, of a coordinate regulation of these two important biological processes. This underlines the importance of arsenite oxidation in the colonization of arsenic-rich ecosystems, a toxic element widespread on Earth. Importantly, our data also illustrate the major contribution of environmental pressure on the evolution of bacterial genomes, which results in a gain and loss of multiple functions, improving the fitness of the strains to extreme ecological niches.
Fig. 7.—

Conceptual representation of the Rhizobium sp. NT-26 response to arsenite exposure. This representation takes into account our genomic, transcriptomic, proteomic, and physiological results as well as data from the literature. Numbers 1 to 9 represent biological functions and the approximate genomic location of the gene clusters encoding their corresponding proteins. 1 and 9: arsenate reduction; 2: phosphate and arsenate transport; 3 and 4: arsenite and sulfur oxidation, respectively; 5: carbon fixation; 6 and 7: nitrate and nitrite reduction, respectively; 8: motility. Block, dashed, dotted, and standard arrows symbolize chemical reactions, electron flow, transport/utilization of molecules and signaling/regulatory pathways, respectively. When highlighted in dark blue, light blue, yellow or gray, elements have been identified as being induced by proteomic, induced by transcriptomic, repressed by transcriptomic, and present in the genome, respectively. For clarity reasons, proteins for which the exact function is still unknown but that are related to the different processes are not shown, the protein complexes of the respiratory chain, that is the NADH dehydrogenase, the fumarate reductase, the cytochrome bc1 and the cytochrome c oxidase are designated by Nuo, Sdh, Cyt bc1, and Cox, respectively, plasmid p2 is not shown and only one flagella is represented. Finally, Cyt c and c552 are for cytochrome c and cytochrome c552, respectively, and NT26v4_4001 is a homolog of qseC.
Supplementary Material
Supplementary figures 1–4, tables S1–S6, and methods S1–S4 are available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org/).
Acknowledgments
This work was supported by the Université de Strasbourg (UdS), the Consortium National de Recherche en Génomique (CNRG), the Centre National de la Recherche Scientifique (CNRS), the Australian Research Council (DP034305), the French Ministère de l’Enseignement Supérieur et de la Recherche to J.A. and J.C.-A., the Direction Générale de l’Armement to L.G., a studentship from the Agence Nationale de la Recherche (ANR) COBIAS project (PRECODD 2007) to M.M., and the Ecole Normale Supérieure (ENS) of Lyon to F.L. The authors thank Mathieu Erhardt for the TEM observations and Ashleigh Clarke for her work on arsenic resistance in strain NT-26. This work was done in part in the frame of the Groupement de Recherche—Métabolisme de l’Arsenic chez les Microorganismes (GDR2909-CNRS) (http://gdr2909.alsace.cnrs.fr/, last accessed April 30, 2013).
Literature Cited
- Altschul SF, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arsène-Ploetze F, et al. Structure, function, and evolution of the Thiomonas spp. genome. PLoS Genet. 2010;6:e1000859. doi: 10.1371/journal.pgen.1000859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aury J-M, et al. High quality draft sequences for prokaryotic genomes using a mix of new sequencing technologies. BMC Genomics. 2008;9:603. doi: 10.1186/1471-2164-9-603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstam L, Nriagu J. Molecular aspects of arsenic stress. J Toxicol Environ Health B Crit Rev. 2000;3:293–322. doi: 10.1080/109374000436355. [DOI] [PubMed] [Google Scholar]
- Bertin PN, et al. Metabolic diversity among main microorganisms inside an arsenic-rich ecosystem revealed by meta- and proteo-genomics. ISME J. 2011;5:1735–1747. doi: 10.1038/ismej.2011.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertin PN, et al. Microbial arsenic response and metabolism in the genomics era. In: Santini JM, Ward SA, editors. The metabolism of arsenite. London: CRC Press; 2012. pp. 99–114. [Google Scholar]
- Bertin PN, Médigue C, Normand P. Advances in environmental genomics: towards an integrated view of micro-organisms and ecosystems. Microbiology. 2008;154:347–359. doi: 10.1099/mic.0.2007/011791-0. [DOI] [PubMed] [Google Scholar]
- Bigot T, Daubin V, Lassalle F, Perrière G. TPMS: a set of utilities for querying collections of gene trees. BMC Bioinformatics. 2012;14:109. doi: 10.1186/1471-2105-14-109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borch T, et al. Biogeochemical redox processes and their impact on contaminant dynamics. Environ Sci Technol. 2010;44:15–23. doi: 10.1021/es9026248. [DOI] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Branco R, Chung A-P, Morais PV. Sequencing and expression of two arsenic resistance operons with different functions in the highly arsenic-resistant strain Ochrobactrum tritici SCII24T. BMC Microbiol. 2008;8:95. doi: 10.1186/1471-2180-8-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryan CG, et al. Carbon and arsenic metabolism in Thiomonas strains: differences revealed diverse adaptation processes. BMC Microbiol. 2009;9:127. doi: 10.1186/1471-2180-9-127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carapito C, et al. Identification of genes and proteins involved in the pleiotropic response to arsenic stress in Caenibacter arsenoxydans, a metalloresistant beta-proteobacterium with an unsequenced genome. Biochimie. 2006;88:595–606. doi: 10.1016/j.biochi.2005.11.004. [DOI] [PubMed] [Google Scholar]
- Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 2000;17:540–552. doi: 10.1093/oxfordjournals.molbev.a026334. [DOI] [PubMed] [Google Scholar]
- Clarke MB, Sperandio V. Transcriptional regulation of flhDC by QseBC and sigma (FliA) in enterohaemorrhagic Escherichia coli. Mol Microbiol. 2005;57:1734–1749. doi: 10.1111/j.1365-2958.2005.04792.x. [DOI] [PubMed] [Google Scholar]
- Cleiss-Arnold J, et al. Temporal transcriptomic response during arsenic stress in Herminiimonas arsenicoxydans. BMC Genomics. 2010;11:709. doi: 10.1186/1471-2164-11-709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creus CM, et al. Nitric oxide is involved in the Azospirillum brasilense-induced lateral root formation in tomato. Planta. 2005;221:297–303. doi: 10.1007/s00425-005-1523-7. [DOI] [PubMed] [Google Scholar]
- Criscuolo A, Gribaldo S. BMGE (block mapping and gathering with entropy): a new software for selection of phylogenetic informative regions from multiple sequence alignments. BMC Evol Biol. 2010;10:210. doi: 10.1186/1471-2148-10-210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danhorn T, Hentzer M, Givskov M, Parsek MR, Fuqua C. Phosphorus limitation enhances biofilm formation of the plant pathogen Agrobacterium tumefaciens through the PhoR-PhoB regulatory system. J Bacteriol. 2004;186:4492–4501. doi: 10.1128/JB.186.14.4492-4501.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daubin V, Lerat E, Perrière G. The source of laterally transferred genes in bacterial genomes. Genome Biol. 2003;4:R57. doi: 10.1186/gb-2003-4-9-r57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dénarié J, Debellé F, Promé JC. Rhizobium lipo-chitooligosaccharide nodulation factors: signaling molecules mediating recognition and morphogenesis. Annu Rev Biochem. 1996;65:503–535. doi: 10.1146/annurev.bi.65.070196.002443. [DOI] [PubMed] [Google Scholar]
- Didelot X, Lawson D, Darling A, Falush D. Inference of homologous recombination in bacteria using whole-genome sequences. Genetics. 2010;186:1435–1449. doi: 10.1534/genetics.110.120121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobbelaere S, Okon Y. The plant growth-promoting effect and plant responses. In: Elmerich C, Newton WE, editors. Associative and endophytic nitrogen-fixing bacteria and cyanobacterial associations. Nitrogen fixation: origins, applications, and research progress V. Dordrecht (The Netherlands): Springer; 2007. pp. 145–170. [Google Scholar]
- Edgar RC. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics. 2004;5:113. doi: 10.1186/1471-2105-5-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eraso JM, et al. Role of the global transcriptional regulator PrrA in Rhodobacter sphaeroides 2.4.1: combined transcriptome and proteome analysis. J Bacteriol. 2008;190:4831–4848. doi: 10.1128/JB.00301-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadd GM. Metals, minerals and microbes: geomicrobiology and bioremediation. Microbiology. 2010;156:609–643. doi: 10.1099/mic.0.037143-0. [DOI] [PubMed] [Google Scholar]
- Gallie DR, Kado CI. Minimal region necessary for autonomous replication of pTAR. J Bacteriol. 1988;170:3170–3176. doi: 10.1128/jb.170.7.3170-3176.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giraud E, et al. Legumes symbioses: absence of Nod genes in photosynthetic bradyrhizobia. Science. 2007;316:1307–1312. doi: 10.1126/science.1139548. [DOI] [PubMed] [Google Scholar]
- Gonzalez V, et al. The partitioned Rhizobium etli genome: genetic and metabolic redundancy in seven interacting replicons. Proc Natl Acad Sci U S A. 2006;103:3834–3839. doi: 10.1073/pnas.0508502103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouy M, Guindon S, Gascuel O. SeaView version 4: a multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol Biol Evol. 2010;27:221–224. doi: 10.1093/molbev/msp259. [DOI] [PubMed] [Google Scholar]
- Gremaud MF, Harper JE. Selection and initial characterization of partially nitrate tolerant nodulation mutants of soybean. Plant Physiol. 1989;89:169–173. doi: 10.1104/pp.89.1.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guibaud G, van Hullebusch E, Bordas F. Lead and cadmium biosorption by extracellular polymeric substances (EPS) extracted from activated sludges: pH-sorption edge tests and mathematical equilibrium modelling. Chemosphere. 2006;64:1955–1962. doi: 10.1016/j.chemosphere.2006.01.012. [DOI] [PubMed] [Google Scholar]
- Guindon S, Delsuc F, Dufayard J-F, Gascuel O. Estimating maximum likelihood phylogenies with PhyML. Methods Mol Biol. 2009;537:113–137. doi: 10.1007/978-1-59745-251-9_6. [DOI] [PubMed] [Google Scholar]
- Guindon S, Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003;52:696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]
- Hall-Stoodley L, Costerton JW, Stoodley P. Bacterial biofilms: from the natural environment to infectious diseases. Nat Rev Microbiol. 2004;2:95–108. doi: 10.1038/nrmicro821. [DOI] [PubMed] [Google Scholar]
- Hao Y, Charles TC, Glick BR. ACC deaminase activity in avirulent Agrobacterium tumefaciens D3. Can J Microbiol. 2011;57:278–286. doi: 10.1139/w11-006. [DOI] [PubMed] [Google Scholar]
- Harrison PW, Lower RPJ, Kim NKD, Young JPW. Introducing the bacterial ‘chromid’: not a chromosome, not a plasmid. Trends Microbiol. 2010;18:141–148. doi: 10.1016/j.tim.2009.12.010. [DOI] [PubMed] [Google Scholar]
- Heinrich-Salmeron A, et al. Unsuspected diversity of arsenite-oxidizing bacteria as revealed by widespread distribution of the aoxB gene in prokaryotes. Appl Environ Microbiol. 2011;77:4685–4692. doi: 10.1128/AEM.02884-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes A, et al. Comparison of two multimetal resistant bacterial strains: Enterobacter sp. YSU and Stenotrophomonas maltophilia ORO2. Curr Microbiol. 2009;59:526–531. doi: 10.1007/s00284-009-9471-2. [DOI] [PubMed] [Google Scholar]
- Hommais F, et al. Effect of mild acid pH on the functioning of bacterial membranes in Vibrio cholerae. Proteomics. 2002;2:571–579. doi: 10.1002/1615-9861(200205)2:5<571::AID-PROT571>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Hynes MF, McGregor NF. Two plasmids other than the nodulation plasmid are necessary for formation of nitrogen-fixing nodules by Rhizobium leguminosarum. Mol Microbiol. 1990;4:567–574. doi: 10.1111/j.1365-2958.1990.tb00625.x. [DOI] [PubMed] [Google Scholar]
- Janssen PJ, et al. The complete genome sequence of Cupriavidus metallidurans strain CH34, a master survivalist in harsh and anthropogenic environments. PLoS One. 2010;5:e10433. doi: 10.1371/journal.pone.0010433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juhas M, et al. Genomic islands: tools of bacterial horizontal gene transfer and evolution. FEMS Microbiol Rev. 2009;33:376–393. doi: 10.1111/j.1574-6976.2008.00136.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang Y-S, Bothner B, Rensing C, McDermott TR. Involvement of RpoN in regulating bacterial arsenite oxidation. Appl Environ Microbiol. 2012;78:5638–5645. doi: 10.1128/AEM.00238-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Toh H. Recent developments in the MAFFT multiple sequence alignment program. Brief Bioinform. 2008;9:286–298. doi: 10.1093/bib/bbn013. [DOI] [PubMed] [Google Scholar]
- Kirov SM, Castrisios M, Shaw JG. Aeromonas flagella (polar and lateral) are enterocyte adhesins that contribute to biofilm formation on surfaces. Infect Immun. 2004;72:1939–1945. doi: 10.1128/IAI.72.4.1939-1945.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koechler S, et al. Multiple controls affect arsenite oxidase gene expression in Herminiimonas arsenicoxydans. BMC Microbiol. 2010;10:53. doi: 10.1186/1471-2180-10-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lassalle F, et al. Genomic species are ecological species as revealed by comparative genomics in Agrobacterium tumefaciens. Genome Biol Evol. 2011;3:762–781. doi: 10.1093/gbe/evr070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le SQ, Gascuel O. An improved general amino acid replacement matrix. Mol Biol Evol. 2008;25:1307–1320. doi: 10.1093/molbev/msn067. [DOI] [PubMed] [Google Scholar]
- Lett M-C, Paknikar KM, Lièvremont D. A simple and rapid method for arsenite and arsenate speciation. In: Ciminelli VST, Garcia O Jr, editors. Biohydrometallurgy: fundamentals, technology and sustainable development, Part B. New York: Elsevier Science; 2001. pp. 541–546. [Google Scholar]
- Lieutaud A, et al. Arsenite oxidase from Ralstonia sp. 22. J Biol Chem. 2010;285:20433–20441. doi: 10.1074/jbc.M110.113761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G, et al. A periplasmic arsenite-binding protein involved in regulating arsenite oxidation. Environ Microbiol. 2012;14:1624–1634. doi: 10.1111/j.1462-2920.2011.02672.x. [DOI] [PubMed] [Google Scholar]
- López-Guerrero MG, et al. Rhizobial extrachromosomal replicon variability, stability and expression in natural niches. Plasmid. 2012;68:149–158. doi: 10.1016/j.plasmid.2012.07.002. [DOI] [PubMed] [Google Scholar]
- Marchal M, Briandet R, Koechler S, Kammerer B, Bertin PN. Effect of arsenite on swimming motility delays surface colonization in Herminiimonas arsenicoxydans. Microbiology. 2010;156:2336–2342. doi: 10.1099/mic.0.039313-0. [DOI] [PubMed] [Google Scholar]
- Marchal M, et al. Subinhibitory arsenite concentrations lead to population dispersal in Thiomonas sp. PLoS One. 2011;6:e23181. doi: 10.1371/journal.pone.0023181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marouga R, David S, Hawkins E. The development of the DIGE system: 2D fluorescence difference gel analysis technology. Anal Bioanal Chem. 2005;382:669–678. doi: 10.1007/s00216-005-3126-3. [DOI] [PubMed] [Google Scholar]
- Martens M, et al. Multilocus sequence analysis of Ensifer and related taxa. Int J Syst Evol Microbiol. 2007;57:489–503. doi: 10.1099/ijs.0.64344-0. [DOI] [PubMed] [Google Scholar]
- McDougald D, Rice SA, Barraud N, Steinberg PD, Kjelleberg S. Should we stay or should we go: mechanisms and ecological consequences for biofilm dispersal. Nat Rev Microbiol. 2012;10:39–50. doi: 10.1038/nrmicro2695. [DOI] [PubMed] [Google Scholar]
- Muller D, et al. A tale of two oxidation states: bacterial colonization of arsenic-rich environments. PLoS Genet. 2007;3:e53. doi: 10.1371/journal.pgen.0030053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller D, Lièvremont D, Simeonova DD, Hubert J-C, Lett M-C. Arsenite oxidase aox genes from a metal-resistant β-proteobacterium. J Bacteriol. 2003;185:135–141. doi: 10.1128/JB.185.1.135-141.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nejidat A, Saadi I, Ronen Z. Effect of flagella expression on adhesion of Achromobacter piechaudii to chalk surfaces. J Appl Microbiol. 2008;105:2009–2014. doi: 10.1111/j.1365-2672.2008.03930.x. [DOI] [PubMed] [Google Scholar]
- Osborne TH, Santini JM. Prokaryotic aerobic oxidation of arsenite. In: Santini JM, Ward SA, editors. The metabolism of arsenite. London: CRC Press; 2012. pp. 61–72. [Google Scholar]
- Parvatiyar K, et al. Global analysis of cellular factors and responses involved in Pseudomonas aeruginosa resistance to arsenite. J Bacteriol. 2005;187:4853–4864. doi: 10.1128/JB.187.14.4853-4864.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penel S, et al. Databases of homologous gene families for comparative genomics. BMC Bioinformatics. 2009;10(6 Suppl):S3. doi: 10.1186/1471-2105-10-S6-S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez-Bahena MH, Nesme X, Muller D. Rapid and simultaneous detection of linear chromosome and large plasmids in proteobacteria. J Basic Microbiol. 2012;52:736–739. doi: 10.1002/jobm.201100278. [DOI] [PubMed] [Google Scholar]
- Rice P, Longden I, Bleasby A. EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet. 2000;16:276–277. doi: 10.1016/s0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- Richardson AE, Barea J-M, McNeill AM, Prigent-Combaret C. Acquisition of phosphorus and nitrogen in the rhizosphere and plant growth promotion by microorganisms. Plant Soil. 2009;321:305–339. [Google Scholar]
- Rigaud J, Puppo A. Indole-3-acetic acid catabolism by soybean bacteroids. J Gen Microbiol. 1975;88:223–228. [Google Scholar]
- Ronquist F, et al. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 2012;61:539–542. doi: 10.1093/sysbio/sys029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santini JM, et al. The NT-26 cytochrome c552 and its role in arsenite oxidation. Biochim Biophys Acta. 2007;1767:189–196. doi: 10.1016/j.bbabio.2007.01.009. [DOI] [PubMed] [Google Scholar]
- Santini JM, Sly LI, Schnagl RD, Macy JM. A new chemolithoautotrophic arsenite-oxidizing bacterium isolated from a gold mine: phylogenetic, physiological, and preliminary biochemical studies. Appl Environ Microbiol. 2000;66:92–97. doi: 10.1128/aem.66.1.92-97.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santini JM, vanden Hoven RN. Molybdenum-containing arsenite oxidase of the chemolithoautotrophic arsenite oxidizer NT-26. J Bacteriol. 2004;186:1614–1619. doi: 10.1128/JB.186.6.1614-1619.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sardiwal S, Santini JM, Osborne TH, Djordjevic S. Characterization of a two-component signal transduction system that controls arsenite oxidation in the chemolithoautotroph NT-26. FEMS Microbiol Lett. 2010;313:20–28. doi: 10.1111/j.1574-6968.2010.02121.x. [DOI] [PubMed] [Google Scholar]
- Slater SC, et al. Genome sequences of three Agrobacterium biovars help elucidate the evolution of multichromosome genomes in bacteria. J Bacteriol. 2009;191:2501–2511. doi: 10.1128/JB.01779-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sourjik V, Muschler P, Scharf B, Schmitt R. VisN and VisR are global regulators of chemotaxis, flagellar, and motility genes in Sinorhizobium (Rhizobium) meliloti. J Bacteriol. 2000;182:782–788. doi: 10.1128/jb.182.3.782-788.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spaepen S, Vanderleyden J, Okon Y. Chapter 7: Plant growth-promoting actions of rhizobacteria. In: Van Loon LC, editor. Advances in botanical research. Vol. 51. Burlington: Academic Press; 2009. pp. 283–320. [Google Scholar]
- Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22:2688–2690. doi: 10.1093/bioinformatics/btl446. [DOI] [PubMed] [Google Scholar]
- Stolz JF. Microbial metal and metalloid metabolism: advances and applications. 1st ed. Washington DC: ASM Press; 2011. [Google Scholar]
- Suyama M, Torrents D, Bork P. PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 2006;34:W609–W612. doi: 10.1093/nar/gkl315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang X, Lu BF, Pan SQ. A bifunctional transposon mini-Tn5gfp-km which can be used to select for promoter fusions and report gene expression levels in Agrobacterium tumefaciens. FEMS Microbiol Lett. 1999;179:37–42. doi: 10.1111/j.1574-6968.1999.tb08704.x. [DOI] [PubMed] [Google Scholar]
- Vallenet D, et al. MaGe: a microbial genome annotation system supported by synteny results. Nucleic Acids Res. 2006;34:53–65. doi: 10.1093/nar/gkj406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallenet D, et al. MicroScope: a platform for microbial genome annotation and comparative genomics. Database (Oxford) 2009;2009 doi: 10.1093/database/bap021. bap021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Lis R, Nitschke W, Duval S, Schoepp-Cothenet B. Evolution of arsenite oxidation. In: Santini JM, Ward SA, editors. The metabolism of arsenite. London: CRC Press; 2012. pp. 125–144. [Google Scholar]
- van Lis R, Nitschke W, Duval S, Schoepp-Cothenet B. Arsenics as bioenergetic substrates. Biochim Biophys Acta. 2013;1827:176–188. doi: 10.1016/j.bbabio.2012.08.007. [DOI] [PubMed] [Google Scholar]
- Weiss S, et al. Enhanced structural and functional genome elucidation of the arsenite-oxidizing strain Herminiimonas arsenicoxydans by proteomics data. Biochimie. 2009;91:192–203. doi: 10.1016/j.biochi.2008.07.013. [DOI] [PubMed] [Google Scholar]
- Wilkins MJ, et al. Proteogenomic monitoring of Geobacter physiology during stimulated uranium bioremediation. Appl Environ Microbiol. 2009;75:6591–6599. doi: 10.1128/AEM.01064-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood DW, et al. The genome of the natural genetic engineer Agrobacterium tumefaciens C58. Science. 2001;294:2317–2323. doi: 10.1126/science.1066804. [DOI] [PubMed] [Google Scholar]
- Young JM, Kuykendall LD, Martínez-Romero E, Kerr A, Sawada H. A revision of Rhizobium Frank 1889, with an emended description of the genus, and the inclusion of all species of Agrobacterium Conn 1942 and Allorhizobium undicola de Lajudie et al. 1998 as new combinations: Rhizobium radiobacter, R. rhizogenes, R. rubi, R. undicola and R. vitis. Int J Syst Evol Microbiol. 2001;51:89–103. doi: 10.1099/00207713-51-1-89. [DOI] [PubMed] [Google Scholar]
- Young JPW, et al. The genome of Rhizobium leguminosarum has recognizable core and accessory components. Genome Biol. 2006;7:R34. doi: 10.1186/gb-2006-7-4-r34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng M, et al. Computation-directed identification of OxyR DNA binding sites in Escherichia coli. J Bacteriol. 2001;183:4571–4579. doi: 10.1128/JB.183.15.4571-4579.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong Z, et al. Nucleotide sequence based characterizations of two cryptic plasmids from the marine bacterium Ruegeria isolate PR1b. Plasmid. 2003;49:233–252. doi: 10.1016/s0147-619x(03)00014-3. [DOI] [PubMed] [Google Scholar]
- Zumft WG. Cell biology and molecular basis of denitrification. Microbiol Mol Biol Rev. 1997;61:533–616. doi: 10.1128/mmbr.61.4.533-616.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.