

Abstract
Transposable elements (TEs) are repetitive DNA sequences that can make new copies of themselves that are inserted elsewhere in a host genome. The abundance and distributions of TEs vary considerably among phylogenetically diverse hosts. With the aim of exploring the basis of this variation, we evaluated correlations between several genomic variables and the presence of TEs and non-TE repeats in the complete genome sequence of the Western clawed frog (Silurana tropicalis). This analysis reveals patterns of TE insertion consistent with gene disruption but not with the insertional preference model. Analysis of non-TE repeats recovered unique features of their genome-wide distribution when compared with TE repeats, including no strong correlation with exons and a particularly strong negative correlation with GC content. We also collected polymorphism data from 25 TE insertion sites in 19 wild-caught S. tropicalis individuals. DNA transposon insertions were fixed at eight of nine sites and at a high frequency at one of nine, whereas insertions of long terminal repeat (LTR) and non-LTR retrotransposons were fixed at only 4 of 16 sites and at low frequency at 12 of 16. A maximum likelihood model failed to attribute these differences in insertion frequencies to variation in selection pressure on different classes of TE, opening the possibility that other phenomena such as variation in rates of replication or duration of residence in the genome could play a role. Taken together, these results identify factors that sculpt heterogeneity in TE distribution in S. tropicalis and illustrate that genomic dynamics differ markedly among TE classes and between TE and non-TE repeats.
Keywords: genome evolution, natural selection, African clawed frogs, gene expression, GC content, Xenopus tropicalis
Introduction
Transposable elements (TEs) are repetitive DNA sequences that are capable of making copies of themselves within a host genome (Wicker et al. 2007; Feschotte 2008). TEs are broadly divided into those that replicate with an RNA intermediate (“Class 1”), such as retrotransposons, and those that do not (“Class 2”), such as DNA transposons. Some retrotransposons have long terminal repeat regions (LTR retrotransposons) and some do not (non-LTR retrotransposons). TEs are associated with chromosomal rearrangements, unequal crossing over, altered gene expression, induction of deleterious mutations, and ectopic (nonhomologous) recombination (Lister et al. 1993; Wright et al. 2003; Kazazian 2004; Feschotte 2008; Hollister and Gaut 2009). TEs can influence gene expression through direct mechanisms such as disruption of the reading frame or promoter region, and by indirect mechanisms such as by facilitating antisense transcription or epigenetic silencing (Casacuberta and González 2013). Although clearly deleterious in some cases, TE insertions also may facilitate adaptive response of host genomes to their dynamic environment, for example, by catalyzing genomic dissemination of environmentally sensitive regulatory elements or by acting as vectors for horizontal transfer of genetic information (Casacuberta and González 2013). Repetitive elements comprise huge proportions of some genomes (Biémont and Vieira 2006; Feschotte 2008), and factors that affect TE abundance, mobility, and distribution are thus prominent determinants of genome evolution.
TEs are unevenly distributed among hosts and within hosts; these distributions also differ for different types of TEs (Pritham 2009). Genomic and demographic variables such as host effective population size (Lynch and Conery 2003), mating systems (Wright et al. 2001; Lockton and Gaut 2010), demographic history (Vieira and Biemont 2004; Lockton and Gaut 2010), and TE deletion rates due to recombination (Vitte and Bennetzen 2006) may play distinct roles in influencing TE distributions in different hosts. It is unclear whether variation among species in TE abundance and distribution is a consequence of equilibrium (a balance between TE replication and removal by natural selection, genetic drift, or other host mechanisms) or nonequilibrium phenomena (Le Rouzic et al. 2007; Lynch 2007).
Models for Genome-Wide TE Heterogeneity
Models that have been proposed to account for the nonuniform distribution of TEs in genomes include the “gene disruption” model, the “insertional preference” model, and the “ectopic exchange” model. Each of these nonmutually exclusive models makes several predictions with respect to the genome wide distribution of TEs. The gene disruption model posits that TEs are deleterious when close to genes and that their distribution in genomes is mainly determined by whether they are in or near a gene, and consequently exposed to removal by natural selection (Wright et al. 2003). TE insertion in or near genes can affect gene function at the nucleotide sequence, transcription, or translation level (Cooley et al. 1988; Han et al. 2004; Smarda et al. 2008; Hollister and Gaut 2009) and can modify the expression of nearby genes (Liu et al. 2004; Hollister and Gaut 2009). Thus, the gene disruption model predicts that TEs should be less common in or near functionally important portions of the genome, such as exons, regulatory regions, or other functional regions, when compared with other parts of the genome that lack important function.
The insertional preference model posits that regions of chromatin that are most frequently unwound (or “open”) are more accessible for TE insertion (Bownes 1990). Because genes that are highly expressed tend to be located in genomic regions with open chromatin, the insertional preference model predicts that TEs should be more abundant near highly expressed genes. This is expected to occur especially upstream of genes where the transcriptional machinery binds and chromatin first unwinds (Bownes 1990; Warnefors et al. 2010). Under this model, TEs should be more prevalent near genes that are highly expressed in the germline because insertion in the germline is necessary for inheritance (Warnefors et al. 2010).
The ectopic exchange model posits that the genomic distribution of TEs is mainly the result of natural selection against ectopic recombination between insertions that are located in nonhomologous regions of the genome (Langley et al. 1988; Montgomery et al. 1991). Under the assumption that the meiotic recombination rate is correlated with the ectopic recombination rate, this model predicts a negative correlation between the local recombination rate and TE abundance (Langley et al. 1988). Because TEs presumably recombine more frequently with other (closely related) TEs that have a similar nucleotide sequence, and because the chances of recombination increase with TE length, this model also predicts that longer TEs should be rarer than shorter TEs (Petrov et al. 2011).
These proposed mechanisms that drive TE heterogeneity (gene disruption, insertional preference, and ectopic recombination) appear to operate to different degrees in different lineages and different TE classes. In the fruit fly Drosophila melanogaster, for example, the ectopic recombination model is supported because TE distribution is negatively correlated with local recombination rate (Fontanillas et al. 2007), and purifying selection is stronger on long TEs than on short TEs (Petrov et al. 2011), but there is relatively weak evidence for selection against gene disruption (Bartolomé et al. 2002). In the plant genus Arabidopsis, in contrast, TEs are not negatively correlated with recombination rate but are negatively correlated with gene density, and TE distribution is influenced by mating system and demographic history (Wright et al. 2003; Lockton and Gaut 2010). In the lizard Anolis carolinensis, the ectopic recombination model is supported because recombination between TEs is common, and because long TE insertions may be subject to negative selection (Novick et al. 2011). The effective population sizes of these study organisms differs over multiple orders of magnitude, and this variation, along with demographic variables such as level of inbreeding or population structure, may be key considerations in efforts to understand the determinants of TE heterogeneity across TE classes and phylogenetically diverse host genomes.
TE Dynamics in the Western Clawed Frog Silurana tropicalis
Not surprisingly, most studies of genome-wide heterogeneity in TE distribution have examined species for which complete genome sequences are available, such as humans (Medstrand et al. 2002), mice (Waterston et al. 2002), fruit flies (Charlesworth and Langley 1989; Fontanillas et al. 2007; Petrov et al. 2011), pufferfish (Neafsey et al. 2004), anolis lizards (Novick et al. 2011), and rockcress (Wright et al. 2003). Recently, the complete genome sequence of the Western clawed frog S. tropicalis (also known as Xenopus tropicalis) was reported, adding a novel and phylogenetically distinct data set for study (Hellsten et al. 2010). Similar to humans, about one-third of the genome of S. tropicalis comprises TEs (Hellsten et al. 2010). All major categories of TEs found in other eukaryotes are also found in S. tropicalis, including DNA transposons, retrotransposons, politons, helitrons, and miniature inverted repeat TEs (Feschotte et al. 2002; Hellsten et al. 2010). However, some features of S. tropicalis TE composition appear to be unusual, including a higher diversity of LTR retrotransposons than most other eukaryotes and a high frequency of DNA transposons (72% of all TEs) (Hellsten et al. 2010). Most other animals and plants, in contrast, tend to be dominated by retrotransposons (Mao et al. 2000; Lander et al. 2001; Waterston et al. 2002; Hellsten et al. 2010). Silurana tropicalis is a diploid species and is closely related to over 20 African clawed frog species, all of which are polyploid (Evans 2008). Chromosomal segregation generally relies on nucleotide similarity between a pair of homologous chromosomes in the context of the entire genome (Charlesworth 1991), a factor that could either be diminished or pronounced by TE insertion after a homologous pair is duplicated. Thus, in addition to providing a novel phylogenetic perspective on TE dynamics, the study of repetitive sequences in S. tropicalis is also potentially relevant to the atypically high incidence of polyploid speciation in African clawed frogs.
In this study, we used the complete genome sequence and expression data from S. tropicalis to test for genomic correlates of TE and non-TE repeat distribution. Our overarching goals were to identify factors that influence heterogeneity in the distribution of TEs in the genome and to explore whether TE dynamics differ among TE classes and between TE and non-TE repeats. To this end, we used logistic regression to jointly evaluate the correlation of multiple genomic variables with the probability of TE or non-TE repeat presence within 2,000 bp windows. To explore whether TE dynamics might vary among TE classes, we also collected insertion polymorphism information from DNA transposons, LTR retrotransposons, and non-LTR retrotransposons from wild-caught individuals. Overall, our analyses provide support for the gene disruption model and demonstrate that dynamics differ dramatically among TE classes and between TE and non-TE repeats.
Materials and Methods
Silurana tropicalis Genome
Version 4.1 of the S. tropicalis genome assembly consists of 19,759 scaffolds (Hellsten et al. 2010). A more recent assembly is now available (version 7.1), but because the annotation was not yet complete when we began this study, we focused our analyses on the older assembly. We used a linkage map developed by Wells et al. (2011) to concatenate adjacent scaffolds for our analysis of nonoverlapping genomic windows spanning 2,000 bp. Because some scaffolds mapped to multiple linkage groups, we only concatenated scaffolds that had a one-to-one mapping with a linkage group (see supplementary table S1, Supplementary Material online). The haploid genome size of S. tropicalis is estimated to be 1.7 Giga base pairs (Gbp); about 1.5 Gbp were present in assembly 4.1 and about 1% of these are “N”s (unknown bases). Unknown bases were not considered when calculating proportions in genomic windows. Windows at the ends of scaffolds that were less than 2,000 bp were excluded from the analysis.
Portions of the version 4.1 assembly that had a high Basic Local Alignment Search Tool (BLAST) match (e value < 10−42) with a primate-specific non-LTR retrotransposon (Alu elements; Longo et al. 2011) were presumed contaminated and discarded. These comprised 59,000 bp (0.03%) of the available genome sequence (see supplementary information, Supplementary Material online, for details). We also discarded from the analysis the 2,000-bp windows that were completely filled with TE sequence, which led to the removal of 2,149 (0.3%) of the windows.
TE and Non-TE Repeats
We used RepeatMasker (Smit et al. 2010) version open 3.2.6 and a S. tropicalis-specific TE library from Repbase (Jurka et al. 2005) to find the genomic locations of TEs. We used the default setting for RepeatMasker except for three variables: 1) the “species” parameter was set to “Xenopus tropicalis,” 2) the “lib” parameter was set to the S. tropicalis TE library from Repbase, and 3) “GC” was set to a genome average of 0.4 that we calculated from the version 4.1 genome sequence. We removed putative TEs less than 40 bp with an aim of decreasing the proportion of putative TEs studied that were not actually derived from TEs. The shortest full-length TE in S. tropicalis is 80 bp (Jurka et al. 2005); the 40-bp cutoff led to the removal of approximately 20% of the putative TEs identified by RepeatMasker. Non-TE repeats were those identified by RepeatMasker as “low complexity” or “simple” repeats. These non-TE repeats included mono-, di-, tri-, and tetranucleotide repeats.
Logistic Regression
We investigated the distribution of all TEs, of various TE categories, and of non-TE repeats in the S. tropicalis genome as a response to genomic variables (hereafter “predictor variables”) using logistic regression. This analysis allowed us to quantify the correlation between repeat presence and each predictor variable, while controlling for the correlations between the predictor variables. To make regression coefficients comparable across different predictors, each predictor x was standardized by subtracting its mean and dividing by its standard deviation. Logistic regressions were performed in R (R Core Development Team 2012) using the “lme4” package (Bates et al. 2011). We performed logistic regression on all TEs, all non-TEs, and also on subsets of the data including non-LTR retrotransposons (6.3% of the genome) and DNA transposons (23.1% of the genome) (Wicker et al. 2007; Hellsten et al. 2010). We were unable to fit our logistic model properly to LTR transposons (at 2.2% of the genome, the smallest group we tried).
To explore how and whether TEs of different lengths are differentially distributed, we performed an additional analysis that included only TEs that are greater than 98% of their reference sequence in Repbase, a class we will call “long TEs,” and TEs that are ≤98% of their reference sequence, a class we will call “short TEs.” A concern with this analysis is that there may be a systematic bias in the diagnosis of “short” TEs related to gaps in the genome sequence and the challenge of assembling repetitive regions. This could potentially increase the apparent frequency of “short TEs” if TEs are frequently poorly assembled or incompletely sequenced. For this reason, we excluded from this analysis TEs that were present at the beginning of a scaffold or flanked by unknown sequence (at least 20 Ns in succession on either side). The caveats discussed below notwithstanding, we considered the possibility that these categories roughly reflect TE age, with long TEs being younger than short TEs.
Predictor Variables
We included eight predictor variables that either related directly to proposed mechanisms that influence genome wide TE insertion heterogeneity or that are simply important genomic variables that are potentially correlated with TE insertions. These predictor variables included the following:
- (i and ii) Upstream and downstream distance with respect to genes: On the basis of studies by Hollister and Gaut (2009) and Medstrand et al. (2002), we had an a priori expectation that the relation between TEs and genes (and therefore the effect of natural selection for or against TEs near genes) is nonlinear with respect to the distance of the TE insertion from the gene. We therefore used a function that reflects a leveling-off effect after a certain threshold distance. We therefore transformed both upstream and downstream distance to the closest gene using the function:
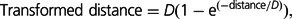
(1)
where D is a characteristic distance chosen a priori. D is calculated from the midpoint of the window to the closest gene either upstream or is downstream. When distance is much less than D, the transformed distance is similar to the distance, but when the distance is large, the transformed distance gradually approaches D rather than increasing without bound. We used a value of D = 1,000 in these statistics under the assumption that regulatory regions tend to occur within approximately 1,000 bp upstream of genes. To be consistent, we used the same value for D for downstream and upstream distances.
(iii and iv) Exon and intron proportions: Hellsten et al. (2010) used homology-based methods with expressed sequence tag (EST) and cDNA data to predict S. tropicalis genes in the version 4.1 genome assembly (http://genome.jgi-psf.org/Xentr4/Xentr4.info.html), resulting in approximately 20,000 gene models. We defined the extent of each gene as the smallest window that included all the components defined about it in this database; these included exons, transcription and/or translation start or stop sites, or codons. We denoted intronic regions as the nucleotide positions between adjacent exons of the same gene.
(v and vi) Somatic and germline expression: To quantify gene expression, we used EST libraries from the National Center for Biotechnology Information (http://www.ncbi.nlm.nih.gov/UniGene). These libraries were generated from 18 different tissue types and six developmental stages, as described in Chain et al. (2011). When more than one library existed for a tissue, we pooled the data for that tissue across libraries. We then BLASTed (Altschul et al. 1997) the ESTs against the transcripts in the gene filtered model from the JGI (http://genome.jgi-psf.org/Xentr4/Xentr4.download.ft.html) to quantify how many transcripts were in each EST library. Our data included EST sequences that did not correspond to predicted genes, as well as predicted genes that did not have any ESTs in the EST libraries. ESTs that did not correspond to predicted genes were excluded from our gene expression analysis. About one-third of the predicted genes did not have any ESTs in any library; we designated their gene expression as zero. Genes were categorized as having “germline” expression if their sequence was present in the EST libraries of ovary, oviduct, testes, or the “embryo_egg” developmental stage. Genes were categorized as having “somatic” expression if their sequence was present in the EST libraries of any other tissues or developmental stages we examined (Chain et al. 2011). A gene therefore could have both “germline” and “somatic” expression. For each gene and tissue, we calculated a total germline and somatic expression (T) across libraries in each category following Chain et al. (2011), where T = ΣLi, and Li is the number of ESTs for the gene divided by the total number of ESTs for a library. Then, for each window, we multiplied the total germline or somatic expression by the number of base pairs of the window that was from an exon of a gene in each category, respectively. For windows that contained exons from more than one gene, these products were summed over all the genes present.
(vii) Conserved regions: As a way of identifying potentially functional noncoding regions, we also included in our analysis conserved regions. These regions were predicted by PhastCon (Siepel et al. 2005) based on a seven-way multiple alignment between human (Homo sapiens), mouse (Mus musculus), rat (Rattus norvegicus), chicken (Gallus gallus), zebrafish (Danio rerio), opposum (Monodelphis domestica), and frog (S. tropicalis). The locations of these regions were obtained from the University of California, Santa Cruz, genome website (http://genome.ucsc.edu/).
(viii) GC content: We calculated the GC content for each window in two ways: 1) as the percentage of the non-N sequence and non-TE sequence that was a G or a C and 2) as the percentage of the non-N sequence that was a G or a C. Because the results from the logistic regressions with each type of GC calculation were similar, we focus on the first and present results from the second approach in supplementary information, Supplementary Material online.
Polymorphism Data
We also quantified TE insertion polymorphism for randomly selected TE insertions sites from the “long TE” category described earlier. TE insertion polymorphism data were collected from 25 insertion sites in 19 wild-caught S. tropicalis individuals, including 15 samples from Ghana, and one each from Nigeria, Sierra Leone, Ivory Coast, and Liberia. For all our assays, TE genotypes (i.e., homozygous for insertion, homozygous for no insertion, or heterozygous) were scored based on the size of at least one polymerase chain reaction (PCR)-amplified product per allele. Put another way, all alleles were genotyped based on at least one successful amplification. For each of nine DNA transposon insertion sites, we were able to use one primer pair with an expected amplification size of approximately 2,000 bp if the insertion was present and a smaller size (∼500 bp) if the insertion was not present. For 12 non-LTR and 4 LTR insertion sites, the TEs were much longer, and we used two or (usually) three nonindependent primer pairs to assay polymorphism. One pair spanned a large (>5 kb) insertion in the S. tropicalis genome sequence. This amplification was expected to produce a product only from alleles that lacked a TE insertion. The other one or two primer pairs were designed from one primer site outside of the TE and the other primer site within the TE, with both amplifying a relatively small (∼800 bp) product if the TE insert was present.
We used a maximum likelihood framework to test whether the TE insertion polymorphism data provided evidence for different selection coefficients on the three TE classes (DNA transposons, LTR retrotransposons, and non-LTR retrotransposons). Here, we assume that TE insertions follow one-way mutational process within an infinite sites framework, wherein each TE insertion occurs in a unique location, and the ultimate evolutionary fate of an inserted TE is either loss or fixation. We do not accommodate the possibility, for example, that a TE might become polymorphic due to a deletion of an allele after fixation.
As detailed in González et al. (2010), the expected population frequency distribution of TE insertions (x) in genomic regions where TE insertions are polymorphic is:
| (2) |
where s is the selection coefficient on TE insertions, which are assumed to have codominant fitness, N is the effective population size, and c is a normalization factor defined, such that the sum of the probabilities of all possible frequencies is 1 (González et al. 2010).
Because the TE insertion sites were initially identified from a single complete genome sequence, we calculated the probability of the observed insertion frequencies conditioning on the observation of an insertion in the genome sequence (González et al. 2010):
| (3) |
We then calculated the binomial probabilities of our observed number of insertions (k), given the number of alleles sampled (n), in an assumed population size of N = 1,000. Thus, the probability T(k) of observing k insertions in n alleles sampled is equal to:
| (4) |
and the normalized probability Θ(k) is:
| (5) |
We repeated the analysis with N = 10,000 to check for consistency. Because the genome sequence was generated from a seventh-generation inbred female (Hellsten et al. 2010), we expect TE insertions to be mostly homozygous. For this reason, we treated the insertion information from the genome sequence as a single allele and, therefore, had polymorphism insertion frequencies from a maximum of 39 alleles for each insertion site (i.e., two alleles from each of 19 diploid wild-caught individuals plus one from the genome sequence). For some insertion sites, there are fewer than 39 alleles genotyped due to PCR failure.
We used a likelihood ratio test to compare two models concerning selection coefficients of TEs. In the first model, the selection coefficient is the same for DNA transposons, LTR retrotransposons, and non-LTR retrotransposons. In the second model, each of these TE classes has a different selection coefficient. Significance of the additional parameters was assessed by comparing −2Δln(L) to the χ2 distribution with two degrees of freedom, where Δln(L) is the difference between the maximum log-likelihood values of the models being compared.
Results
Data
We examined TE presence in 2,000 bp windows in a draft genome sequence of the Western clawed frog S. tropicalis. There was a pronounced disparity in the abundance and length of TE and non-TE repeats in this genome, with DNA transposons comprising the most abundant (∼650,000 fragments) and largest portion of the genome (∼127.9 Mbp). Non-LTR, LTR, and non-TE fragments, by comparison, were fewer in number (∼88,000, ∼9,000, and ∼82,000, respectively) and spanned smaller proportions of the genome (∼27.6 Mbp, 4.7 Mbp, and 6.8 Mbp, respectively). LTR retrotransposon fragments were longer on average (515 bp) than DNA transposons, non-LTR retrotransposons, or non-TE repeats (197 bp, 313 bp, and 83 bp, respectively). Additional descriptions of data, R-scripts for analysis, and input files are provided in supplementary information, Supplementary Material online, and in Dryad (http://dx.doi.org/10.5061/dryad.76487).
Logistic Regression of All TEs, DNA Transposons, and Non-LTR Retrotransposons
Our central goal is to evaluate alternative models for TE heterogeneity (gene disruption and insertional preference) and to test for evidence of distinct dynamics among TE classes. To this end, we used logistic regression to quantify the relationship between a binary variable—the presence or absence of a TE insertion within genomic windows—and various “predictor” variables that characterize features of these genomic windows such as GC content, level of gene expression, distance from genes, and whether the window included a conserved region. Some of these variables, such as the distance from genes, have a direct prediction discussed above associated with a particular model. Other variables, such as GC content, do not necessarily have a prediction associated with a model but are nonetheless potentially important genomic features that may be correlated with heterogeneity in TE insertions. We performed analyses on the entire TE data set and also on TE categories based on the mechanism of transposition, including DNA transposons and non-LTR retrotransposons (LTR retrotransposons were excluded because of a fitting error with the logistic model). Results of the logistic regressions that excluded TEs in the GC calculation are presented in table 1 and figure 1, and results that included TEs in the GC calculation are presented in supplementary table S2 and fig. S1, Supplementary Material online. Null and residual deviances of these analyses are presented in supplementary table S3, Supplementary Material online. All our predictor variables were correlated in some way with abundance of all TEs, a class of TEs, non-TE repeats, or some combination of these, and the correlations were generally similar in magnitude, sign, and significance whether TEs were included in the calculation of GC content. The difference between the null and residual deviances indicates that for all analyses, the predictors provided a significantly improved fit (χ2 test, 8 degrees of freedom).
Table 1.
Coefficients of Logistic Regression
| Predictor | All TEs | TEs by Type |
TEs by Size |
Non-TE Repeat | ||
|---|---|---|---|---|---|---|
| DNA transposon | Non-LTR Retrotransposons | Long | Short | |||
| Exon | −0.14421* | −0.10878* | −0.23647* | −0.08214* | −0.14448* | 0.00180* |
| Intron | 0.00330 | 0.02476* | −0.06380* | 0.01551* | 0.00236 | −0.01565* |
| Downstream distance | 0.07326* | 0.06659* | 0.01910* | 0.08695* | 0.06266* | 0.02147* |
| Upstream distance | 0.06494* | 0.06270* | 0.00589 | 0.08392* | 0.05458* | −0.00894 |
| Germline expression | 0.00588 | 0.00908* | −0.03113* | −0.02406 | 0.00633 | −0.01477* |
| Somatic expression | −0.01789* | −0.01542* | −0.00950 | −0.03282 | −0.01617* | 0.00008 |
| Conserved | −0.14227* | −0.19235* | 0.09414* | −0.17893* | −0.13198* | 0.10243* |
| GC | −0.22590* | −0.22798* | −0.14974* | −0.21848* | −0.22172* | −0.34670* |
Note.—GC content is calculated without including TE.
*Individually significant departure from zero (P < 0.05).
Fig. 1.—

Logistic regression coefficients between genomic variables and the three TE classes and non-TE repeats. GC content was calculated excluding the GC content of TEs. Bars indicate two standard deviations of the correlation coefficients and in most cases are small enough to be hidden by the symbols.
When all TEs were considered collectively, the strongest relation was a negative correlation between TE presence and GC content (table 1). There were also strong negative correlations with the proportion of the window that was exon, and the proportion of the window that was conserved. Positive correlations were observed with distance upstream and downstream from genes. A slightly negative correlation was observed with somatic expression, and the correlation with germline expression was not significant (table 1). When TEs were divided into categories based on mechanism of transposition (DNA transposons and non-LTR retrotransposons), both had strong negative correlations with GC content. Both also had a negative correlation with the proportion of the window that is exon and a positive correlation with distance downstream of genes. However, there were key differences that illustrate distinct genomic dynamics. Unlike DNA transposons, non-LTR retrotransposons had 1) no significant correlation with the distance upstream of genes and a comparatively small positive correlation with distance downstream of genes, 2) a positive correlation with conserved regions, and 3) a negative correlation with germline expression.
Logistic Regression of Long and Short TEs
With an aim of better understanding temporal and structural dynamics of TEs, we categorized TEs into two classes (“long” and “short”) based on their length relative to a full-length consensus sequence. Comparison of the distributions of young and old TEs has the potential to offer insights into mechanisms governing TE distributions (reviewed in Lynch [2007]). Comparison of full length and fragmented TEs potentially also offers insights into temporal dynamics of TE evolution because full length TEs can be converted into fragmented TEs, but fragmented TEs presumably are rarely ever converted into full length TEs. The genome-wide distribution of full length TEs, therefore, might be sculpted by a shorter period of natural selection than fragmented length. However, the age disparity between full length and fragmented TEs is potentially reduced or eliminated by truncation of TEs immediately upon insertion (Ostertag and Kazazian 2001) and by natural selection favoring insertions of truncated as opposed to full length TEs to limit deleterious effects.
For the logistic regression analysis of short and long TEs, 49,343 of 1,077,503 short TE fragments (4.58%) were excluded because they were either flanked by an N or at the beginning of a scaffold, whereas 205 of 35,640 long TE fragments (0.58%) were excluded for this reason. Most (92.7%) of the remaining TEs in the S. tropicalis genome were short, and long and short TEs had significantly distinct correlations with our predictor variables. The long TE class was enriched for DNA transposons, which comprised 93% of the long TEs when compared with 72% of all TEs in the genome (Hellsten et al. 2010). Under-representation of non-LTR retrotransposon in the long TE class is consistent with the observation that 5′-ends of these TEs are frequently truncated upon insertion (Luan et al. 1993). Short TEs were significantly negatively correlated with somatic expression, but no significant correlation was recovered for germline expression for long or short TEs (table 1, supplementary table S2, Supplementary Material online). Short TEs had a more negative correlation with the proportion of windows that is exon than long TEs, although the opposite was true for conserved regions. Long TEs had a more positive correlation with upstream and downstream distance from genes than short TEs. When TEs were excluded from the GC content calculation, the correlation with GC content was negative and of similar magnitude for long and short TEs (table 1, fig. 1). However, when TEs were included in the GC content calculation, the correlation with GC content was positive for long TEs but negative for short TEs (supplementary table S2 and fig. S1, Supplementary Material online), suggesting that the long TEs were GC rich.
Logistic Regression of Non-TE Repeats
To further contextualize heterogeneity of repetitive genomic regions that originate by different mechanisms than TEs, we also performed a logistic regression using non-TE repeats. Non-TE repetitive elements encompass a wide range of nucleotide sequences, including simple sequence repeats that involve the repetition of nucleotide repeats of a few to hundreds of base pairs in length (Richard et al. 2008). Non-TE repeats generally form via slippage of DNA replication (Schlotterer and Tautz 1992) and, similar to TEs, could destabilize the genome due to ectopic recombination (Wang and Leung 2006) or disrupt genes by causing frameshift mutations (Metzgar et al. 2000). If both of these repeat types tend to be deleterious in similar ways, for example, because of gene disruption or ectopic exchange, then they should both be under-represented in similar parts of the genome, which presumably are subject to purifying selection. However, if heterogeneity in TE distributions derives in large part from insertion biases, we would expect to see different distributions of TEs and non-TE repeats. Differences in TE and non-TE repeat distributions also could be related to differences in length or nucleotide composition, which could have unique and difficult to predict fitness consequences.
There were several differences between TE and non-TE repeat distributions in terms of the sign, magnitude, and significance of their correlation with the genomic predictor variables. The most striking difference was that non-TE repeats were not significantly negatively correlated with the proportion of the window that was exon. Another distinction from most of the TE analyses was that the correlation of non-TE repeats with upstream distance was not significant. The correlation with germline expression was similar for TE and for non-TE repeats and was near zero. Also similar to TEs, non-TE repeats had a strong negative correlation with GC content, although the magnitude of this negative correlation was larger for non-TE repeats.
Insertion Polymorphism
To further characterize dynamics in these two TE classes, we collected insertion polymorphism data from long TEs in wild-caught individuals (table 2). Although all the TEs for which insertion polymorphism was quantified were full length (≥98%) with respect to a consensus sequence, the size of the TE insertions depended on the TE class, with DNA transposons being substantially smaller (mean length = 359 bp) than the LTR or non-LTR retrotransposons (mean length = 5,149 bp and 5,064 bp, respectively). As expected, the TE insertion genotypes of an individual from Nigeria (XEN231) were most similar to the genome sequence, which was also generated from an individual from Nigeria. We observed a substantial difference between the frequencies of TE insertion polymorphisms in retrotransposons (LTR and non-LTR) compared with DNA transposons. In DNA transposons, eight of nine sites were fixed for an insertion, and one site had an insertion almost fixed (an insertion was present in 37 of 39 alleles). In non-LTR retrotransposons, only 4 of 12 sites had fixed insertions, and eight had a rare insertion (which we arbitrarily categorized as an insertion with frequency ≤ 10%), including three genomic regions in which none of the wild-caught individuals had an insertion. In LTR retrotransposons, none of four sites had fixed insertions, two had a common insertion, and two had a rare insertion. The average frequency of DNA transposon insertions was 99%, of non-LTR retrotransposon insertions 37%, and of LTR retrotransposon insertions 51%.
Table 2.
TE Insertion Polymorphism Data for Wild-Caught Silurana tropicalis Individuals
| Ghana |
||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sample ID | UWBM5959 | UWBM5961 | UWBM5958 | UWBM5957 | UWBM5962 | UWBM5960 | UWBM5963 | MVZ 254093 | MVZ 245094 | MVZ 245095 | MVZ 245096 | MVZ 245097 | MVZ 245420 | MVZ 252599 | MVZ 252600 | XEN 231 Nigeria | AMNH 17271 Sierra Leone | XEN 228 Ivory Coast | L 1720 Liberia | Summary |
| DNA transposons | ||||||||||||||||||||
| DNA transposon 2 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | NA | F/100 |
| DNA transposon 3 | NA | NA | NA | 1 | NA | 1 | 1 | 1 | NA | 1 | NA | NA | 1 | 1 | 1 | 1 | 1 | 1 | NA | F/100 |
| DNA transposon 5 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 0/1 | 1 | 1 | 0/1 | 1 | 1 | 1 | 1 | C/95 |
| DNA transposon 6 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | F/100 |
| DNA transposon 10 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | F/100 |
| DNA transposon 11 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | F/100 |
| DNA transposon 12 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | F/100 |
| DNA transposon 14 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | F/100 |
| DNA transposon 15 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | F/100 |
| Non-LTR Retrotransposons | ||||||||||||||||||||
| Non-LTR 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | NA | 0 | 0 | 0 | 0 | 0 | 0 | 0/1 | 0 | NA | NA | R/3 |
| Non-LTR 3 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | NA | R/3 |
| Non-LTR 4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | R /3 |
| Non-LTR 5 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | R/3 |
| Non-LTR 6 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | R/8 |
| Non-LTR 7 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0/1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | R/10 |
| Non-LTR 9 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | F/100 |
| Non-LTR 10 | 1 | 1 | 1 | 1 | NA | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | NA | F/100 |
| Non-LTR 11 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0/1 | 0 | 0 | 0 | R/5 |
| Non-LTR 12 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | F/100 |
| Non-LTR 14 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | NA | R/8 |
| Non-LTR 15 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | F/100 |
| LTR retrotransposons | ||||||||||||||||||||
| LTR 3 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 0 | C/95 |
| LTR 4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | R/8 |
| LTR 6 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 0/1 | 1 | 1 | 1 | 1 | 1 | 1 | 0/1 | 0/1 | 0 | C/92 |
| LTR 7 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0/1 | 0/1 | 0 | 0 | R/8 |
Note.—For each locus, individuals are either homozygous for a TE insertion (I), homozygous for no insertion (0), or heterozygous (0/1). Some loci for some individuals did not amplify (NA). A summary of insertion genotype frequencies across all individuals, including the genome sequence (not shown) includes rare (R), intermediate (I), common (C), or fixed (F), followed by the percentage of alleles surveyed that have an insertion. For these calculations, the genome sequence was treated as haploid, so a maximum of 39 alleles were surveyed.
A posteriori justification for assuming a one-way mutation model is provided in supplementary information, Supplementary Material online. We did not recover a significant improvement in the likelihood of the TE polymorphism data when the selection coefficient was estimated independently for each TE class (with N = 1,000 or 10,000, –ln(L) = 45.386 or 45.407) compared with when one selection coefficient was estimated across all TE classes (with N = 1,000 or 10,000, –ln(L) = 47.009 or 47.009, and P = 0.197 or 0.202). Thus, the polymorphism data did not provide evidence for a significant difference in the selection coefficient for different TE classes. Another study that surveyed insertion polymorphism of DNA transposon in S. tropicalis also found a high frequency of fixed insertions (six of eight sites surveyed in five individuals and the genome sequence were fixed for an insertion), with two polymorphic sites having either a rare (27%) or intermediate (64%) frequency insertion (Hikosaka et al. 2007). Including these data in our statistical analysis did not change the result of no significant improvement for the more parameterized model (data not shown).
Discussion
Support for the Gene Disruption Model
We used logistic regression to evaluate the relationships between TE insertions in 2,000 bp windows in the genome of the frog S. tropicalis and genomic attributes including the presence of exons and introns, level of gene expression, distance from genes, GC content, and whether the window included a conserved region. We also collected insertion polymorphism data from a total of 25 TE insertion sites for three TE classes (DNA transposons, LTR and non-LTR retrotransposons). Together this information offers insight into the applicability of various proposed mechanisms that drive heterogeneity in TE distributions and also sheds light on whether genomic dynamics differ between different TE classes. Overall, our analyses provided support for the gene disruption model because TEs tended to be rare in or near functional regions such as exons, upstream regulatory regions, and conserved regions (table 1, supplementary table S2, Supplementary Material online). This pattern was evident in the analysis of all TEs, of long and short TEs, in DNA transposons, and partially in non-LTR retrotransposons (see later). When we compared results from long and short TEs, if the first category is indeed younger than the second, under the gene disruption model we expected (i) a significantly more negative correlation with the proportion of windows that is exon or conserved for short TEs compared with long TEs due removal of TE insertions near functional regions by natural selection. We also expected (ii) a significantly more positive correlation with upstream or downstream distance from genes for short TEs compared with long TEs. Expectation (i) was met for exons and conserved regions when TEs were not included in the calculation of GC content but not when they were included. Expectation (ii) was not met for either gene distance, irrespective of how the GC content was calculated. Differences in these correlations may be driven by natural selection on TE length, differences in the age of long and short TEs, or some combination of these possibilities.
For non-LTR retrotransposons, there was not a significant positive correlation with upstream distance, which is inconsistent with the gene disruption model. The difference between non-LTR retrotransposons and DNA transposons points to distinct but counterintuitive dynamics of each of these TE classes: Non-LTR retrotransposons are approximately 5 times larger than DNA transposons, which could make them more deleterious near genes, yet they were not positively correlated with upstream distance from genes. This is surprising and could be explained by any of many phenomena including a recent increased rate of non-LTR TE transposition or beneficial regulatory consequences of non-LTR TEs upstream of genes. Non-TE repeats were also not positively correlated with distance upstream from genes and additionally were not negatively correlated with exons. This suggests that gene disruption plays a less prominent role in their distribution.
No Support for the Insertional Preference Model
These results do not support the insertional preference model because expression had a small effect on TE presence and because, for non-LTR retrotransposons, a larger negative correlation existed between germline expression and TE presence than between somatic expression and TE presence. If long TEs are younger than short TEs, under the insertional preference model, we expected a more positive correlation with expression intensity (especially germline expression) in long TEs compared with putatively older short TEs, due to the loss of TE insertions near genes over time by natural selection. This expectation also was not met. Analysis of non-TE repeats further undermines the insertional preference model, because the correlation with germline expression was similar, and near zero, for both TE and for non-TE repeats. Overall, these results suggest a negligible role for levels of gene expression in driving differences in the respective distributions of TE and non-TE repeats in S. tropicalis.
Ectopic Recombination Model
It is not possible to conclusively evaluate the strength of the ectopic recombination model here because we lack data on variation in recombination rates in the S. tropicalis genome. If GC content is positively correlated with recombination rates in S. tropicalis, as it appears to be in humans, mice, and fruit flies (Fullerton et al. 2001; Jensen-Seaman et al. 2004; Singh et al. 2005), then the generally observed negative correlation between GC and TE and non-TE repeats is consistent with the ectopic exchange model (table 1, supplementary table S2, Supplementary Material online). Importantly, however, a correlation between GC content and recombination has not, to our knowledge, been demonstrated in amphibians. Furthermore, GC content in other species is known to be correlated with various genomic features that may be independent of ectopic recombination rates, including CpG islands (Jensen-Seaman et al. 2004) and the rate of gene conversion (Galtier 2003). Some TE families have insertion site biases with respect to GC content (Liao et al. 2000), which could further limit the utility of GC content for inferring relationships between TEs and rates of recombination. In any case, a negative association between recombination and TE presence can arise via mechanisms other than the ectopic recombination model: Namely that selection on deleterious TE insertions is less effective in regions with low recombination due to linkage to beneficial mutations (the Hill–Robertson effect; Bartolomé et al. 2002). The much higher frequency of short TEs (∼11-fold higher) than long TEs also is consistent with the ectopic recombination model but could also arise from selection against deleterious effects of large insertions that are not related to ectopic recombination.
Distinct Dynamics among Repeat Classes
The logistic regression recovered substantial differences in genomic dynamics between different TE classes (DNA transposons and non-LTR retrotransposons) and between TE and non-TE repeats. For example, logistic regression results provide strong evidence that TE insertions in exons are deleterious, but we do not find strong evidence that non-TE repeats in exons are deleterious. Non-LTR retrotransposon insertions in exons seem to be more strongly selected against than DNA transposon insertions in exons, but the opposite was true for insertions in conserved regions, which had a strong positive correlation with non-LTR retrotransposons, and for upstream distance, which was not significantly correlated with non-LTR retrotransposons. TE insertion polymorphism data suggest that DNA transposons have a higher frequency of fixed insertions but failed to provide evidence for a significant difference in the selection coefficient among TE classes based on analysis of polymorphic insertions. The difference in the proportion of fixed insertions in each class suggests that 1) natural selection against LTR and non-LTR retrotransposon insertions is stronger than that against DNA transposons but that we lack statistical power to detect this, 2) that our one-way mutation model is not a good approximation for this system, or that 3) nonequilibrium dynamics are at play, such as changes over time in the rate of replication of a TE class.
Conclusions
TEs play a central role in genome evolution by influencing a myriad of factors including genome size, gene expression, and recombination. With a goal of examining TE dynamics in S. tropicalis, we used logistic regression to evaluate the relationship between various genomic features and the presence of TEs and non-TE repeats in the genome sequence of the frog S. tropicalis. Our results point to substantially distinct relationships between different repeat types and these genomic variables, a result that is reinforced by polymorphism data from different TE classes that we collected using a PCR assay. Overall, these findings argue most strongly for a gene disruption model wherein TE insertions in or near genes are generally deleterious, although this model appears to be less applicable to non-LTR retrotransposons than to DNA transposons. We did not recover support for the insertional preference model. Interestingly, a recent study of TEs in Drosophila concluded that variation in the selection coefficient on different TE classes is largely attributable to physical properties of TEs such as length and copy number, as opposed to their mechanism of replication (Petrov et al. 2011), an interpretation that is also not consistent with the insertional preference model.
Repetitive sequences in polyploid genomes can lead to the formation of multivalents during meiosis and to inappropriate chromosomal segregation. The history of genome duplication in African clawed frogs (Evans 2008) thus provides motivation to understand the drivers of genome-wide heterogeneity in TE distribution of S. tropicalis, the only diploid species in this group. If polyploidization is associated with a population bottleneck, ectopic recombination could increase because mildly deleterious TE insertions that were polymorphic in a diploid ancestor could drift to fixation in a polyploid descendant (Hazzouri et al. 2008). However, differential fixation of TEs in paralogous chromosomes in a polyploid genome could also contribute to divergence and facilitate “diploidization"—the formation of bivalents rather than multivalents during meiosis (Wolfe 2001). Additional information on variation in genome-wide levels of recombination in S. tropicalis would permit further evaluation of the ectopic recombination model and thus potentially increase understanding of the unusually high incidence of polyploid speciation in African clawed frogs.
Supplementary Material
Supplementary tables S1–S3 and figure S1 are available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org/).
Acknowledgments
The authors thank Ben Bolker, Philipp Messer, Brian Charlesworth, and two anonymous reviewers for very helpful discussion, and the Museum of Vertebrate Zoology at the University of California at Berkeley and the Burke Museum at the University of Washington for providing tissue samples. This work was supported by grants from the National Science and Engineering Research Council of Canada to B.J.E. and J.D., an Early Researcher Award to B.J.E. from the Ontario Ministry of Economic Development and Innovation, and McMaster University.
Literature Cited
- Altschul SF, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartolomé C, Maside X, Charlesworth B. On the abundance and distribution of transposable elements in the genome of Drosophila melanogaster. Mol Biol Evol. 2002;19:926–937. doi: 10.1093/oxfordjournals.molbev.a004150. [DOI] [PubMed] [Google Scholar]
- Bates D, Maechler M, Bolker B. lme4: linear mixed-effects model using S4 classes. 2011. Available from: http://cran.r-project.org/web/packages/lme4/index.html, CRAN.
- Biémont C, Vieira C. Junk DNA as an evolutionary force. Nature. 2006;443:521–524. doi: 10.1038/443521a. [DOI] [PubMed] [Google Scholar]
- Bownes M. Preferential insertion of P elements into genes expressed in the germ-line of Drosophila melanogaster. Mol Gen Genet. 1990;222:457–460. doi: 10.1007/BF00633856. [DOI] [PubMed] [Google Scholar]
- Casacuberta E, González J. The impact of transposable elements in environmental adaptation. Mol Ecol. 2013;22:1503–1517. doi: 10.1111/mec.12170. [DOI] [PubMed] [Google Scholar]
- Chain FJJ, Dushoff J, Evans BJ. The odds of duplicate gene persistence after polyploidization. BMC Genomics. 2011;12:599. doi: 10.1186/1471-2164-12-599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth B. The evolution of sex chromosomes. Science. 1991;251:1030–1033. doi: 10.1126/science.1998119. [DOI] [PubMed] [Google Scholar]
- Charlesworth B, Langley CH. The population genetics of Drosophila transposable elements. Annu Rev Genet. 1989;23:251–287. doi: 10.1146/annurev.ge.23.120189.001343. [DOI] [PubMed] [Google Scholar]
- Cooley L, Kelley R, Spradling A. Insertional mutagenesis of the Drosophila genome with single P elements. Science. 1988;239:1121–1128. doi: 10.1126/science.2830671. [DOI] [PubMed] [Google Scholar]
- Evans BJ. Genome evolution and speciation genetics of clawed frogs (Xenopus and Silurana) Front Biosci. 2008;13:4687–4706. doi: 10.2741/3033. [DOI] [PubMed] [Google Scholar]
- Feschotte C. Transposable elements and the evolution of regulatory networks. Nat Rev Genet. 2008;9:397–405. doi: 10.1038/nrg2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feschotte C, Zhang X, Wessler SR. Miniature inverted-repeat transposable elements and their relationship to established DNA transposons. In: Craig NL, Craigie R, Gellert M, Lambowitz AM, editors. Mobile DNA II. Washington (DC): ASM Press; 2002. pp. 1147–1209. [Google Scholar]
- Fontanillas P, Hartl DL, Reuter M. Genome organization and gene expression shape the transposable element distribution in the Drosophila melanogaster euchromatin. PLoS Genet. 2007;3:e210. doi: 10.1371/journal.pgen.0030210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fullerton SM, Bernardo Carvalho A, Clark AG. Local rates of recombination are positively correlated with GC content in the human genome. Mol Biol Evol. 2001;18:1139–1142. doi: 10.1093/oxfordjournals.molbev.a003886. [DOI] [PubMed] [Google Scholar]
- Galtier N. Gene conversion drives GC content evolution in mammalian histones. Trends Genet. 2003;19:65–68. doi: 10.1016/s0168-9525(02)00002-1. [DOI] [PubMed] [Google Scholar]
- González J, Karasov TL, Messer PW, Petrov DA. Genome-wide patterns of adaptation to temperate environments associated with transposable elements in Drosophila. PLoS Genet. 2010;6:e1000905. doi: 10.1371/journal.pgen.1000905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han JS, Szak ST, Boeke JD. Transcriptional disruption by the L1 retrotransposon and implications for mammalian transcriptomes. Nature. 2004;429:268–274. doi: 10.1038/nature02536. [DOI] [PubMed] [Google Scholar]
- Hazzouri KM, Mohajer A, Dejak SI, Otto SP, Wright SI. Contrasting patterns of transposable-element insertion polymorphism and nucleotide diversity in autotetraploid and allotetraploid Arabidopsis species. Genetics. 2008;179:581–592. doi: 10.1534/genetics.107.085761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellsten U, et al. The genome of the Western clawed frog Xenopus tropicalis. Science. 2010;328:633–636. doi: 10.1126/science.1183670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hikosaka A, Kobayashi T, Saito Y, Kawahara A. Evolution of the Xenopus piggyBac transposon family TxpB: domesticated and untamed strategies of transposon subfamilies. Mol Biol Evol. 2007;24:2648–2656. doi: 10.1093/molbev/msm191. [DOI] [PubMed] [Google Scholar]
- Hollister JD, Gaut BS. Epigenetic silencing of transposable elements: a trade-off between reduced transposition and deleterious effects on neighboring gene expression. Genome Res. 2009;19:1419–1428. doi: 10.1101/gr.091678.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen-Seaman MI, et al. Comparative recombination rates in the rat, mouse, and human genomes. Genome Res. 2004;14:528–538. doi: 10.1101/gr.1970304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurka J, et al. Repbase update, a database of eukaryotic repetitive elements. Cytogenet Genome Res. 2005;110:462–467. doi: 10.1159/000084979. [DOI] [PubMed] [Google Scholar]
- Kazazian HH., Jr Mobile elements: drivers of genome evolution. Science. 2004;303:1626–1632. doi: 10.1126/science.1089670. [DOI] [PubMed] [Google Scholar]
- Lander ES, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- Langley CH, Montgomery E, Hudson R, Kaplan N, Charlesworth B. On the role of unequal exchange in the containment of transposable element copy number. Genet Res. 1988;52:223–235. doi: 10.1017/s0016672300027695. [DOI] [PubMed] [Google Scholar]
- Le Rouzic A, Boutin TS, Capy P. Long-term evolution of transposable elements. Proc Natl Acad Sci U S A. 2007;104:19375–19380. doi: 10.1073/pnas.0705238104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao G, Rehm EJ, Rubin GM. Insertion site preferencesof the P transposable element in Drosophila melanogaster. Proc Natl Acad Sci U S A. 2000;97:3347–3351. doi: 10.1073/pnas.050017397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister C, Jackson D, Martin C. Transposon-induced inversion in Antirrhinum modifies nivea gene expression to give a novel flower color pattern under the control of cycloidearadialis. Plant Cell. 1993;5:1541–1553. doi: 10.1105/tpc.5.11.1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, He Y, Amasino R, Chen X. siRNAs targeting an intronic transposon in the regulation of natural flowering behavior in Arabidopsis. Genes Dev. 2004;18:2873–2878. doi: 10.1101/gad.1217304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockton S, Gaut BS. The evolution of transposable elements in natural populations of self-fertilizing Arabidopsis thaliana and its outcrossing relative Arabidopsis lyrata. BMC Evol Biol. 2010;10:10. doi: 10.1186/1471-2148-10-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longo MS, O'Neill MJ, O'Neill RJ. Abundant human DNA contamination identified in non-primate genome databases. PLoS One. 2011;6:e16410. doi: 10.1371/journal.pone.0016410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luan DD, Korman MH, Jakubczak JL, Eickbush TH. Reverse transcription of R2Bm RNA is primed by a nick at the chromosomal target site: a mechanism for non-LTR retrotransposition. Cell. 1993;72:595–605. doi: 10.1016/0092-8674(93)90078-5. [DOI] [PubMed] [Google Scholar]
- Lynch M. The origins of genome architecture. Sunderland (MA): Sinauer; 2007. [Google Scholar]
- Lynch M, Conery JS. The origins of genome complexity. Science. 2003;320:140–144. doi: 10.1126/science.1089370. [DOI] [PubMed] [Google Scholar]
- Mao L, et al. Rice transposable elements: a survey of 73,000 sequence-tagged-connectors. Genome Res. 2000;10:982–990. doi: 10.1101/gr.10.7.982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medstrand P, van de Lagemaat LN, Mager DL. Retroelement distributions in the human genome: variations associated with age and proximity to genes. Genome Res. 2002;12:1483–1495. doi: 10.1101/gr.388902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzgar D, Bytof J, Wills C. Selection against frameshift mutations limits microsatellite expansion in coding DNA. Genome Res. 2000;10:72–80. [PMC free article] [PubMed] [Google Scholar]
- Montgomery EA, Huang S-M, Langley CH, Judd BH. Chromosome rearrangement by ectopic recombination in Drosophila melanogaster: genome structure and evolution. Genetics. 1991;129:1085–1098. doi: 10.1093/genetics/129.4.1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neafsey DE, Blumenstiel JP, Hartl DL. Different regulatory mechanisms underlie similar transposable element profiles in pufferfish and fruitflies. Mol Biol Evol. 2004;21:2310–2318. doi: 10.1093/molbev/msh243. [DOI] [PubMed] [Google Scholar]
- Novick PA, Smith JD, Floumanhaft M, Ray DA, Boissinot S. The evolution and diversity of DNA transposons in the genome of the lizard Anolis carolinensis. Genome Biol Evol. 2011;3:1–14. doi: 10.1093/gbe/evq080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostertag EM, Kazazian HH. Biology of mammalian L1 retrotransposons. Annu Rev Genet. 2001;35:501–538. doi: 10.1146/annurev.genet.35.102401.091032. [DOI] [PubMed] [Google Scholar]
- Petrov DA, Fiston-Lavier AS, Lipatov M, Lenkov K, Gonzalez J. Population genomics of transposable elements in Drosophila melanogaster. Mol Biol Evol. 2011;28:1633–1644. doi: 10.1093/molbev/msq337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritham EJ. Transposable elements and factors influencing their success in eukaryotes. J Hered. 2009;100:648–655. doi: 10.1093/jhered/esp065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Development Team. Vienna (Austria): R Foundation for Statistical Computing; 2012. R: a language and environment for statistical computing. [Google Scholar]
- Richard GF, Kerrest A, Dujon B. Comparative genomics and molecular dynamics of DNA repeats in eukaryotes. Microbiol Mol Biol Rev. 2008;72:686–727. doi: 10.1128/MMBR.00011-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlotterer C, Tautz D. Slippage synthesis of simple sequence DNA. Nucleic Acids Res. 1992;20:211–215. doi: 10.1093/nar/20.2.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siepel A, et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005;15:1034–1050. doi: 10.1101/gr.3715005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh ND, Davis JC, Petrov DA. Codon bias and noncoding GC content correlate negatively with recombination rate on the Drosophila X chromosome. J Mol Evol. 2005;61:315–324. doi: 10.1007/s00239-004-0287-1. [DOI] [PubMed] [Google Scholar]
- Smarda P, Bures P, Horova L, Foggi B, Rossi G. Genome size and GC content evolution of Festuca: ancestral expansion and subsequent reduction. Ann Bot. 2008;101:421–433. doi: 10.1093/aob/mcm307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit AFA, Huble R, Green P. RepeatMasker Open 3.0. 2010. Available from: http://www.repeatmasker.org.
- Vieira C, Biemont C. Transposable element dynamics in two sibling species: Drosophila melanogaster and Drosophila simulans. Genetica. 2004;120:115–123. doi: 10.1023/b:gene.0000017635.34955.b5. [DOI] [PubMed] [Google Scholar]
- Vitte C, Bennetzen JL. Analysis of retrotransposon structural diversity uncovers properties and propensities in angiosperm genome evolution. Proc Natl Acad Sci U S A. 2006;103:17638–17643. doi: 10.1073/pnas.0605618103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Leung FC. Long inverted repeats in eukaryotic genomes: recombinogenic motifs determine genomic plasticity. FEBS Lett. 2006;580:1277–1284. doi: 10.1016/j.febslet.2006.01.045. [DOI] [PubMed] [Google Scholar]
- Warnefors M, Pereira V, Eyre-Walker A. Transposable elements: insertion pattern and impact on gene expression evolution in hominids. Mol Biol Evol. 2010;27:1955–1962. doi: 10.1093/molbev/msq084. [DOI] [PubMed] [Google Scholar]
- Waterston RH, et al. Initial sequencing and comparative analysis of the mouse genome. Nature. 2002;420:520–562. doi: 10.1038/nature01262. [DOI] [PubMed] [Google Scholar]
- Wells DE, et al. A genetic map of Xenopus tropicalis. Dev Biol. 2011;354:1–8. doi: 10.1016/j.ydbio.2011.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wicker T, et al. A unified classification system for eukaryotic transposable elements. Nat Rev Genet. 2007;8:973–982. doi: 10.1038/nrg2165. [DOI] [PubMed] [Google Scholar]
- Wolfe K. Yesterday's polyploids and the mystery of diploidization. Nat Rev Genet. 2001;2:333–341. doi: 10.1038/35072009. [DOI] [PubMed] [Google Scholar]
- Wright SI, Agrawal N, Bureau TE. Effects of recombination rate and gene density on transposable element distributions in Arabidopsis thaliana. Genome Res. 2003;13:1897–1903. doi: 10.1101/gr.1281503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright SI, Le QH, Schoen DJ, Bureau TE. Population dynamics of an Ac-like transposable element in self- and cross-pollinating Arabidopsis. Genetics. 2001;158:1279–1288. doi: 10.1093/genetics/158.3.1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.